
Abstract
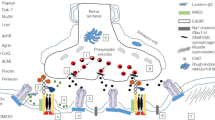
The congenital myasthenic syndromes have now been traced to an array of molecular targets at the neuromuscular junction encoded by no fewer than 11 disease genes. The disease genes were identified by the candidate gene approach, using clues derived from clinical, electrophysiological, cytochemical, and ultrastructural features. For example, electrophysiologic studies in patients suffering from sudden episodes of apnea pointed to a defect in acetylcholine resynthesis and CHAT as the candidate gene (Ohno et al., Proc Natl Acad Sci USA 98:2017–2022, 2001); refractoriness to anticholinesterase medications and partial or complete absence of acetylcholinesterase (AChE) from the endplates (EPs) has pointed to one of the two genes (COLQ and ACHE T ) encoding AChE, though mutations were observed only in COLQ. After a series of patients carrying mutations in a disease gene have been identified, the emerging genotype–phenotype correlations provided clues for targeted mutation analysis in other patients. Mutations in EP-specific proteins also prompted expression studies that proved pathogenicity, highlighted important functional domains of the abnormal proteins, and pointed to rational therapy.








Similar content being viewed by others

References
Barisic, N., Muller, J. S., Paucic-Kirincic, E., et al. (2005). Clinical variability of CMS-EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. European Journal of Paediatric Neurology, 9, 7–12.
Beeson, D., Hantai, D., Lochmuller, H., & Engel, A. G. (2005). Report of the 126th International Workshop: Congenital myasthenic syndromes. Neuromuscular Disorders, 15, 498–512.
Beeson, D., Higuchi, O., Palace, J., et al. (2006). Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science, 313, 1975–1978.
Bon, S., Coussen, F., & Massoulié, J. (1997). Quaternary associations of acetylcholinesterase. II. The polyproline attachment domain of the collagen tail. Journal of Biological Chemistry, 272, 3016–3021.
Byring, R. F., Pihko, H., Shen, X.-M., et al. (2002). Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscular Disorders, 12, 548–553.
Cai, Y., Cronin, C. N., Engel, A. G., Ohno, K., Hersh, L. B., & Rodgers, D. (2004). Choline acetyltransferase structure reveals distribution of mutations that cause motor disorders. EMBO Journal, 23, 2047–2058.
Chevessier, F., Faraut, B., Ravel-Chapuis, A., et al. (2004). MUSK, a new target for mutations causing congenital myasthenic syndrome. Human Molecular Genetics, 13, 3229–3240.
Croxen, R., Newland, C., Beeson, D., et al. (1997). Mutations in different functional domains of the human muscle acetylcholine receptor α subunit in patients with the slow-channel congenital myasthenic syndrome. Human Molecular Genetics, 6, 767–774.
Di Castro, A., Martinello, K., Grassi, F., Eusebi, F., & Engel, A. G. (2007). Pathogenic point mutations in a transmembrane domain of the ε subunit increase the Ca2+ permeability of the human endplate ACh receptor. Journal of Physiology (London), 579, 671–677.
Donger, C., Krejci, E., Serradell, P., et al. (1998). Mutation in the human acetylcholinesterase-associated gene, COLQ, is responsible for congenital myasthenic syndrome with end-plate acetylcholinesterase deficiency. American Journal of Human Genetics, 63, 967–975.
Engel, A. G. (2004). Congenital myasthenic syndromes. In A. G. Engel & C. Franzini-Armstrong (Eds.), Myology (3rd ed., pp. 1801–1844). New York: McGraw-Hill.
Engel, A. G. (2007). The therapy of congenital myasthenic syndromes. Neurotherapeutics, 4, 252–257.
Engel, A. G., & Lambert, E. H. (1987). Congenital myasthenic syndromes. Electroencephalography and Clinical Neurophysiology Supplement, 39, 91–102.
Engel, A. G., Lambert, E. H., & Gomez, M. R. (1977). A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Annals of Neurology, 1, 315–330.
Engel, A. G., Lambert, E. H., Mulder, D. M., et al. (1982). A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Annals of Neurology, 11, 553–569.
Engel, A. G., Ohno, K., Bouzat, C., Sine, S. M., & Griggs, R. G. (1996a). End-plate acetylcholine receptor deficiency due to nonsense mutations in the ε subunit. Annals of Neurology, 40, 810–817.
Engel, A. G., Ohno, K., Milone, M., et al. (1996b). New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Human Molecular Genetics, 5, 1217–1227.
Engel, A. G., Ohno, K., & Sine, S. M. (2003a). Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nature Reviews. Neuroscience, 4, 339–352.
Engel, A. G., Ohno, K., & Sine, S. M. (2003b). Congenital myasthenic syndromes: A diverse array of molecular targets. Journal of Neurocytology, 32, 1017–1037.
Fucile, S., Sucapane, A., Grassi, A., Eusebi, F., & Engel, A. G. (2006). The human adult subtype AChR channel has high Ca2+ permeability. Journal of Physiology (London), 573, 35–43.
Fukudome, T., Ohno, K., Brengman, J. M., & Engel, A. G. (1998). Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. NeuroReport, 9, 1907–1911.
Gomez, C. M., Maselli, R., Gammack, J., et al. (1996). A beta-subunit mutation in the acetylcholine receptor gate causes severe slow-channel syndrome. Annals of Neurology, 39, 712–723.
Gomez, C. M., Maselli, R., Vohra, B. P. S., et al. (2002). Novel delta subunit mutation in slow-channel syndrome causes severe weakness by novel mechanism. Annals of Neurology, 51, 102–112.
Harper, C. M., & Engel, A. G. (1998). Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Annals of Neurology, 43, 480–484.
Harper, C. M., Fukudome, T., & Engel, A. G. (2003). Treatment of slow channel congenital myasthenic syndrome with fluoxetine. Neurology, 60, 170–173.
Hatton, C. J., Shelley, C., Brydson, M., Beeson, D., & Colquhoun, D. (2003). Properties of the human muscle nicotinic receptor, and of the slow-channel myasthenic syndrome mutant εL221F, inferred from maximum likelihood fits. Journal of Physiology (London), 547, 729–760.
Horovitz, A., & Fersht, A. (1990). Strategy for analyzing the co-operativity of intramolecular interactions in peptides and proteins. Journal of Molecular Biology, 214, 613–617.
Kim, A.-R., Rylett, R. J., & Shilton, B. H. (2006). Substrate binding and catalytic mechanism of human acetylcholinesterase. Biochemistry, 45, 14621–14631.
Kimbell, L. M., Ohno, K., Engel, A. G., & Rotundo, R. L. (2004). C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. Journal of Biological Chemistry, 279, 10997–11005.
Kraner, S., Lufenberg, I., Strassburg, H. M., Sieb, J. P., & Steinlein, O. K. (2003). Congenital myasthenic syndrome with episodic apnea in patients homozygous for a CHAT missense mutation. Archives of Neurology, 60, 761–763.
Maselli, R. A., Chen, D., Mo, D., Bowe, C., Fenton, G., & Wollman, R. L. (2003). Choline acetyltransferase mutations in myasthenic syndrome due to deficient acetylcholine resynthesis. Muscle and Nerve, 27, 180–187.
Milone, M., Wang, H.-L., Ohno, K., et al. (1997). Slow-channel syndrome caused by enhanced activation, desensitization, and agonist binding affinity due to mutation in the M2 domain of the acetylcholine receptor alpha subunit. Journal of Neuroscience, 17, 5651–5665.
Mora, M., Lambert, E. H., & Engel, A. G. (1987). Synaptic vesicle abnormality in familial infantile myasthenia. Neurology, 37, 206–214.
Mukhtasimova, N., & Sine, S. M. (2007). An inter-subunit trigger of channel gating in the muscle nicotinic receptor. Journal of the Neurological Sciences, 27, 4110–4119.
Ohno, K., Hutchinson, D. O., Milone, M., et al. (1995). Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the ε subunit. Proceedings of the National Academy of Sciences of the United States of America, 92, 758–762.
Ohno, K., Wang, H.-L., Milone, M., et al. (1996). Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor ε subunit. Neuron, 17, 157–170.
Ohno, K., Brengman, J. M., Tsujino, A., & Engel, A. G. (1998a). Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proceedings of the National Academy of Sciences of the United States of America, 95, 9654–9659.
Ohno, K., Milone, M., Brengman, J. M., Lo Monaco, M., Evoli, A., Tonali, P., et al. (1998b). Slow-channel congenital myasthenic syndrome caused by a novel mutation in the acetylcholine receptor ε subunit. Neurology, 50, A432. Ref Type: Abstract.
Ohno, K., Wang, H.-L., Shen, X.-M., et al. (2000). Slow-channel mutations in the center of the M1 transmembrane domain of the acetylcholine receptor α subunit. Neurology, 54(Suppl 3), A183.
Ohno, K., Tsujino, A., Brengman, J. M., et al. (2001). Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proceedings of the National Academy of Sciences of the United States of America, 98, 2017–2022.
Okada, K., Inoue, A., Okada, M., et al. (2006). The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science, 312, 1802–1805.
Rothbart, H. B. (1937). Myasthenia gravis. Familial occurrence. JAMA, 108, 715–717.
Schmidt, C., Abicht, A., Krampfl, K., et al. (2003). Congenital myasthenic syndrome due to a novel missense mutation in the gene encoding choline acetyltransferase. Neuromuscular Disorders, 13, 245–251.
Selcen, D., Milone, M., Shen, X.-M., et al. (2008). Dok-7 myasthenia: Phenotypic and molecular genetic studies in 16 patients. Annals of Neurology, 64, 71–87.
Shen, X.-M., Ohno, K., Adams, C., & Engel, A. G. (2002). Slow-channel congenital myasthenic syndrome caused by a novel epsilon subunit mutation in the second AChR transmembrane domain. Journal of the Neurological Sciences, 199(Suppl. 1), S96.
Shen, X.-M., Ohno, K., Sine, S. M., & Engel, A. G. (2005). Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle AChR M3–M4 linker. Brain, 128, 345–355.
Shen, X.-M., Deymeer, F., Sine, S. M., & Engel, A. G. (2006). Slow-channel mutation in AChR αM4 domain and its efficient knockdown. Annals of Neurology, 60, 128–136.
Shen, X.-M., Fukuda, T., Ohno, K., Sine, S. M., & Engel, A. G. (2008). Congenital myasthenia-related AChR ä subunit mutation interferes with intersubunit communication essential for channel gating. Journal of Clinical Investigation, 118, 1867–1876.
Sine, S. M., & Engel, A. G. (2006). Recent advances in Cys-loop receptor structure and function. Nature, 440, 448–455.
Sine, S. M., Ohno, K., Bouzat, C., et al. (1995). Mutation of the acetylcholine receptor α subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron, 15, 229–239.
Slater, C. R., Fawcett, P. R. W., Walls, T. J., et al. (2006). Pre- and postsynaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain, 127, 2061–2076.
Wang, H.-L., Auerbach, A., Bren, N., Ohno, K., Engel, A. G., & Sine, S. M. (1997). Mutation in the M1 domain of the acetylcholine receptor alpha subunit decreases the rate of agonist dissociation. Journal of General Physiology, 109, 757–766.
Wang, H.-L., Ohno, K., Milone, M., et al. (2000). Fundamental gating mechanism of nicotinic receptor channel revealed by mutation causing a congenital myasthenic syndrome. Journal of General Physiology, 116, 449–460.
Acknowledgements
Work in our laboratories was supported by NIH grants to A.G. Engel (NS-6277) and to S.M. Sine (NS-31744) and by a Muscular Dystrophy Association grant to A.G. Engel.
Author information
Authors and Affiliations
Corresponding author
Additional information
Proceedings of the XIII International Symposium on Cholinergic Mechanisms
Rights and permissions
About this article
Cite this article
Engel, A.G., Shen, XM., Selcen, D. et al. What Have We Learned from the Congenital Myasthenic Syndromes. J Mol Neurosci 40, 143–153 (2010). https://doi.org/10.1007/s12031-009-9229-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-009-9229-0