
Abstract
Fish aquaculture is considered to be one of the most sustainable sources of protein for humans. Many different species are cultured worldwide, but among them, marine flatfishes comprise a group of teleosts of high commercial interest because of their highly prized white flesh. However, the aquaculture of these fishes is seriously hampered by the scarce knowledge on their biology. In recent years, various experimental ‘omics’ approaches have been applied to farmed flatfishes to increment the genomic resources available. These tools are beginning to identify genetic markers associated with traits of commercial interest, and to unravel the molecular basis of different physiological processes. This article summarizes recent advances in flatfish genomics research in Europe. We focus on the new generation sequencing technologies, which can produce a massive amount of DNA sequencing data, and discuss their potentials and applications for de novo genome sequencing and transcriptome analysis. The relevance of these methods in nutrigenomics and foodomics approaches for the production of healthy animals, as well as high quality and safety products for the consumer, is also briefly discussed.
Similar content being viewed by others

Introduction
Aquaculture is becoming an increasingly important source of fish protein available for human consumption. Fish meal is characterized by its high content in proteins and lipids (carbohydrates represent lower than 0.5 %) (Huss 1995). Although the chemical composition is highly dependent on the species, age, sex, environment, migratory behaviour or season, fish proteins represent a valuable source of essential amino acids including lysine, methionine and cysteine. By contrast, lipid content ranges between 0.3 and 45 % (w/w), but particularly in cold-water marine species of fish includes an important fraction (~40 %) of omega-3 polyunsaturated fatty acids (PUFAs) that have been associated with the prevention and treatment of cardiovascular disease, cancer and some other inflammatory disorders in humans (Hibbeln et al. 2006; Kolakowska et al. 2003). Moreover, fish meal carries other essential nutrients such as B vitamins as well as vitamin A and D in fatty fish, minerals (calcium and phosphorus) and some micronutrients essential for metabolism and endocrinological regulation such as iodine, fluorine and selenium.
Different freshwater and marine fish species are being cultured worldwide, and among these, the flatfishes represent an important food resource. Flatfishes are considered as low-fat fish (2–4 % fat) with a firm, white, mild tasting flesh highly accepted by the consumers. As in other cold-water marine species of fish, they possess a high content of PUFAs such as eicosapentaenoic (EPA) and docosahexaenoic (DHA) acids (71–93 mg EPA/100 g and 106–292 mg DHA/100 g) (Hibbeln et al. 2006). Certain flatfishes, such as some flounders (Hippoglossoides dubius, H. pinetorum, Glyptocephalus stelleri) and plaice (Acanthopsetta nadeshnyi), have a high percentage of very long-chain 24:6(n-3) fatty acids (6–9 % of total fatty acids in the flesh) associated with their diet based on invertebrate such as polychaeta, crustaceans and molluscs (Ota et al. 1994). The possibility to transform them into fillets for multiple commercial preparations represents an added value that converts flatfish into a highly appreciated seafood product.
Flatfishes comprise a relatively large group of fishes, mostly marine, which show unique developmental and reproductive processes. This includes a remarkable metamorphic alteration in bauplan during the larval to juvenile transition, and sophisticated courtship behaviours or unusual gametogenesis in the adults. However, the lack of basic knowledge of the control of these mechanisms hampers the farming of flatfishes and the establishment of a sustainable and profitable aquaculture industry. In recent years, different ‘omic’ technologies have been applied to flatfish research to enhance the knowledge of the biology of these species by unravelling the complex genetic control underlying different physiological processes. Although we are still some way from developing wide applications for flatfish aquaculture, it is expected that these ‘omics’ approaches will have a profound impact in the near future. In this short review, we summarize the advances in genomic research in flatfishes currently being cultured in Europe, and discuss the potentials and applications of new DNA sequencing technologies.
The aquaculture of flatfish
A total of 716 flatfish species belonging to 123 genera and about 11 families have been reported worldwide (reviewed by Munroe 2005). In the Northeast Atlantic area, there exist a total of 11 relevant species for fisheries belonging to the teleost order Pleuronectiformes. This includes Pleuronectidae, such as North Sea plaice (Pleuronectes platessa), Atlantic halibut (Hippoglossus hippoglossus) and winter flounder (Pseudopleuronectes americanus), Soleidae, such as the common sole (Solea solea) and Senegalese sole (S. senegalensis), and Bothidae, such as turbot (Scophthalmus maximus), brill (Scophthalmus rhombus) and megrim (Lepidorhombus whiffiagonis). In 1998, the number of flatfish landings in the Atlantic region reached 104,671 metric tons (MT) for plaice, 31,194 MT for common sole and 5,431 MT for turbot (Millner et al. 2005). However, due to their economic importance, increased fishing pressure has resulted in a drastic drop of flatfish landings with some difficulties to meet the current demands (ICES 2008). Also, overexploitation of wild stocks has reduced genetic diversity in plaice (Hoarau et al. 2005), and is thought to underlie modified life-history traits with a shift towards earlier sexual maturation at smaller size in sole and plaice (Mollet et al. 2007; van Walraven et al. 2010). As a consequence, the development of aquaculture for some of these species has been proposed to supplement the demands for human consumption while reducing the pressure on natural populations.
Currently, flatfish production is still much lower than that of salmonids or sea basses and sea breams. Within Europe, the main flatfishes being cultured are the turbot and Atlantic halibut, and a lesser extent the common sole and the Senegalese sole. The aquaculture production of turbot is the highest among flatfishes, reaching 9,142 MT in 2009 (FEAP 2010), and it is predicted to double in size by 2014. The Atlantic halibut is the largest of pleuronectid flatfishes and has been identified as an ideal species for farming at higher latitudes because of its high growth rate in the relatively cold northern waters (0–14 °C) (Bromage et al. 2000). However, although the production of halibut is now successfully underway, improvements in efficiency remain a major challenge (Naylor and Burke 2005). The Senegalese sole is also a promising candidate, particularly in Southern Europe, because of its fast growth rates. A significant increase in the production of this species has occurred in the last years mainly due to the development of recirculation technologies and advances in larval culture and husbandry procedures. Nevertheless, its production is still low around 200 MT/year (Imsland et al. 2003; APROMAR 2011). The production of common sole has similarly remained stable in central Europe at around 40 MT/year.
The sustainability of flatfish aquaculture, as well as of other fishes, relies on a better understanding of the biology and nutritional requirements of each target species. Thus, identification and characterization of the genes and genetic networks controlling traits of commercial interest such as growth rates, reproduction, larval development and disease resistance, would allow for a better optimization of production and management procedures in the industry. Limited implementation of effective selective breeding programs, as well as the poor knowledge concerning pathologies and its prevention, has been highlighted as some of the major problems in turbot culture (Millán et al. 2011). Similarly, in Atlantic halibut and soles, current obstacles associated with the control of reproduction in captivity, ongrowing diets and optimization of larval culture, and nutrition to reduce malformations and pigmentation anomalies and improve growth and disease resistance are hard to overcome due to the scarce knowledge of the physiological mechanisms involved (Weltzien et al. 2004; Agulleiro et al. 2006; Conceição et al. 2007).
The progressive replacement of proteins and lipids in commercial fish feeds from sources other than fish is another essential issue for the continued growth and intensification of aquaculture production in order to decrease the exploitation of wild populations (Naylor et al. 2009; Welch et al. 2010; Ardura et al. 2012). Flatfish occupy a high trophic position (trophic-level value = 3.5) similar to that of Atlantic salmon (Salmo salar), Atlantic cod (Gadus morhua) or hake (Merluccius sp.) (Duarte et al. 2009) with an estimated demand for fish meal and fish oil ranging from 50 to 70 % and 8 to 12 %, respectively (Tacon and Metian 2008). There has been some success in replacing fish meal and fish oil in commercial feeds by using plant feedstuffs in fish feeds, including oilseeds, legumes and cereal grains, which traditionally have been used as protein or energy concentrates, as well as novel products developed through various processing technologies (Day and Plascencia-González 2000; Choi et al. 2004; Grisdale-Helland et al. 2002; Gatlin et al. 2007; Naylor et al. 2009; Bengtson and Nardi 2010; Valente et al. 2011). However, although important advances have been achieved in terms of percentage of protein substitution and establishment of non-lipid-rich diets without affecting growth or nutrient utilization of flatfish, further research is required to improve diets and avoid some adverse effects associated with toxic factors or unbalanced nutrients provoking hepatic damage (Valente et al. 2011). Moreover, the role of alternative food products such as zooplankton (i.e. krill) and micro- and macro-algae needs to be explored to determine their effects on digestion, absorption, growth and the immune system to attain a more sustainable flatfish aquaculture.
Flatfish genomics research
During the last years, an important effort has been directed towards the use of functional genomics, proteomics and metabolomics to better characterize reproduction, development, nutrition, immunity and toxicology of flatfishes (reviewed by Cerdà et al. 2008, 2010; Forné et al. 2010). Such comprehensive functional analyses aim at identifying the critical genes and molecules that control these physiological processes in order to improve current flatfish farming technologies. In addition, ‘omics’ approaches can identify suitable biomarkers to monitor the welfare of cultured fish and the quality of aquaculture products. However, because farmed flatfishes do not represent biological models for basic or biomedical research, the genomic information for these species has remained very limited and it has thus been necessary to generate the data de novo.
Transcriptome sequencing employing automated classical chain-termination or dideoxy methods, Sanger sequencing, has been the most utilized approach in flatfish genomics to date, allowing the construction of expressed sequence tag (EST) databases. These platforms have been successfully used in turbot, Atlantic halibut as well as in Senegalese sole to investigate the expression of individual genes, or to design and construct cDNA- and oligo-based, species-specific microarrays for the holistic analysis of changes in gene expression (Cerdà et al. 2010; Millán et al. 2010). EST databases are also used as a source of genetic markers, including microsatellites or single nucleotide polymorphisms (SNPs), which may be useful for genome mapping studies (Cerdà et al. 2010; Sha et al. 2010). Microarray-based gene expression profiling is expanding our knowledge of the different genes involved in gametogenesis, larval development and nutrition, immunology and pathology, toxicology and even in population genetics (reviewed by Cerdà et al. 2010). However, due to the high cost and time-consuming nature of the Sanger sequencing methods, EST databases were very limited, and microarrays for turbot, Atlantic halibut and soles could only analyse about 10,000 genes at the most. This number is low considering that a whole transcriptome in vertebrates is composed of approximately 20,000–25,000 protein coding RNAs. To these figures, we have to add alternative splicing of protein-coding RNAs, small and non-coding RNAs (ncRNA) (approximately 16,000 in mice), and several different types of antisense RNA. Such RNA variants make up about three quarters of a transcriptionally active mammalian genome (Frith et al. 2005; Suzuki and Hayashizaki 2004). It is thus evident that the genomic resources currently available for flatfishes are still too scarce to gather a profound knowledge of the biology of these species, and therefore they should be augmented. To achieve this goal, the use of new sequencing technologies that allow massive-scale DNA sequencing is now being applied.
Next-generation sequencing (NGS) technologies
Next-generation sequencing technologies have drastically transformed the way researchers can address genomic questions on non-model species, including flatfishes. The distinct NGS platforms can quickly generate (from hours to days) an enormous bulk of genomic information (from megabases to gigabases) even for those species with limited or no previous genomic information available. In contrast to the Sanger sequencing approach, it is possible to generate massive transcriptome and genome data in a feasible and cost-effective way. Moreover, the rapid development of new and improved equipment, consumables and methodologies has led to a rapid reduction in cost per base pair sequenced, making it easier to incorporate NGS technologies as routine laboratory techniques. Although bioinformatics and database design still remain grand challenges, the high number of applications of NGS, from gene expression and genome mapping to microRNA (miRNA) characterization and epigenetics, makes these new technologies the preferred tools for flatfish genomic research.
Currently, several NGS platforms that differ in chemistry, yield of genomic information and costs have been developed. These can be divided into two major types: second (SGS)- and third (TGS)-generation sequencing platforms. The SGS requires a PCR amplification step (emulsion PCR or bridge PCR) prior to the sequencing reaction. Thereafter, the clusters of DNA templates are sequenced by synthesis or ligation in a phased approach. There are three main SGS platforms (reviewed by Glenn 2011; Metzker 2010; Voelkerding et al. 2009): (a) the 454 platform (Roche Applied Science), based on emulsion PCR followed by pyrosequencing reactions to produce approximately 0.4–0.5 gigabases (Gb) per run rendering sequences of 300–400 base pairs (bp) mean length (although a new chemistry is ongoing to reach 600–700 bp mean length) (e.g. 454 FLX Titanium sequencer); (b) the Solexa-Illumina® platform (Illumina, Inc.), which uses bridge PCR for DNA amplification and dye-labelled terminators in a polymerase-mediated reaction for sequencing, and produces 200–300 Gb/run with a sequence mean length of 100 bp (e.g. HiSeq 2000 sequencer); and (c) the Solid™ (Life Technologies), based on a ligase reaction of dye-labelled octamers with a 2-bases recognition code, and results in 50–75 Gb/run and a mean read length of 50–75 bp (e.g. Solid-5500 sequencer).
To overcome potential biases introduced by the PCR amplification step in the SGS, the TGS technologies have emerged which are based on single-molecule DNA sequencing. The higher simplicity of these latter techniques improves the sequencing speed and reduces the error read rates, the amounts of starting material and the costs (reviewed by Schadt et al. 2010). The main TGS platforms are PacBio (Pacific Biosciences of California, Inc.), GridION (Oxford Nanopore Technologies Ltd.) and HeliScope™ (Helicos BioSciences Corporation), although new platforms are under development (Schadt et al. 2010; Pareek et al. 2011). An additional emerging technology is the ion torrent, considered as an intermediate approach between the SGS and TGS because it does not require any laser, fluorescent dyes or cameras, reducing drastically the costs and generating an important volume of genomic information, although it is still dependent on a PCR amplification step (Schadt et al. 2010). For more technical and methodological aspects about SGS and TGS platforms, extensive reviews describing and comparing chemical costs, read performance and some other characteristics are available (Metzker 2010; Schadt et al. 2010; Glenn 2011; Pareek et al. 2011; Zhang et al. 2011a).
Applications of NGS in flatfish genomics
The generation of large numbers of sequence reads at high competitive costs has favoured the use of the NGS platforms for many applications. These include whole-genome sequencing, bacterial artificial chromosome (BAC) analysis and target re-sequencing, single nucleotide polymorphism (SNP) and mutation identification, transcriptome analysis (RNA-seq), small RNA profiling, analysis of epigenetic markers and chromatin structure using seq-based methods (ChIP-seq, methyl-seq and Dnase-seq) or metagenomics (Metzker 2010; Glenn 2011). Each NGS platform shows advantages or disadvantages depending on the application of interest. In general, those platforms that produce long reads are optimal for de novo genome and transcriptome characterization since long sequences are more amenable for computational analysis and assembly, whereas those producing shorter reads are often targeted for re-sequencing, expression profiling and metagenomics. However, the continuous development of advanced NGS technologies has resulted in most of them being used for a wide range of applications (Glenn 2011). Although the application of NGS in flatfish genomics research has just started, we will summarize the main applications in which these technologies are currently being used.
De novo genome sequencing
Whole-genome sequencing is one of the most challenging applications of NGS to accelerate biological and applied research in non-model fish species. Fish genome size greatly varies between taxonomic groups. Massive genomes have been described in lungfishes with C values, amount of DNA contained within a haploid nucleus, comprised between 81 and 225 pg (Hardie and Hebert 2004). In the most evolved group of ray-finned fishes, the smallest genome size was reported for pufferfish (Tetraodon nigroviridis) with 0.35 pg/haploid genome, and the largest for the shortnose sturgeon (Acipenser brevirostrum) with 13.8 pg (Hardie and Hebert 2004). In flatfishes, the mean C values are 0.6–0.7 pg, ranging from 0.4 pg in Plaice to 1.1 pg in California tonguefish (Symphurus atricaudus) (www.genomesize.com). In these teleosts, the number of chromosomes ranges between 21 pairs in Soleidae and Cynoglossidae, and 24 for Pleuronectidae and Paralichthyidae (Table 1). Altogether, these data indicate that flatfish genomes are small and compact, which should facilitate de novo whole-genome sequencing, assembly and further characterization. However, if we consider the mean flatfish genome size indicated above, and we assume neither bias in sequencing reads nor the time required for the preparation of genomic libraries, the mean time to obtain a number of reads equivalent to a flatfish genome with a 20× coverage by using NGS would be 1–11 days (estimated mean speed of 50 Mb/h for 454 platform or ~1,000 Mb/h for Illumina) (Glenn 2011). However, although a high volume of genomic information can be generated in a short time with NGS methods, the strategy followed for the construction of the genomic libraries, sequence read length and quality, and genome structure (repetitive sequences, gene duplications, etc.) still represents major issues for bioinformatics, data management and de novo assembly.
Massive genome sequencing of some flatfishes is underway. In 2010, the 1,000 Plant and Animal Reference Genomes Project (http://ldl.genomics.cn/) began the whole-genome sequencing of the Tongue sole (Cynoglossus semilaevis) and Japanese (olive) flounder (Paralichthys olivaceus), and in 2011, the International Consortium AQUAGENET (www.aquagenet.eu) initiated the sequencing of the Senegalese sole genome. Probably, new initiatives will soon emerge for the sequencing of the genome of other farmed flatfishes. Although limited information about sequencing strategies and assemblies is still available, length reads and strategies to orientate adequately the scaffolds and contigs are critical. For the Senegalese sole, as in other de novo eukaryotic genomes published until now, a two-step strategy is being followed: scaffolding using matepair (Illumina®) and long paired-end libraries (454) with insert sizes comprised between 2 kb and 20 kb, followed by massive whole-genome shotgun sequencing (454 and Illumina paired-ends) to be assembled into smaller contigs (Huang et al. 2009; Dalloul et al. 2010; Li et al. 2010; Suen et al. 2011; Woycicki et al. 2011). However, de novo assembly of large and complex eukaryotic genomes is still challenging due to repetitive DNA and, in fish, by additional whole-genome duplication (WGD) events that occurred in the teleost lineage (3R and 4R in salmonids). In this regard, BACs represent a useful tool for superscaffolding due to their large size (40–200 kb), and the unbiased and deep haploid genome coverage (Schulte et al. 2011). There are BAC libraries available for most flatfishes of commercial interest with adequate size and coverage to be used for de novo assembly and NGS sequencing data (Table 1). Moreover, these BACs can also be very useful for complementary techniques such as fluorescent in situ hybridization (FISH), as employed in the Senegalese sole to validate relative BAC positioning between scaffolds and anchor them to specific chromosomes (Ponce et al. 2011).
Transcriptome sequencing
Next-generation sequencing of the transcriptome or RNA-seq is one the main applications in flatfish aquaculture because it opens new possibilities for the analysis of the transcriptome complexity under particular experimental or production situations. RNA-seq can evaluate the complete set of transcripts in a given species in one sample, including mRNAs, their splicing and gene boundaries, ncRNAs and small RNAs, such as microRNAs (miRNAs), PIWI-interacting RNAs (piRNAs) and small nucleolar (snoRNAs) as well as small interfering (siRNAs) RNAs. Although most RNA-seq analyses in fish have been applied to gene discovery, identification of splice variants and quantification of gene expression (Meijer et al. 2005; Aanes et al. 2011; Coppe et al. 2010; Fu et al. 2011; Vesterlund et al. 2011; Zhang et al. 2011b), other applications such as SNP identification (Canovas et al. 2010; Yang et al. 2011), allele-specific gene expression (Degner et al. 2009), or interaction transcriptome-epigenome (Elling and Deng 2009), are also possible and will probably be explored in the near future.
As mentioned earlier, most farmed fish lack a reference genome and in most cases the transcriptome information available is very scarce. However, this is not limiting for RNA-seq approaches since both Illumina or 454 platforms are powerful tools for de novo assembly and transcriptome profiling of different eukaryotes including non-model fish species (Coppe et al. 2010; Crawford et al. 2010; Mu et al. 2010; Surget-Groba and Montoya-Burgos 2010; Zhang et al. 2011b). The potential of RNA-seq for whole-transcriptome analysis has been demonstrated recently in zebrafish (Danio rerio), a well-established model for genetics and developmental biology with an annotated transcriptome of 16,416 genes (www.vega.sanger.ac.uk). In this species, RNA-seq analysis during embryonic development generated an average of 108,216 novel transcribed regions (NTRs) in each library, of which an average of 4,067 had no previous supporting information (Aanes et al. 2011). The identification of NTRs is an interesting application of NGS-based RNA-seq in flatfishes, as demonstrated in the Senegalese sole after 454 transcriptome analysis and comparison with a BAC of 57,915 kb bearing three main coding genes (g-type lysozyme, peptidoglycan recognition protein II and mucolipin 1), which allowed for the identification of 24 new NTRs (Ponce et al. 2011).
Despite these advances, the RNA-seq approach still presents some challenging issues. For example, library construction is a critical step that will depend on the RNA-seq application. For small RNA quantification, only the ligation of an adaptor sequence is required, whereas for long mRNAs fragmentation into smaller pieces (200–500 bp) is necessary. Moreover, the discovery of new transcripts or the detection of rare transcripts requires an additional step of normalization and removal of highly abundant transcripts (Christodoulou et al. 2011). In some other cases, a strand-specific library is required to accurately identify antisense transcripts, boundaries of adjacent genes transcribed on opposite strands or to discriminate adequately between the expression of coding or non-coding overlapping transcripts (Wang et al. 2009; Levin et al. 2010). A second critical point in RNA-seq is the depth of coverage. In humans, protein-coding regions (exome) represent approximately 1 % of the human genome corresponding to approximately 30 Mb (Ng et al. 2009). For this transcriptome size, it is estimated that less than 10 million reads of 35 nt in length (~10× coverage) would accurately quantify mRNA expression levels for more than 80 % of the genes, 200 million reads would be required to quantify splicing levels for 80 % of genes (~200× coverage) and approximately 700 million reads would be necessary to obtain accurate quantification for more than 95 % of the expressed transcripts (~800× coverage) (Blencowe et al. 2009; Tarazona et al. 2011). However, these studies have been carried out in humans with a reference genome that requires less sequencing depth to reconstruct full-length transcripts. For the assembly of de novo transcriptomes, sequencing with a 30× coverage might be required for the same task (Martin et al. 2010). Finally, the assembly and mapping are other major issues in RNA-seq. Unlike small RNAs, reconstruction of the full-length transcripts previous to mapping requires an adequate bioinformatic platform and the development of assembly algorithms especially designed to identify and resolve unambiguous transcript variants from the same gene. Until now, three strategies have been used for this purpose: a reference-based strategy, a de novo strategy or a combination of both. The former is only possible in model-species where the genome is well-annotated and curated, which demands lower bioinformatic effort. The de novo strategy has been successfully applied in prokaryotes and lower eukaryotic transcriptomes. However, in higher eukaryotes, this approach is more challenging requiring higher computing resources to identity alternatively spliced variants. In the combined strategy, a reference genome helps for sequence assembly and data mining for de novo assemblers to detect novel and trans-spliced transcripts (reviewed by Martin and Wang 2011).
RNA-seq and microarrays
Both RNA-seq and microarrays allow for a more holistic evaluation of gene expression in a given organism. However, the high volume of information generated by RNA-seq, the progressive reduction of costs and the capacity to evaluate different RNA families, makes this technique a powerful method that can complement or, in some cases, surpass the information generated with microarrays. The main advantages and disadvantages of RNA-seq and microarrays are depicted in Table 2, where technical constraints including sensitivity, specificity or requirements for previous transcriptome information, as well as some other more pragmatic issues such as difficulty, costs or bioinformatics, have been considered. The high performance of RNA-seq to produce sequences without previous genomic information makes these techniques the most powerful approaches for gene discovery and de novo characterization of a whole transcriptome. In this way, the information generated by RNA-seq can be used for the development of custom microarrays for systematic transcriptome profiling (Wong et al. 2011), and other high-throughput tools for quantitative real-time PCR (qPCR), such as the OpenArray® Real-Time PCR System (Life technologies) based on nanoliter qPCR (Morrison et al. 2006; Dixon et al. 2009). The information generated by RNA-seq and microarrays is thus complementary, highly congruent and in most cases allows for an integrative analysis of cellular pathways. A high correlation for mRNA abundance (0.7–0.9) between microarray- and RNA-seq-based gene expression analyses has been demonstrated (Malone and Oliver 2011; Marioni et al. 2008), and matches differentially expressed genes ranges between 62 and 81 % (Brunskill et al. 2011; Malone and Oliver 2011; Marioni et al. 2008). Small discrepancies usually have been associated with sequencing depth and the number of reads to satisfactorily cover the genome in RNA-seq, and therefore a higher correlation between microarray and RNA-seq data is found at higher sequencing depth (genes mapped by more than 32 reads) (Marioni et al. 2008). As a consequence, arrays may have an advantage in measuring differential gene expression for low-abundance transcripts when RNA-seq is not deep enough (Bloom et al. 2009). Moreover, due to their intrinsic methodological properties, microarrays and RNA-seq approaches usually identify different sets of differentially expressed genes (Bradford et al. 2010; Brunskill et al. 2011; Malone and Oliver 2011; Marioni et al. 2008), which suggests that a combination of RNA-seq and custom microarrays represents a powerful strategy to uncover wider transcriptome responses.
Changes in transcriptome during larval development and nutrigenomics
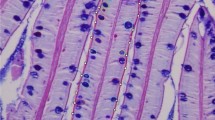
The aquaculture of flatfishes requires the optimization of the culture and nutritional conditions to assure the mass production of juveniles. Currently, high mortality of larvae on start-feeding diets or elevated incidences of skeletal malformations and pigment abnormalities remain bottlenecks for the development of the industry. This is in part due to the unique and critical process of metamorphosis that flatfishes experience during development when larvae shift from a planktonic to a benthic mode of life (Power et al. 2008). This process not only involves morphological changes in eye migration and skin pigmentation but also important modifications of physiology and behaviour. However, the basic biological mechanisms involved, as well as the nutritional requirements that flatfishes demand during metamorphosis, are largely unknown, although it is recognized that this process is controlled by thyroid hormones (Miwa et al. 1988; Solbakken et al. 1999; Power et al. 2008; Manchado et al. 2008). In support of this is the observation that goitrogens, substances that suppress the function of the thyroid gland, can block metamorphosis and even provoke some malformations and malpigmentations (Manchado et al. 2008; Fig. 1). In addition, dietary components, such as lipids and vitamins, and environmental conditions, such as photoperiod, can also affect the metamorphic process (e.g. Cañavate et al. 2006; Lall and Lewis-McCrea 2007).

Skeletal malformations and abnormal skin pigmentation in farmed Senegalese sole. a Normal juvenile. b Albino juveniles produced in a hatchery. Complete (c) or partial (d) malpigmentation after experimental treatment of pre-metamorphic larvae with thiourea (Manchado et al. 2008). e Individual showing a head malformation
The optimization of species-specific diets for flatfishes, the replacement of fish proteins by plant proteins in the feeds for flatfish larvae or juveniles, as well as the substitution of live feeds by artificial microdiets in larvae, would benefit the aquaculture industry. The development of species-specific diets is important since not all flatfishes show the same nutritional requirements, i.e. they can be fish-feeders, crustacean-feeders and polychaete/mollusc-feeders (de Groot 1971). The design of microdiets is also of high interest because with these methods the amount and quality of the dietary components can be controlled. In this regard, microencapsulation, a technique whereby stable particles with minimal nutrient leaching are produced, has been used for the production of larval diets (Langdon 2003). The digestibility of the capsules, the ability to retain nutrients, the nutrient load in the encapsulated particle, the durability and the minimal use of toxic substances, are the key qualities in the design of microencapsulated diets (Yúfera et al. 2005).
Oligo-based microarray analysis has been performed in Atlantic halibut to assess gene expression changes in response to dietary modifications (e.g. replacement of fish meal protein by soy proteins), or whether the expression of genes involved in digestion and absorption of macronutrients are altered in the larvae as a result of the introduction of a microencapsulated diet (Murray et al. 2009, 2010). The first study showed that Atlantic halibut juveniles tolerate up to 30 % soy protein in the diet, although microarray analysis showed that the expression of several immune markers and genes involved in detoxification increased (Murray et al. 2009). Microdiet particles can be readily consumed by halibut larvae after a period of adaptation, providing an adequate source of nutrients with no significant increase in mortality (Murray et al. 2010). However, growth of the larvae seems to be impaired and accompanied by an increased incidence of malpigmentation of the eye and the skin, which correlates with changes in the expression of genes involved in pigmentation and eye development (Murray et al. 2010). These studies thus suggest that microarray analysis can be a suitable approach to optimize nutritional manipulations in flatfishes and to establish associated physiological alterations.
RNA-seq methods have been recently used to investigate changes in gene expression during flatfish development. In one study on the Japanese flounder, Solexa sequencing of a small RNA library was conducted to identify miRNAs potentially involved during metamorphosis (Fu et al. 2011), since these small non-coding RNA molecules are known to regulate gene expression during fish development (Begemann 2008). This study discovered 140 conserved miRNAs and 57 miRNA: miRNA* pairs, and a further validation using miRNA microarrays identified 66 differentially expressed miRNAs at two different metamorphic stages (Fu et al. 2011). Some of these miRNA is related to muscle development and skin pigmentation indicating that these processes are likely to be highly regulated during flounder metamorphosis. This work suggests that the analysis of flatfish miRNA expression would be helpful for further understanding of the functions of miRNAs in flatfish metamorphic development, which could help to explain the complex genetic network that controls the process of flatfish metamorphosis. Additional RNA-seq analyses during larval metamorphosis are ongoing for Senegalese sole to determine changes in gene expression during head remodelling (Power DM, personal communication) or in the whole larvae during different metamorphic stages (M. Manchado, personal communication), in turbot to study larval development (P. Martinez, personal communication), and in common sole to study the effects of retinoic acid, a metabolite of vitamin A (X. Cousin, personal communication).
The analysis of the transcriptome of the innate immune and osmoregulatory systems has also been recently carried out in Senegalese sole larvae by using 454 sequencing (Manchado et al. 2011). De novo assembly yielded 117,152 contigs with approximately 28,000 annotated genes for the innate immune system library, and 73,026 contigs and 35,000 annotated genes for the osmoregulation library. Global assembly generated a total of 164,741 contigs with a coverage of ~60 Mb, suggesting the presence of a high number of specific transcripts in both libraries. A database containing all this information is available (http://www.juntadeandalucia.es/agriculturaypesca/ifapa/aquagenet/soleaDB), which is being used to design a novel and improved oligomicroarray for Senegalese sole.
Genetic maps and detection of quantitative trait loci (QTLs)
Genetic linkage maps are powerful tools in breeding programs and genome evolution studies. They allow for the identification and location of genomic regions associated with monogenic or complex traits of aquaculture interest to be applied in marker-assisted selection programs (Canario et al. 2008). Moreover, genetic maps provide a useful tool to study genome organization and for gathering information about syntenic relationships across species and identification of putative candidate genes associated with QTLs (Castaño-Sanchez et al. 2010; Sarropoulou et al. 2008). Although genetic maps have been developed for turbot and Atlantic halibut (Reid et al. 2007; Bouza et al. 2007), the recent application of NGS has allowed the identification of a high number of genetic markers useful for high-density maps. In the turbot, for instance, the first generation map had 26 linkage groups (LG) based on 242 anonymous microsatellites and with an average marker distance of 6.5 centimorgan (cM) (Bouza et al. 2007). Later, a second map using 158 different anonymous markers identified 21 and 30 LG in the male and female maps, respectively, although with higher intermarker spacing (9.9 and 12.9 cM, respectively) (Ruan et al. 2010). A new high-density map for turbot using new codominant markers identified from NGS analysis is ongoing (P. Martínez, personal communication).
Genetic maps are essential for the identification of QTLs to improve flatfish aquaculture. Several QTLs linked to growth, sex differentiation and disease resistance have been described. In turbot, up to eleven significant QTLs for weight (LG5, LG14, LG15 and LG16), length (LG5, LG6, LG12, LG14 and LG15) and Fulton’s condition factor (LG3 and LG16) (Sánchez-Molano et al. 2011) have been identified. Also, a QTL associated with turbot body length was detected on male LG7 (Ruan et al. 2010). The identification of sex markers is also a major issue in flatfish aquaculture because of the higher size of females. Some flatfishes such as the tongue sole exhibit a pair of morphologically distinct sex chromosomes (WZ/ZZ) (Zhuang et al. 2006). However, no sex-linked chromosome heteromorphisms have been described in turbot (Bouza et al. 2007) or Senegalese sole (Cross et al. 2006). Recently, a sex determining region was located at the proximal end of LG5 in turbot, close to the SmaUSC-E30 marker (Martinez et al. 2009). This marker has been associated with comparative genomics to other sex-associated QTLs in tilapia (Oreochromis sp), and indirectly to the LG8 of the three-spined stickleback (Gasterosteus aculeatus) (Martinez et al. 2009; Sarropoulou et al. 2008). In the tongue sole, a female-specific DNA marker, located in the sex-linked LG5 (Liao et al. 2009), has also been identified (Chen et al. 2007). Regarding disease resistance, Aeromonas salmonicida resistance-related traits and a major locus on LG15 (marker Poli.9-8TUF) for resistance to lymphocystis disease have been reported in turbot (Rodríguez-Ramilo et al. 2011) and Japanese flounder (Fuji et al. 2006, 2007), respectively. The latter trait in Japanese flounder has been successfully applied as a selection breeding marker. In this species, the toll-like receptor 2 has recently been reported as a putative candidate linked to the Poli.9-8TUF marker responsible for lymphocystis resistance (Hwang et al. 2011). In addition, some loci associated with resistance against streptococcal disease caused by Streptococcus iniae (LG7, LG10, LG11 and LG17), and some alleles of MHC class IIB gene that confer protection against Vibrio anguillarum (Xu et al. 2008) have also been found in this species.
Future prospects
It is generally agreed that the application of ‘omics’ technologies in flatfish aquaculture, as in other farming activities, may significantly contribute to promote this industry. However, in most cases, there will be still some time until practical applications can be developed purely based on advances in genomics, since current knowledge on flatfish genetics and physiology remains scarce. For some purposes, such as genetic breeding programs, the application of genomics might be faster since de novo sequencing of genomes and further identification of genes and genetic markers, and the construction of genetic linkage maps, may allow a relatively rapid recognition of QTLs for marker-assisted selection in flatfish. Therefore, the sequencing of the genomes of cultured flatfish must be a priority for the upcoming years. In this scenario, the development of NGS and their multiple applications, which can generate an enormous volume of genomic information in a short time, will undoubtedly speed up the discovery of genes, markers and genetic traits of commercial interest.
The use of NGS in flatfish genomics is however facing important challenges related to bioinformatics. Specialized bioinformatic platforms are essential to manage, store and analyse genomic data efficiently, and therefore they will become indispensable when more fish genomes are sequenced. The optimization of algorithms for de novo assembly and statistical analyses of sequencing data, the creation of databases for commercially important flatfish species, as well as the development of specific bioinformatic applications adapted to the new emerging sequencing technologies, are thus crucial tasks if we are to exploit fully the potentials of genomics in flatfish aquaculture.
The NGS technologies and bioinformatic tools are also extremely useful for functional genomics approaches since they can allow a complete evaluation of the transcriptome in a given tissue under specific experimental conditions, thus obtaining a broader picture of the physiological regulatory factors involved. In this regard, a particular area of growing interest in flatfish genomics is nutrigenomics. The nutritional conditions have a marked effect on larval development and metamorphosis of flatfishes, as well as in juvenile growth and disease resistance, and therefore they need to be optimized for each species and developmental stage. Transcriptome analysis can be a powerful approach to optimize the dietary components. However, the influence of the feeding and nutritional conditions of fish on the quality and nutritive value of the products for human consumption, which can be included in the recently proposed new discipline ‘Foodomics’ (Cifuentes 2009), remains poorly investigated. Foodomics has been defined as a discipline that studies food and nutrition domains through the application of advanced ‘omics’ technologies in which mass spectrometry techniques are considered indispensable (Cifuentes 2009). Thus, nutrigenomics and foodomics approaches, which can be used to produce healthy animals as well as high-quality and safe products for the consumer, are likely to emerge as promising research areas for a sustainable and profitable flatfish aquaculture.
References
Aanes H, Winata CL, Lin CH, Chen JP, Srinivasan KG, Lee SG, Lim AY, Hajan HS, Collas P, Bourque G, Gong Z, Korzh V, Alestrom P, Mathavan S (2011) Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res 21(8):1328–1338
Agulleiro MJ, Anguis V, Cañavate JP, Martínez-Rodríguez G, Mylonas CC, Cerdà J (2006) Induction of spawning of captive-reared Senegal sole (Solea senegalensis) using different delivery systems for gonadotropin-releasing hormone agonist. Aquaculture 257(1–4):511–524
Ardura A, Horreo JL, Hernandez E, Jardon A, Pola IG, Martinez JL, Garcia-Vazquez E (2012) Forensic DNA analysis reveals use of high trophic level marine fish in commercial aquaculture fish meals. Fish Res 115–116:115–120
Asociación Empresarial de Productores de Cultivos Marinos de España (APROMAR) (2011) La Acuicultura Marina de Peces en España. Ministerio de Medio Ambiente y Medio Rural y Marino, 76 pp (in Spanish)
Begemann G (2008) MicroRNAs and RNA interference in zebrafish development. Zebrafish 5(2):111–119
Bengtson D, Nardi G (2010) Culture of summer flounder. In: Daniels HV, Watanabe WO (eds) Practical flatfish culture and stock enhancement. Wiley, New York, pp 65–81
Blencowe BJ, Ahmad S, Lee LJ (2009) Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes. Genes Dev 23(12):1379–1386
Bloom JS, Khan Z, Kruglyak L, Singh M, Caudy AA (2009) Measuring differential gene expression by short read sequencing: quantitative comparison to 2-channel gene expression microarrays. BMC Genomics 10:221
Bouza C, Sanchez L, Martinez P (1994) Karyotypic characterization of turbot (Scophthalmus maximus) with conventional, fluorochrome, and restriction endonuclease banding techniques. Mar Biol 120(4):609–613
Bouza C, Hermida M, Pardo BG, Fernandez C, Fortes GG, Castro J, Sanchez L, Presa P, Pérez M, Sanjuan A, de Carlos A, Alvarez-Dios JA, Ezcurra S, Cal RM, Piferrer F, Martínez P (2007) A microsatellite genetic map of the turbot (Scophthalmus maximus). Genetics 177(4):2457–2467
Bradford JR, Hey Y, Yates T, Li Y, Pepper SD, Miller CJ (2010) A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling. BMC Genomics 11:282
Bromage N, Mazorra C, Bruce M, Brown N, Shields R (2000) Halibut culture. In: Stickney RR (ed) Encyclopedia of aquaculture. Wiley, New York, pp 425–432
Brown NP, Bromage NR, Penman DJ, Shields RJ (1997) The karyotype of the Atlantic halibut, Hippoglossus hippoglossus (Linnaeus). Aquac Res 28(7):489–491
Brunskill EW, Lai HL, Jamison DC, Potter SS, Patterson LT (2011) Microarrays and RNA-Seq identify molecular mechanisms driving the end of nephron production. BMC Dev Biol 11:15
Canario AVM, Bargelloni L, Volckaert F, Houston RD, Massault C, Guiguen Y (2008) Genomics toolbox for farmed fish. Rev Fish Sci 16(Supp 1):3–15
Cañavate JP, Zerolo R, Fernández-Diaz C (2006) Feeding and development of Senegal sole (Solea senegalensis) larvae reared in different photoperiods. Aquaculture 258(1–4):368–377
Canovas A, Rincon G, Islas-Trejo A, Wickramasinghe S, Medrano JF (2010) SNP discovery in the bovine milk transcriptome using RNA-Seq technology. Mamm Genome 21(11–12):592–598
Castaño-Sanchez C, Fuji K, Ozaki A, Hasegawa O, Sakamoto T, Morishima K, Nakayama I, Fujiwara A, Masaoka T, Okamoto H, Hayashida K, Tagami M, Kawai J, Hayashizaki Y, Okamoto N (2010) A second generation genetic linkage map of Japanese flounder (Paralichthys olivaceus). BMC Genomics 11:554
Cerdà J, Bobe J, Babin PJ, Admon A, Lubzens E (2008) Functional genomics and proteomic approaches for the study of gamete formation and viability in farmed finfish. Rev Fish Sci 16(Supp 1):54–70
Cerdà J, Douglas S, Reith M (2010) Genomic resources for flatfish research and their applications. J Fish Biol 77(5):1045–1070
Chen SL, Li J, Deng SP, Tian YS, Wang QY, Zhuang ZM, Sha ZX, Xu JY (2007) Isolation of female-specific AFLP markers and molecular identification of genetic sex in half-smooth tongue sole (Cynoglossus semilaevis). Marine Biotechnol 9(2):273–280
Choi SM, Wang X, Park GJ, Lim SR, Kim KW, Bai SC, Shin IS (2004) Dietary dehulled soybean meal as a replacement for fish meal in fingerling and growing olive flounder Paralichthys olivaceus (Temminck et Schlegel). Aquac Res 35(4):410–418
Christodoulou DC, Gorham JM, Herman DS, Seidman JG (2011) Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Curr Protoc Mol Biol Chapter 4 Unit 4 12
Cifuentes A (2009) Food analysis and foodomics. J Chromatogr A 1216(43):7109
Conceição LCE, Ribeiro L, Engrola S, Aragão C, Morais S, Lacuisse M, Soares F, Dinis MT (2007) Nutritional physiology during development of Senegalese sole (Solea senegalensis). Aquaculture 268(1–4):64–81
Coppe A, Pujolar JM, Maes GE, Larsen PF, Hansen MM, Bernatchez L, Zane L, Bortoluzzi S (2010) Sequencing, de novo annotation and analysis of the first Anguilla anguilla transcriptome: EeelBase opens new perspectives for the study of the critically endangered European eel. BMC Genomics 11:635
Crawford JE, Guelbeogo WM, Sanou A, Traore A, Vernick KD, Sagnon N, Lazzaro BP (2010) De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology. PLoS ONE 5(12):e14202
Cross I, Merlo A, Manchado M, Infante C, Canavate JP, Rebordinos L (2006) Cytogenetic characterization of the sole Solea senegalensis (Teleostei: Pleuronectiformes: Soleidae): Ag-NOR, (GATA)n, (TTAGGG)n and ribosomal genes by one-color and two-color FISH. Genetica 128(1–3):253–259
Cuñado N, Terrones J, Sanchez L, Martinez P, Santos JL (2001) Synaptonemal complex analysis in spermatocytes and oocytes of turbot, Scophthalmus maximus (Pisces, Scophthalmidae). Genome 44(6):1143–1147
Dalloul RA, Long JA, Zimin AV, Aslam L, Beal K, Le Blomberg A, Bouffard P, Burt DW, Crasta O, Crooijmans RP, Cooper K, Coulombe RA, De S et al (2010) Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): genome assembly and analysis. PLoS Biol 8(9):e1000475
Day OJ, Plascencia-González HG (2000) Soybean protein concentrate as a protein source for turbot Scophthalmus maximus L. Aquac Nutr 6(4):221–228
de Groot SJ (1971) On the interrelationships between morphology of the alimentary tract, food and feeding behaviour in flatfishes (Pisces: Pleuronectiformes). Nether J Sea Res 5(2):121–196
Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK (2009) Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25(24):3207–3212
Dixon JM, Lubomirski M, Amaratunga D, Morrison TB, Brenan CJ, Ilyin SE (2009) Nanoliter high-throughput RT-qPCR: a statistical analysis and assessment. Biotechniques 46(6):2–8
Duarte CM, Holmer M, Olsen Y, Soto D, Marba N, Guiu J, Black K, Karakassis I (2009) Will the oceans help feed humanity? Bioscience 59(11):967–976
Elling AA, Deng XW (2009) Next-generation sequencing reveals complex relationships between the epigenome and transcriptome in maize. Plant Signal Behav 4(8):760–762
FEAP (2010) Production and Price reports of member associations of the FEAP. Federation of European Aquaculture Producers. http://www.aquamedia.org/home/default2_en.asp
Forné I, Abián J, Cerdà J (2010) Fish proteome analysis: model organisms and non-sequenced species. Proteomics 10(4):858–872
Frith MC, Pheasant M, Mattick JS (2005) The amazing complexity of the human transcriptome. Eur J Hum Genet 13(8):894–897
Fu Y, Shi Z, Wu M, Zhang J, Jia L, Chen X (2011) Identification and differential expression of microRNAs during metamorphosis of the Japanese flounder (Paralichthys olivaceus). PLoS ONE 6(7):e22957
Fuji K, Kobayashi K, Hasegawa O, Coimbra M, Sakamoto T, Okamoto N (2006) Identification of a single major genetic locus controlling the resistance to lymphocystis disease in Japanese flounder (Paralichthys olivaceus). Aquaculture 254(1–4):203–210
Fuji K, Hasegawa O, Honda K, Kumasaka K, Sakamoto T, Okamoto N (2007) Marker-assisted breeding of a lymphocystis disease-resistant Japanese flounder (Paralichthys olivaceus). Aquaculture 272(1–4):291–295
Gatlin DM III, Barrows FT, Brown P, Dabrowski K, Gaylord TG, Hardy RW, Herman E, Hu G, Krogdahl Å, Nelson R, Overturf K, Rust M, Sealey W, Skonberg D, Souza EJ, Stone D, Wilson R, Wurtele E (2007) Expanding the utilization of sustainable plant products in aquafeeds: a review. Aquac Res 38:551–579
Glenn TC (2011) Field guide to next-generation DNA sequencers. Mol Ecol Res 11(5):759–769
Grisdale-Helland B, Helland SJ, Baeverfjord G, Berge GM (2002) Full-fat soybean meal in diets for Atlantic halibut: growth, metabolism and histology. Aquac Nutr 8(4):265–270
Hardie DC, Hebert PDN (2004) Genome-size evolution in fishes. Can J Fish Aquat Sci 61:1636–1646
Hibbeln JR, Nieminen LRG, Blasbalg TL, Riggs JA, Lands WEM (2006) Healthy intakes of n-3 and n-6 fatty acids: estimations considering worldwide diversity. Am J Clin Nutr 83(6):1483S–1493S
Hoarau G, Boon E, Jongma DN, Ferber S, Palsson J, Van der Veer HW, Rijnsdorp AD, Stam WT, Olsen JL (2005) Low effective population size and evidence for inbreeding in an overexploited flatfish, plaice (Pleuronectes platessa L.). Proc R Soc B 272:497–503
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P, Ren Y, Zhu H, Li J et al (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41(12):1275–1281
Huss HH (1995) Quality and quality changes in fresh fish. FAO Fisheries Technical Paper 348, pp 195
Hwang SD, Fuji K, Takano T, Sakamoto T, Kondo H, Hirono I, Aoki T (2011) Linkage Mapping of Toll-Like Receptors (TLRs) in Japanese Flounder, Paralichthys olivaceus. Mar Biotechnol 13(6):1086–1091
ICES (2008) Report of the study group on mixed fisheries management (SGMixMan). ICES CM 2008/ACOM:23
Imsland AK, Foss A, Conceição LEC, Dinis MT, Delbare D, Schram E, Kamstra A, Rema P, White P (2003) A review of the culture potential of Solea solea and S. senegalensis. Rev Fish Biol Fish 13(4):379–407
Kolakowska A, Olley J, Dunstan GA (2003) Fish lipids. In: Sikorski ZE, Kolakowska A (eds) Chemical and functional properties of food lipids. CRC press, New York, pp 228–271
Lall SP, Lewis-McCrea LM (2007) Role of nutrients in skeletal metabolism and pathology in fish—an overview. Aquaculture 267(1–4):3–19
Langdon C (2003) Microparticle types for delivering nutrients to marine fish larvae. Aquaculture 227(1–4):259–275
Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A (2010) Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7(9):709–715
Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, Zhang Z, Zhang Y, Wang W et al (2010) The sequence and de novo assembly of the giant panda genome. Nature 463(7279):311–317
Liao X, Ma HY, Xu GB, Shao CW, Tian YS, Ji XS, Yang JF, Chen SL (2009) Construction of a genetic linkage map and mapping of a female-specific DNA marker in half-smooth tongue sole (Cynoglossus semilaevis). Mar Biotechnol 11(6):699–709
Malone JH, Oliver B (2011) Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol 9:34
Manchado M, Infante C, Asensio E, Planas JV, Cañavate JP (2008) Thyroid hormones down-regulate thyrotropin beta subunit and thyroglobulin during metamorphosis in the flatfish Senegalese sole (Solea senegalensis Kaup). Gen Comp Endocrinol 155(2):447–455
Manchado M, Infante C, Ponce M, Planas JV, Cañavate JP (2011) Massive transcriptome sequencing in sole. In: Howell B (ed) Sole Workshop V. CCMAR, University of the Algarve, Faro, Portugal
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18(9):1509–1517
Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nat Rev Genet 12(10):671–682
Martin J, Bruno VM, Fang Z, Meng X, Blow M, Zhang T, Sherlock G, Snyder M, Wang Z (2010) Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genomics 11:663
Martinez P, Bouza C, Hermida M, Fernandez J, Toro MA, Vera M, Pardo B, Millan A, Fernandez C, Vilas R, Viñas A, Sanchez L, Felip A, Piferre F, Ferreiro I, Cabaleiro S (2009) Identification of the major sex-determining region of turbot (Scophthalmus maximus). Genetics 183(4):1443–1452
Mechaly AS, Viñas J, Murphy C, Reith M, Piferrer F (2010) Gene structure of the Kiss1 receptor-2 (Kiss1r-2) in the Atlantic halibut: insights into the evolution and regulation of Kiss1r genes. Mol Cell Endocrinol 317(1–2):78–89
Meijer AH, Verbeek FJ, Salas-Vidal E, Corredor-Adamez M, Bussman J, van der Sar AM, Otto GW, Geisler R, Spaink HP (2005) Transcriptome profiling of adult zebrafish at the late stage of chronic tuberculosis due to Mycobacterium marinum infection. Mol Immunol 42(10):1185–1203
Metzker ML (2010) Sequencing technologies - the next generation. Nat Rev Genet 11(1):31–46
Millán A, Gomez-Tato A, Fernandez C, Pardo BG, Alvarez-Dios JA, Calaza M, Bouza C, Vázquez M, Cabaleiro S, Martinez P (2010) Design and performance of a turbot (Scophthalmus maximus) oligo-microarray based on ESTs from immune tissues. Mar Biotechnol 12(4):452–465
Millán A, Gómez-Tato A, Pardo BG, Fernández C, Bouza C, Vera M, Alvarez-Dios JA, Cabaleiro S, Lamas J, Lemos ML, Martínez P (2011) Gene expression profiles of the spleen, liver, and head kidney in turbot (Scophthalmus maximus) along the infection process with Aeromonas salmonicida using an immune-enriched oligo-microarray. Mar Biotechnol 13(6):1099–1114
Millner R, Walsh SJ, Díaz de Astarloa JM (2005) Atlantic flatfish fisheries. In: Gibson RN (ed) Flatfishes: biology and exploitation. Blackwell Publishing, Oxford, pp 240–271
Miwa S, Tagawa M, Inui Y, Hirano T (1988) Thyroxine surge in metamorphosing flounder larvae. Gen Comp Endocrinol 70:158–163
Mollet FM, Kraak SBM, Rijnsdorp AD (2007) Fisheries-induced evolutionary changes in maturation reaction norms in North Sea sole Solea solea. Mar Ecol Prog Ser 351:189–199
Morrison T, Hurley J, Garcia J, Yoder K, Katz A, Roberts D, Cho J, Kanigan T, Ilyin SE, Horowitz D, Dixon JM, Brenan CJ (2006) Nanoliter high throughput quantitative PCR. Nucleic Acids Res 34(18):e123
Mu Y, Ding F, Cui P, Ao J, Hu S, Chen X (2010) Transcriptome and expression profiling analysis revealed changes of multiple signaling pathways involved in immunity in the large yellow croaker during Aeromonas hydrophila infection. BMC Genomics 11:506
Munroe TA (2005) Systematic diversity of the Pleuronectiformes. In: Gibson RN (ed) Flatfishes: biology and exploitation. Blackwell Publishing, Oxford, pp 10–41
Murray HM, Lall SP, Rajaselvam R, Boutilier LA, Blanchard B, Flight RM, Colombo S, Mohindra V, Douglas SE (2009) A nutrigenomic analysis of intestinal response to partial soybean meal replacement in diets for juvenile Atlantic halibut, Hippoglossus hippoglossus L. Aquaculture 298(3–4):282–293
Murray HM, Lall SP, Rajaselvam R, Boutilier LA, Flight RM, Blanchard B, Colombo S, Mohindra V, Yúfera M, Douglas SE (2010) Effect of early introduction of microencapsulated diet to larval Atlantic halibut, Hippoglossus hippoglossus L. assessed by microarray analysis. Mar. Biotechnol 12(2):214–229
Naylor R, Burke M (2005) Aquaculture and ocean resources: raising tigers of the sea. Annu Rev Environ Resour 30:185–218
Naylor RL, Hardy RW, Bureau DP, Chiu A, Elliott M, Farrell AP, Forster I, Gatlin DM, Goldburg RJ, Hua K, Nichols PD (2009) Feeding aquaculture in an era of finite resources. Proc Natl Acad Sci USA 106:15103–15110
Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461(7261):272–276
Ota T, Chihara Y, Itabashi Y, Takagi T (1994) Occurrence of all-cis-6, 9, 12, 15, 18, 21-Tetracosahexaenoic acid in flatfish lipids. Fish Sci 60:171–175
Pareek CS, Smoczynski R, Tretyn A (2011) Sequencing technologies and genome sequencing. J Appl Genet 52(4):413–435
Ponce M, Salas-Leiton E, Garcia-Cegarra A, Boglino A, Coste O, Infante C, Gisbert E, Rebordinos L, Manchado M (2011) Genomic characterization, phylogeny and gene regulation of g-type lysozyme in sole (Solea senegalensis). Fish Shellfish Immunol 31(6):925–937
Power DM, Einarsdóttir IE, Pittman K, Sweeney GE, Hildahl J, Campinho MA, Silva N, Sæle Ø, Galay-Burgos M, Smáradóttir H, Björnsson BT (2008) The molecular and endocrine basis of flatfish metamorphosis. Rev Fish Sci 16(Supp 1):95–111
Reid DP, Smith CA, Rommens M, Blanchard B, Martin-Robichaud D, Reith M (2007) A genetic linkage map of Atlantic halibut (Hippoglossus hippoglossus L.). Genetics 177:1193–1205
Rodríguez-Ramilo ST, Toro MA, Bouza C, Hermida M, Pardo BG, Cabaleiro S, Martínez P, Fernández J (2011) QTL detection for Aeromonas salmonicida resistance related traits in turbot (Scophthalmus maximus). BMC Genomics 12:541
Ruan X, Wang W, Kong J, Yu F, Huang X (2010) Genetic linkage mapping of turbot (Scophthalmus maximus L.) using microsatellite markers and its application in QTL analysis. Aquaculture 308(3–4):89–100
Sánchez-Molano E, Cerna A, Toro MA, Bouza C, Hermida M, Pardo BG, Cabaleiro S, Fernández J, Martinez P (2011) Detection of growth-related QTL in turbot (Scophthalmus maximus). BMC Genomics 12:473
Sarropoulou E, Nousdili D, Magoulas A, Kotoulas G (2008) Linking the genomes of nonmodel teleosts through comparative genomics. Mar Biotechnol 10(3):227–233
Schadt EE, Turner S, Kasarskis A (2010) A window into third-generation sequencing. Human Mol Genet 19(R2):R227–R240
Schulte D, Ariyadasa R, Shi B, Fleury D, Saski C, Atkins M, deJong P, Wu CC, Graner A, Langridge P, Stein N (2011) BAC library resources for map-based cloning and physical map construction in barley (Hordeum vulgare L.). BMC Genomics 12:247
Sha Z, Wang S, Zhuang Z, Wang Q, Wang Q, Li P, Ding H, Wang N, Liu Z, Chen S (2010) Generation and analysis of 10,000 ESTs from the half-smooth tongue sole Cynoglossus semilaevis and identification of microsatellite and SNP markers. J Fish Biol 76(5):1190–1204
Solbakken JS, Norberg B, Watanabe K, Pittman K (1999) Thyroxine as a mediator of metamorphosis of Atlantic halibut (Hippoglossus hippoglossus L.). Environ Biol Fish 56:53–65
Suen G, Teiling C, Li L, Holt C, Abouheif E, Bornberg-Bauer E, Bouffard P, Caldera EJ, Cash E, Cavanaugh A, Denas O, Elhaik E, Fave MJ et al (2011) The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet 7(2):e1002007
Surget-Groba Y, Montoya-Burgos JI (2010) Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res 20(10):1432–1440
Suzuki M, Hayashizaki Y (2004) Mouse-centric comparative transcriptomics of protein coding and non-coding RNAs. BioEssays 26(8):833–843
Tacon AGJ, Metian M (2008) Global overview on the use of fish meal and fish oil in industrially compounded aquafeeds: trends and future prospects. Aquaculture 285(1–4):146–158
Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A (2011) Differential expression in RNA-seq: a matter of depth. Genome Res 21(12):2213–2223
Valente LMP, Linares F, Villanueva JLR, Silva JMG, Espe M, Escorcio C, Pires MA, Saavedra MJ, Borges P, Medale F, Alvárez-Blázquez B, Peleteiro JB (2011) Dietary protein source or energy levels have no major impact on growth performance, nutrient utilization or flesh fatty acids composition of market-sized Senegalese sole. Aquaculture 318(1–2):128–137
van Walraven L, Mollet FM, van Damme CJG, Rijnsdorp AD (2010) Fisheries-induced evolution in growth, maturation and reproductive investment of the sexually dimorphic North Sea plaice (Pleuronectes platessa L.). J Sea Res 64(1–2):85–93
Vesterlund L, Jiao H, Unneberg P, Hovatta O, Kere J (2011) The zebrafish transcriptome during early development. BMC Dev Biol 11(1):30
Voelkerding KV, Dames SA, Durtschi JD (2009) Next-generation sequencing: from basic research to diagnostics. Clin Chem 55(4):641–658
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1):57–63
Welch A, Hoenig R, Stieglitz J, Benetti D, Tacon A, Sims N, O’Hanlon B (2010) From fishing to the sustainable farming of carnivorous marine finfish. Rev Fish Sci 18:235–247
Weltzien FA, Andersson E, Andersen Ø, Shalchian-Tabrizi K, Norberg B (2004) The brain-pituitary-gonad axis in male teleosts, with special emphasis on flatfish (Pleuronectiformes). Comp Biochem Physiol A Mol Integr Physiol 137(3):447–477
Wong CE, Zhao YT, Wang XJ, Croft L, Wang ZH, Haerizadeh F, Mattick JS, Singh MB, Carroll BJ, Bhalla PL (2011) MicroRNAs in the shoot apical meristem of soybean. J Exp Bot 62(8):2495–2506
Woycicki R, Witkowicz J, Gawronski P, Dabrowska J, Lomsadze A, Pawelkowicz M, Siedlecka E, Yagi K, Plader W, Seroczynska A, Smiech M, Gutman W, Niemirowicz-Szczytt K et al (2011) The genome sequence of the North-European cucumber (Cucumissativus L.) unravels evolutionary adaptation mechanisms in plants. PLoS ONE 6(7):e22728
Xu TJ, Chen SL, Ji XS, Tian YS (2008) MHC polymorphism and disease resistance to Vibrio anguillarum in 12 selective Japanese flounder (Paralichthys olivaceus) families. Fish Shellfish Immunol 25(3):213–221
Yang SS, Tu ZJ, Cheung F, Xu WW, Lamb JF, Jung HJ, Vance CP, Gronwald JW (2011) Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genomics 12:199
Yúfera M, Fernández-Diaz C, Pascual E (2005) Food microparticles for larval fish prepared by internal gelation. Aquaculture 248(1–4):253–262
Zhang J, Chiodini R, Badr A, Zhang G (2011a) The impact of next-generation sequencing on genomics. J Genet Genomics 38(3):95–109
Zhang Y, Stupka E, Henkel CV, Jansen HJ, Spaink HP, Verbeek FJ (2011b) Identification of common carp innate immune genes with whole-genome sequencing and RNA-Seq Data. J Integr Bioinform 8(2):169
Zhuang ZM, Wu D, Zhang SC, Pang QX, Wang CL, Wan RJ (2006) G-banding patterns of the chromosomes of tonguefish Cynoglossus semilaevis Günther, 1873. J App Ichthyol 22(5):437–440
Acknowledgments
This work has been partially financed by the project AQUAGENET (SOE2/P1/E287), Territorial Cooperation Programme of Southeast Europe (INTERREG IVB SUDOE) to M.M.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special section: “Foodomics”; Guest Editors Dr. A. Bordoni and F. Capozzi.
Rights and permissions
About this article
Cite this article
Cerdà, J., Manchado, M. Advances in genomics for flatfish aquaculture. Genes Nutr 8, 5–17 (2013). https://doi.org/10.1007/s12263-012-0312-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12263-012-0312-8