
Abstract
The epithelial sodium channel (ENaC) is a heteromeric channel composed of three similar but distinct subunits, α, β and γ. This channel is an end-effector in the rennin-angiotensin-aldosterone system and resides in the apical plasma membrane of the renal cortical collecting ducts, where reabsorption of Na+ through ENaC is the final renal adjustment step for Na+ balance. Because of its regulation and function, the ENaC plays a critical role in modulating the homeostasis of Na+ and thus chronic blood pressure. The development of most forms of hypertension requires an increase in Na+ and water retention. The role of ENaC in developing high blood pressure is exemplified in the gain-of-function mutations in ENaC that cause Liddle's syndrome, a severe but rare form of inheritable hypertension. The evidence obtained from studies using animal models and in human patients indicates that improper Na+ retention by the kidney elevates blood pressure and induces salt-sensitive hypertension.
Similar content being viewed by others


Introduction
Several studies have classified humans who are suffering from hypertension as salt-sensitive or salt-resistant based upon blood pressure (BP) responses to differences in sodium balance1, 2. The increment in BP that is driven by a salt load is characteristic of salt-sensitive hypertension, a condition affecting more than two thirds of individuals with essential hypertension who are older than 60 years3. Salt-sensitive hypertension may exacerbate mortality rates and worsen the manifestations of target organ damage1, 2. The discovery of mutations in the β- and γ-subunits of epithelial sodium channel (ENaC) to understand Liddle's syndrome4, 5, a severe form of low-renin hypertension6, was followed by a search for common genetic variants in ENaC subunits. Several variants were identified7. Interestingly, these variants were almost universally more common in black individuals, which correlate nicely with higher prevalence of low-renin, salt-sensitive hypertension in black individuals. The questions and issues addressed in the current review are whether ENaC that resides in the renal distal nephron plays a role in the development of hypertension, particularly in salt-sensitive hypertension, how ENaC variants segregate with high BP, and whether high-salt intake induces oxidative stress and whether oxidative stress could activate ENaC, resulting in over reabsorption of Na+. We also briefly discuss the role of ENaC expressed in vascular endothelia and the central nervous system in the development of hypertension.
The topology and physiology of ENaC
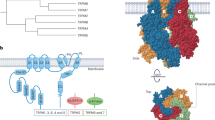
Since 1994, when ENaC was initially cloned from the rat colon8, the biophysical properties and molecular structure of ENaC have been extensively studied. ENaC consists of at least three subunits including α, β, and γ, each of which possesses two transmembrane domains, a large extracellular loop, a cytoplasmic C-terminal domain and a N-terminal domain. All three subunits are required to form a functional α-, β-, γ-ENaC channel complex (Figure 1)8, 9, 10, 11, 12, 13, 14, 15. ENaC belongs to a member of the ENaC/Deg superfamily of ion channels that are responsible for sodium transport. The channel is typically located at the apical membrane of epithelial tissues throughout the body, including the colon, the sweat glands, the salivary duct, the airway, and the cortical collecting duct (CCD) of the kidney, and this channel regulates sodium transport in tissues16, 17, 18, 19, 20. Recent studies have shown that ENaC subunits are also present in the endothelial cells of the artery and may function as a vascular mechanosensor21, 22, 23.

The topology of ENaC. ENaC consists of at least three subunits including α, β, and γ subunits, each of which possesses two transmembrane domains, a large extracellular loop, a cytoplasmic C-terminal domain and an N-terminal domain.
ENaC α, β, and γ subunits share approximately 30% homology at the amino acid level, and each subunit corresponds to a molecular mass of 70–85 kDa. The three ENaC subunits are inserted into the plasma membrane with a proposed stoichiometry of 2:1:124 or 3:3:325, 26. The α-subunit is critical to the formation of the ion permeating pore, whereas the β and γ subunits are required for the maximal channel activity and may play regulatory role. While α-ENaC is knocked out in mice, the mice would die within 40 h of birth because failure of pulmonary fluid clearance. This result clearly demonstrates the pivotal role of α-ENaC in forming a functional Na+ channel complex in vivo27. Moreover, decreased α-ENaC expression in mice causes a respiratory distress syndrome, whereas the β and γ have only a modest effect on pulmonary fluid clearance28. The α-, β-, γ-ENaC channel complex is highly selective for Na+ and mediates Na+ entry through the apical membrane of distal renal epithelial cells with a slope single-channel conductance of approximately 5 pS. ENaC accounts for a small proportion of distal renal sodium reabsorption (<5%). However, there appears to be no further downstream sodium transport system beyond CCD, which places ENaC in a very critical position for the regulation (or homeostasis) of the extracellular fluid volume, electrolyte balance and long term BP7. The factors such as high-salt intake that affect ENaC activity and the ENaC expression level at the apical membrane of CCD may constitute the critical role of ENaC in Na+ over reabsorption and water retention.
The experiments assessed in animal models reveal the role of ENaC in developing salt-sensitive hypertension
The strains of rats were bred by Dr Lewis K Dahl for sensitivity or resistance to the hypertensive effect of a high-salt diet in the 1960s and were named Dahl salt-sensitive (DS) or Dahl salt-resistance (DR)29, 30. When DS rats were placed on a high-salt (8% NaCl) diet at 21–23 d of age, they rapidly developed hypertension. All of the DS rats died by the 16th week of salt feeding. With similarly treated DR rats, the BP remained in the normotensive range, and 80% of animals survived to the 48th week on a high-salt diet30. Since then the kidney has been the focus of considerable attention in DS and DR rats because of the results obtained from the renal cross-transplantation assays, which involve the transplantation of a kidney from a DS rat into a DR rat that had the original kidneys removed, the results suggest that the DR rats develop a higher BP than DR rats with transplanted DR kidneys or DR rats with a unilateral nephrectomy. Conversely, a DR kidney that was transplanted into a DS rat ameliorated the increase in the BP that was seen in DR rats with transplanted DS kidneys or DS rats with a unilateral nephrectomy31, 32, 33. Later studies revealed that the plasma renin and aldosterone concentrations were normal or lower in DS rats compared with that in control rats34, 35. Aoi et al investigated the mechanisms by which quercetin, a plant extract, exerted an anti-hypertensive effect, and they found that quercetin diminished the αENaC mRNA expression in the kidney, which was associated with the reduction of the systolic BP that was elevated by a high-salt diet in DS rats. These results suggest a role for ENaC in salt-sensitive hypertension36. The same group attempted to determine whether a high-salt diet in DR rats stimulated the expression level of ENaC. These investigators divided the DS and DR rats into several salt diet groups as follows: DS and DR rats that were fed with a low-sodium diet (0.005% NaCl), a normal-sodium diet (0.3% NaCl), or a high-sodium diet (8% NaCl). Four weeks after the high-salt diet in DS rats, an increase in the systolic BP was observed. However, the BP was not altered in any of the other groups. Subsequently, these investigators examined the expression level of α-ENaC mRNA and serum and glucocorticoid-regulated kinase 1 (SGK1) mRNA. They found that the expression level of α-, β-, γ-ENaC, and the SGK1 mRNAs was significantly enhanced by the high-sodium diet in DS rats. Interestingly, the expression of SGK1 mRNA was down-regulated in DR rats that were fed a high sodium diet. These observations suggest that the expression of ENaC and SGK1 mRNAs is abnormally regulated by the dietary sodium in salt-sensitively hypertensive rats and that this abnormal expression may be a factor that causes salt-sensitive hypertension37. Convincing evidence indicates that the aldosterone activated mineralocorticoid receptor increases SGK1 gene transcription in the CCD, and consequently, SGK1 strongly stimulates the activity and expression of the ENaC and renal Na+/H+ exchanger (NHE)38, 39, 40, 41, 42, 43. High-salt intake may up-regulate both ENaC and SGK1 in DS rats. However, functional studies that examine ENaC activity are required to evaluate the role of ENaC and SGK1 in salt-sensitive hypertension in DS rats and the mechanisms by which the high-salt intake regulates ENaC and SGK1.
Fenton and co-authors found that none of the ENaC subunits was increased in abundance in the inner medullas of DS rats compared with that of DR rats44. In fact, the α-subunit was strongly down regulated, which may be a consequence of the marked increase in 11β-HSD2 expression in the cells of the inner medulla. Consistent with this view, the protein abundance of α-ENaC was markedly elevated following the carbenoxolone-induced inhibition of the 11β-HSD2 activity. These investigators also examined whether the ENaC subunits may be upregulated by a high-salt diet in DS rats. However, Husted and co-workers showed that the ENaC activity is doubled in the IMCD cells of DS rats versus in that of DR rats40, 45. The reasons for this variability in ENaC activity and/or expression level in DS versus DR rats are poorly understood at the present time. Shehata and co-workers examined the complete coding sequences of three ENaC subunits and showed that there were no genetic differences within the 5' and 3' flanking regions in DS vs DR rats46. The alternative splicing of α-ENaC may regulate α-ENaC by formation of coding RNA species (α-ENaC-a and -b) and non-coding RNA species (α-ENaC-c and -d). The α-ENaC-a and -b mRNA levels are significantly higher in DR versus DS rats. After 4 weeks of the high-salt intake, the level of α-ENaC-b was dramatically elevated compared to that in DR rats fed a normal-salt diet. These results suggest that α-ENaC-b is a salt-sensitive transcript. Furthermore, among the four α-ENaC transcripts (-a, -b, -c, and -d), α-ENaC-b is a predominant transcript that exceeds α-ENaC-wt abundance by approximately 32 fold. α-ENaC-b may potentially act as a dominant negative protein for ENaC activity and rescue DR rats from developing salt-sensitive hypertension on a high-salt diet47.
The molecular variations in ENaC and the risk for developing hypertension in humans
The search for common genetic variants in ENaC subunits that affect the susceptibility in less rare forms of hypertension took place soon after the discovery of mutations in α-, β-, and γ-ENaC that cause Liddle's syndrome (Table 1). The first variant that was associated with hypertension was T594M in the C-terminus of β-ENaC in black individuals. This variant was found in approximately 8% of hypertensive individuals, whereas the variant was detected in only approximately 2% of normotensive individuals48. In another study, seven variants in β-ENaC, including G589S, T594M, R597H, R624C, E632G (last exon), G442V, and V434M (exon 8) were identified and almost found in black individuals49. The functional properties of the variants were evaluated in Xenopus oocytes expressing these mutants. Interestingly, small but not significant differences were detected between the variants and wild-type ENaC. The clinical evaluation of the family bearing the G589S variant, which provided the highest relative ENaC activity, did not show any cosegregation between the mutation and hypertension49. However, the lack of a significant increase in the Na+ current that was observed in Xenopus oocytes that overexpressed these variants cannot completely rule out any functional impact of the ENaC mutants in developing hypertension49. One possibility to explain why the association studies of ENaC variants are often inconclusive is that many factors influence the ENaC activity. Therefore, a variant that affects ENaC function in vitro may not necessarily cause Na+ retention, unless at the same time, the regulatory factors do not adjust accordingly. The variants identified in the β- and γ-subunits of ENaC were almost exclusively identified in individuals of African origin. However, the physiological significance of the β- and γ-subunit polymorphisms may partially explain the high incidence of salt-sensitive hypertension in African Americans. Among the black hypertensive population, approximately 75% is salt-sensitive, characterized by a BP increase after dietary salt intake2.
To determine whether SCNN1B or SCNN1G, which encode β and γ subunits, respectively, were present in a patient who was clinically suspected to have Liddle's syndrome with no familial history of hypertension, Wang and coworkers identified a mutation causing Liddle's syndrome. They demonstrated that a frameshift mutation of the γ subunit resulted in a new termination site at the 585 codon of the γ subunit and the deletion of its PY motif. Moreover, the parents of the patient, the other 50 randomly selected hypertensive patients, and 50 controls did not have the mutation that causes Liddle's syndrome. These results suggest that this frameshift mutation is a de novo mutation and not a common genetic variant50.
Several α-ENaC variants at the residues 334, 618 and 663 are possibly associated with the abnormal Na+handling by the kidney and the salt-sensitive hypertension that is prevalent in black populations51, 52, 53, 54. Several groups have studied whether these variants segregate with BP, and the outcomes are controversial51, 53. Ambrosius and coworkers reported that the allele of T663A was twice as common in whites and that T663A was associated with being normotensive in black and white populations51. The expression of T663A did not alter the basal Na+ current51. Kleyman's group used Xenopus oocytes expressing a mouse/human chimera (m(1-678)/h(650-669)/T663A), which was generated by the replacement of the distal C terminus of the mouse α-subunit with the distal C terminus of the human α-subunit, and determined that the human αT663βγ ENaC has increased activity in Xenopus oocytes when compared with human αT663Aβγ ENaC. The increase in the channel activity in human αT663βγ reflected an increase in surface expression55. Stockand's group has reported that the polymorphic C618F and A663T ENaCs had greater activity compared with the wild-type channels in the excised patches with activity of channels increased 3.8- and 2.6-fold, respectively56. This increase in the channel activity is associated with an increase in the surface expression of the polymorphisms. The results obtained by these studies are consistent with the C618F and A663T polymorphisms leading to an elevated ENaC activity with the possibility that these polymorphisms facilitate the altered Na+handling by the kidney55, 56. Iwai et al reported that a polymorphism in the promoter region of the α-ENaC gene G2139A is associated with BP status and that the G2139 allele significantly increased the risk of hypertension in the general Japanese population57.
Does oxidative stress induced by high-salt intake activate ENaC?
ENaC activity depends upon the number of channels in the apical membrane, the permeation properties, and the open probability of the channel (Po). One of the best examples is the salt-sensitive hypertension of Liddle's syndrome, in which a gain of function ENaC mutant enhances its trafficking to the plasma membrane and thereby increases its cell surface expression4, 5, 6, 58, 59, 60, 61, which leads to over reabsorption of Na+ and water.
Evidence obtained in rats and humans suggest that high-salt diets also cause oxidative stress. This high-salt intake induced increase in oxidative stress is more obvious in salt-sensitive hypertension62, 63. Furthermore, high-salt intake may also target the tissues and organs independent of hypertension via a mechanism of elevating reactive oxygen species (ROS)64. In the absence of the prominent elevations of BP after salt-loading, salt sensitivity may be revealed by the structural and functional injuries of the targeting organs such as the heart and kidney65. Recent studies have shown that hydrogen peroxide (H2O2), an isoform of ROS, stimulates ENaC and that high NaCl elevates ROS in CCD cells66. However, the mechanism by which a high-salt diet induces an increase in ROS to stimulate ENaC is not known. A high-salt intake is known to induce the compensatory natriuresis to maintain sodium homeostasis. Reduced plasma aldosterone causes a decrease in α-ENaC mRNA level, which suggests an important role in the compensatory natriuresis40. Previous electrophysiological experiments assessed in renal CCD have indicated that dietary sodium intake and variations in aldosterone plasma levels regulates the abundance of functional ENaC in the apical plasma membrane39. A high or low Na+ diet for three weeks also influenced the distribution pattern of ENaC in the mouse kidney. The regulation of ENaC function in vivo involves shifting the β- and γ-subunits from the cytoplasm to the apical plasma membrane and vice versa, respectively12. The insertion of these subunits into the apical plasma membrane coincides with the upregulation of the α-subunit and its insertion into the apical plasma membrane67. These studies together suggest that dietary salt modulates the expression pattern of ENaC subunits in the kidney and may stimulate its activity via enhanced ROS level, which in turn leading to an increase in Na+ reabsorption.
Several studies have demonstrated that there is increased oxidative stress in animals with high-salt intake68, 69, 70, 71. In experimental models of salt-sensitive hypertension, high-salt intake increased the markers of vascular and systemic oxidative stress1. Studies in essential hypertensive patients have suggested that high-salt intake and/or salt sensitivity is associated with impaired endothelial function72, 73, 74, 75. Miyoshi et al76 reported a decrease in acetylcholine-induced forearm vasodilation in salt-sensitive hypertensive subjects regardless of the level of salt intake. Increased ROS have a critical role in the initiation of hypertension and may be generated by the hypertension itself, suggesting a positive-feedback mechanism. In addition to the systemic effects of ROS, recent evidence demonstrated that oxidative stress within the kidney plays a central role in the pathophysiology of sodium retention by inducing the tubulointerstitial accumulation of Ang II-positive cells. The prohypertensive role of intrarenal ROS is suggested by the strong correlation between the renal superoxide-positive cells and the severity of hypertension in the spontaneously hypertensive rats (SHR)77. However, there is a lack of information at the present time regarding whether oxidative stress induced by high-salt intake affects the BP via influencing ENaC activity. In our preliminary studies, we found that the high-salt intake decreased the expression level of α-ENaC in the CCD cells of DR rats, but not that in DS rats (unpublished observations). When cultured CCD cells are treated with high NaCl, ROS accumulated within these cells. Using patch-clamp experiments, we found that H2O2 stimulates ENaC activity. These results suggest that high-salt intake may activate ENaC through an elevation of ROS [unpublished observations].
Does altered activity of ENaC affect the function of the vascular endothelium and sympathetic nervous system to influence BP?
Although ENaC was known as the typical sodium channel in the kidney, the colon and the lung, vascular endothelial cells were also shown to express ENaC and mineralocorticoid receptors21, 78, 79. Endothelial cells are targets for aldosterone, which activates the apically located ENaC, and its activity modifies the biomechanical properties of the endothelium. Therefore, ENaC is proposed as the key mediator of aldosterone-dependent BP control in the endothelium80. Several studies, in different cell types including CCD and endothelial cells, have suggested that ENaC may function as a mechanosensor and that mechanical stimuli may activate ENaC22, 81, 82. Because endothelial ENaC inhibition may activate nitric oxide (NO) synthase83, it is completely possible that altered blood flow (shear stress), which is caused by over reabsorption of Na+ via ENaC located at distal nephron, may affect the NO production in endothelia. High-salt intake may cause an increase in plasma [Na+], which may or may not be detectable depending upon the extent of water intake and the timing of blood sampling relative to high-salt intake. Fang and coworkers showed that four days after 8% high-salt diet exposure, plasma [Na+] increased by 3–4 mmol/L in SHR and Wistar Kyoto rats84. In normotensive rats, when salt intake increased from 10 to 250 mmol/d over 5 d, the plasma [Na+] increased by 3 mmol/L. In addition, reducing the salt intake from 350 to 12–20 mmol/d lowered the plasma [Na+] to a similar extent by 3–4 mmol/L85. Huang and coworkers showed that high-salt intake increased [Na+] in the cerebrospinal fluid (CSF) up to 5 mmol/L in DS rats and SHR rats but not in DR rats86. Similar to the results obtained from the animal models, [Na+] in CSF was increased by 2–3 mmol/L in patients with both salt-sensitive and non-salt-sensitive hypertension after a 7-d high-salt diet (16–18 g/d) compared to those given a low-salt diet (1–3 g/d)87. Nevertheless, high-salt diets elevated the arterial pressure in salt-sensitive individuals. These results suggest that increases in plasma [Na+] may trigger this effect84 via a mechanism that has not been elucidated.
Previous studies have suggested that DS rats have abnormalities in the sympathetic nervous system (SNS)88, 89 and endothelial function90, 91, which causes significant vascular resistance. In addition, there is evidence that supports the hypothesis that abnormal modulation of SNS is involved in salt-inducd hypertension. Salt loading has been shown to augment the sympathetic activity in DS rats but not in DR rats92, 93, 94. The intracerebroventricular (ICV) infusion of sodium caused sympathoexcitatory and pressor responses to a greater degree in DS rats than in DR rats85, 95. The strict regulation of [Na+] in the CSF is crucial for the normal function of neurons. An increase in CSF [Na+] by as little as 2 mmol/L can increase the firing rate of neurons. A chronic 5 mmol/L increase in CSF [Na+] causes sympathetic hyperactivity and hypertension96, 97, 98. Increases in CSF [Cl−] or the osmolarity of CSF did not cause such sympathoexcitation and hypertension99. Because the role of ENaC in regulating sodium transport across the epithelia is important, investigators started to study whether ENaC in neural components also plays a role in salt-sensitive hypertension. Stoichiometrically different populations of ENaC may be present in both epithelial and neural components in the brain, which may contribute to the regulation of CSF and interstitial Na+ concentrations and neuronal excitation90, 97. ENaC subunits are also expressed in sensory nerve endings in the rat foot pad100 and in the trigeminal mechanosensory neurons101. However, the function of the ENaC subunits in these tissues has not yet been elucidated. Functional studies have suggested the presence of specific Na+ channels, presumably ENaC, in the brain that are activated by aldosterone or a high-salt diet and blocked by amiloride or benzamil. In Wistar rats, ICV infusion of aldosterone or Na+-rich artificial CSF increased BP and renal sympathetic nerve activity. In DS rats but not DR rats, a high-salt diet or ICV infusion of aldosterone caused sympathoexcitation and hypertension. The blood-brain barrier in DS rats is five to eight times more permeable to Na+ than that in DR rats102. Increases in CSF [Na+] are observed in DS rats but not DR rats on a high-salt diet and precede changes in BP by 1–2 d86. Importantly, the responses to aldosterone or Na+-rich artificial CSF in Wistar rats and to aldosterone or a high-salt diet in DS rats can be prevented by ICV infusion of benzamil or spironolactone103, 104, 105. These findings suggest that the mineralocorticoid receptor (MR)-mediated activation of sodium channels in the brain is responsible for the mechanisms leading to increased sympathetic outflow and hypertension.
Conclusion
We present evidence that places ENaC in a central position for Na+ retention, which is necessary to achieve a state of high BP in the salt-sensitive population. The Na+ reabsorptive site (ENaC) does not act alone in the mechanisms for developing hypertension. The emerging evidence is compelling for the consideration of ENaC as the additional requisite participant in endothelia and SNS (Figure 2). However, the mechanisms by which the activation of ENaC to induce Na+ retention and the consequences in the vascular compartment and SNS require further investigation.

A schematic that illustrates the central role of ENaC in the development of salt-sensitive hypertension after the loss of compensatory natriuresis. High-salt intake in the salt-sensitive population induces oxidative stress in the kidney, which enhances the apical membrane expression of ENaC and ENaC activity with an unknown mechanism at the present time. This increased activity eventually causes Na+ over-reabsorption in CCD followed by water retention and elevation of BP. The volume expansion in the vascular compartment alters blood flow (shear stress) and directly affects endothelial function by reducing the synthesis of NO109, 110. ENaC may act as a mechanosensor in endothelial cells and may sense changes in shear stress. Alterations in shear stress may activate ENaC residing at the apical membrane of endothelial cells and may affect the regulation of vasoactive substances. Na+ over-reabsorption in CCD elevates [Na+] in CSF, which in turn triggers sympathetic activity in neurons and contributes to hypertension. Stoichiometrically different populations of ENaC may be present in epithelial cells and neurons in the brain, which may contribute to the regulation of CSF and interstitial [Na+] as well as neuronal excitation. CCD: cortical collecting duct; CSF: cerebrospinal fluid; and NO: nitric oxide. Black solid lines with arrows: already known; grey dash lines with arrows and words in gray: open questions.
References
Campese VM . Salt sensitivity in hypertension. Renal and cardiovascular implications. Hypertension 1994; 23: 531–50.
Weinberger MH . Salt sensitivity of blood pressure in humans. Hypertension 1996; 27: 481–90.
Staessen JA, Wang J, Bianchi G, Birkenhäger WH . Essential hypertension. Lancet 2003; 361: 1629–41.
Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, et al. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 1994; 79: 407–14.
Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet 1995; 11: 76–82.
Liddle GW, Bledsoe T, Coppage WS Jr . Hypertension reviews. J Tenn Med Assoc 1974; 67: 669.
Pratt JH . Central role for ENaC in development of hypertension. J Am Soc Nephrol 2005; 16: 3154–9.
Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994; 367: 463–7.
Lingueglia E, Voilley N, Waldmann R, Lazdunski M, Barbry P . Expression cloning of an epithelial amiloride-sensitive Na+ channel. A new channel type with homologies to Caenorhabditis elegans degenerins. FEBS Lett 1993; 318: 95–9.
McDonald FJ, Snyder PM, McCray PB Jr, Welsh MJ . Cloning, expression, and tissue distribution of a human amiloride-sensitive Na+ channel. Am J Physiol 1994; 266: L728–34.
McDonald FJ, Price MP, Snyder PM, Welsh MJ . Cloning and expression of the beta- and gamma-subunits of the human epithelial sodium channel. Am J Physiol 1995; 268: C1157–63.
Firsov D, Gautschi I, Merillat AM, Rossier BC, Schild L . The heterotetrameric architecture of the epithelial sodium channel (ENaC). EMBO J 1998; 17: 344–52.
Fyfe GK, Canessa CM . Subunit composition determines the single channel kinetics of the epithelial sodium channel. J Gen Physiol 1998; 112: 423–32.
Fyfe GK, Quinn A, Canessa CM . Structure and function of the Mec-ENaC family of ion channels. Semin Nephrol 1998; 18: 138–51.
Horisberger JD . Amiloride-sensitive Na channels. Curr Opin Cell Biol 1998; 10: 443–9.
Garty H, Benos DJ . Characteristics and regulatory mechanisms of the amiloride-blockable Na+ channel. Physiol Rev 1988; 68: 309–73.
Palmer LG . Epithelial Na channels: function and diversity. Annu Rev Physiol 1992; 54: 51–66.
Garty H, Palmer LG . Epithelial sodium channels: function, structure, and regulation. Physiol Rev 1997; 77: 359–96.
Benos DJ, Stanton BA . Functional domains within the degenerin/epithelial sodium channel (Deg/ENaC) superfamily of ion channels. J Physiol 1999; 520: 631–44.
Snyder PM . The epithelial Na+ channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension. Endocr Rev 2002; 23: 258–75.
Golestaneh N, Klein C, Valamanesh F, Suarez G, Agarwal MK, Mirshahi M . Mineralocorticoid receptor-mediated signaling regulates the ion gated sodium channel in vascular endothelial cells and requires an intact cytoskeleton. Biochem Biophys Res Commun 2001; 280: 1300–6.
Drummond HA, Gebremedhin D, Harder DR . Degenerin/epithelial Na+ channel proteins: components of a vascular mechanosensor. Hypertension 2004; 44: 643–8.
Oberleithner H, Ludwig T, Riethmüller C, Hillebrand U, Albermann L, Schäfer C, et al. Human endothelium: target for aldosterone. Hypertension 2004; 43: 952–6.
Rossier BC, Pradervand S, Schild L, Hummler E . Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu Rev Physiol 2002; 64: 877–97.
Snyder PM, Cheng C, Prince LS, Rogers JC, Welsh MJ . Electrophysiological and biochemical evidence that DEG/ENaC cation channels are composed of nine subunits. J Biol Chem 1998; 273: 681–4.
Eskandari S, Snyder PM, Kreman M, Zampighi GA, Welsh MJ, Wright EM . Number of subunits comprising the epithelial sodium channel. J Biol Chem 1999; 274: 27281–6.
O'Brodovich HM . Immature epithelial Na+ channel expression is one of the pathogenetic mechanisms leading to human neonatal respiratory distress syndrome. Proc Assoc Am Physicians 1996; 108: 345–55.
Egli M, Duplain H, Lepori M, Cook S, Nicod P, Hummler E, et al. Defective respiratory amiloride-sensitive sodium transport predisposes to pulmonary oedema and delays its resolution in mice. J Physiol 2004; 560: 857–65.
Dahl LK, Heine M, Tassinari L . Role of genetic factors in susceptibility to experimental hypertension due to chronic excess salt ingestion. Nature 1962; 194: 480–2.
Dahl LK, Heine M, Tassinari L . Effects of chronia excess salt ingestion. Evidence that genetic factors play an important role in susceptibility to experimental hypertension. J Exp Med 1962; 115: 1173–90.
Dahl LK, Heine M, Thompson K . Genetic influence of renal homografts on the blood pressure of rats from different strains. Proc Soc Exp Biol Med 1972; 140: 852–6.
Dahl LK, Heine M, Thompson K . Genetic influence of the kidneys on blood pressure. Evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ Res 1974; 40: 94–101.
Dahl LK, Heine M . Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res 1975; 36: 692–6.
Rapp JP, Tan SY, Margolius HS . Plasma mineralocorticoids, plasma renin, and urinary kallikrein in salt-sensitive and salt-resistant rats. Endocr Res Commun 1978; 5: 35–41.
Baba K, Mulrow PJ, Franco-Saenz R, Rapp JP . Suppression of adrenal renin in Dahl salt-sensitive rats. Hypertension 1986; 8: 1149–53.
Aoi W, Niisato N, Miyazaki H, Marunaka Y . Flavonoid-induced reduction of ENaC expression in the kidney of Dahl salt-sensitive hypertensive rat. Biochem Biophys Res Commun 2004; 315: 892–6.
Aoi W, Niisato N, Sawabe Y, Miyazaki H, Tokuda S, Nishio K, et al. Abnormal expression of ENaC and SGK1 mRNA induced by dietary sodium in Dahl salt-sensitively hypertensive rats. Cell Biol Int 2007; 31: 1288–91.
Verrey F . Transcriptional control of sodium transport in tight epithelial by adrenal steroids. J Membr Biol 1995; 144: 93–110.
Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, et al. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci U S A 1999; 96: 2514–9.
Náray-Fejes-Tóth A, Canessa C, Cleaveland ES, Aldrich G, Fejes-Tóth G . sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial Na+ channels. J Biol Chem 1999; 274: 16973–8.
Pearce D . The role of SGK1 in hormone-regulated sodium transport. Trends Endocrinol Metab 2001; 12: 341–7.
Yun CC, Chen Y, Lang F . Glucocorticoid activation of Na+/H+ exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. J Biol Chem 2002; 277: 7676–83.
Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, et al. Aberrant ENaC activation in Dahl salt-sensitive rats. J Hypertens 2009; 27: 1679–89.
Fenton RA, Chou CL, Ageloff S, Brandt W, Stokes JB, Knepper MA . Increased collecting duct urea transporter expression in Dahl salt-sensitive rats. Am J Physiol Renal Physiol 2003; 285: F143–51.
Husted RF, Takahashi T, Stokes JB . IMCD cells cultured from Dahl S rats absorb more Na+ than Dahl R rats. Am J Physiol 1996; 271: F1029–36.
Shehata MF, Leenen FH, Tesson F . Sequence analysis of coding and 3′ and 5′ flanking regions of the epithelial sodium channel alpha, beta, and gamma genes in Dahl S versus R rats. BMC Genet 2007; 8: 35.
Shehata MF . Characterization of the epithelial sodium channel alpha subunit coding and non-coding transcripts and their corresponding mRNA expression levels in Dahl R versus S rat kidney cortex on normal and high salt diet. Int Arch Med 2009; 2: 5.
Baker EH, Dong YB, Sagnella GA, Rothwell M, Onipinla AK, Markandu ND, et al. Association of hypertension with T594M mutation in beta subunit of epithelial sodium channels in black people resident in London. Lancet 1998; 351: 1388–92.
Persu A, Barbry P, Bassilana F, Houot AM, Mengual R, Lazdunski M, et al. Genetic analysis of the beta subunit of the epithelial Na+ channel in essential hypertension. Hypertension 1998; 32: 129–37.
Wang Y, Zheng Y, Chen J, Wu H, Zheng D, Hui R . A novel epithelial sodium channel gamma-subunit de novo frameshift mutation leads to Liddle syndrome. Clin Endocrinol (Oxf) 2007; 67: 801–4.
Ambrosius WT, Bloem LJ, Zhou L, Rebhun JF, Snyder PM, Wagner MA, et al. Genetic variants in the epithelial sodium channel in relation to aldosterone and potassium excretion and risk for hypertension. Hypertension 1999; 34: 631–7.
Su YR, Menon AG . Epithelial sodium channels and hypertension. Drug Metab Dispos 2001; 29: 553–6.
Sugiyama T, Kato N, Ishinaga Y, Yamori Y, Yazaki Y . Evaluation of selected polymorphisms of the Mendelian hypertensive disease genes in the Japanese population. Hypertens Res 2001; 24: 515–21.
Swift PA, Macgregor GA . Genetic variation in the epithelial sodium channel: a risk factor for hypertension in people of African origin. Adv Ren Replace Ther 2004; 11: 76–86.
Samaha FF, Rubenstein RC, Yan W, Ramkumar M, Levy DI, Ahn YJ, et al. Functional polymorphism in the carboxyl terminus of the alpha-subunit of the human epithelial sodium channel. J Biol Chem 2004; 279: 23900–7.
Tong Q, Menon AG, Stockand JD . Functional polymorphisms in the alpha-subunit of the human epithelial Na+ channel increase activity. Am J Physiol Renal Physiol 2006; 290: F821–7.
Iwai N, Baba S, Mannami T, Ogihara T, Ogata J . Association of a sodium channel alpha subunit promoter variant with blood pressure. J Am Soc Nephrol 2002; 13: 80–5.
Firsov D, Schild L, Gautschi I, Mérillat AM, Schneeberger E, Rossier BC . Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci U S A 1996; 93: 15370–5.
Goulet CC, Volk KA, Adams CM, Prince LS, Stokes JB, Snyder PM . Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle's syndrome. J Biol Chem 1998; 273: 30012–7.
Abriel H, Loffing J, Rebhun JF, Pratt JH, Schild L, Horisberger JD, et al. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle's syndrome. J Clin Invest 1999; 103: 667–73.
Rotin D, Kanelis V, Schild L . Trafficking and cell surface stability of ENaC. Am J Physiol Renal Physiol 2001; 281: F391–9.
Bayorh MA, Ganafa AA, Socci RR, Silvestrov N, Abukhalaf IK . The role of oxidative stress in salt-induced hypertension. Am J Hypertens 2004; 17: 31–6.
Laffer CL, Bolterman RJ, Romero JC, Elijovich F . Effect of salt on isoprostanes in salt-sensitive essential hypertension. Hypertension 2006; 47: 434–40.
Ritz E, Mehls O . Salt restriction in kidney disease — a missed therapeutic opportunity. Pediatr Nephrol 2009; 24: 9–17.
Frohlich ED, Varagic J . Sodium directly impairs target organ function in hypertension. Curr Opin Cardiol 2005; 20: 424–9.
Bayorh MA, Ganafa AA, Eatman D, Walton M, Feuerstein GZ . Simvastatin and losartan enhance nitric oxide and reduce oxidative stress in salt-induced hypertension. Am J Hypertens 2005; 18: 1496–502.
Loffing J, Vallon V, Loffing-Cueni D, Aregger F, Richter K, Pietri L, et al. Altered renal distal tubule structure and renal Na+ and Ca2+ handling in a mouse model for Gitelman's syndrome. J Am Soc Nephrol 2004; 15: 2276–88.
Swei A, Lacy F, DeLano FA, Schmid-Schönbein GW . Oxidative stress in the Dahl hypertensive rat. Hypertension 1997; 30: 1628–33.
Zhou MS, Adam AG, Jaimes EA, Raij L . In salt-sensitive hypertension, increased superoxide production is linked to functional upregulation of angiotensin II. Hypertension 2003; 42: 945–51.
Tian N, Moore RS, Braddy S, Rose RA, Gu JW, Hughson MD, et al. Interactions between oxidative stress and inflammation in salt-sensitive hypertension. Am J Physiol Heart Circ Physiol 2007; 293: H3388–95.
Jaimes EA, Zhou MS, Pearse DD, Puzis L, Raij L . Upregulation of cortical COX-2 in salt-sensitive hypertension: role of angiotensin II and reactive oxygen species. Am J Physiol Renal Physiol 2008; 294: F385–92.
Stein CM, Nelson R, Brown M, Wood M, Wood AJ . Dietary sodium intake modulates vasodilation mediated by nitroprusside but not by methacholine in the human forearm. Hypertension 1995; 25: 1220–3.
Campese VM, Tawadrous M, Bigazzi R, Bianchi S, Mann AS, Oparil S, et al. Salt intake and plasma atrial natriuretic peptide and nitric oxide in hypertension. Hypertension 1996; 28: 335–40.
Facchini FS, DoNascimento C, Reaven GM, Yip JW, Ni XP, Humphreys MH . Blood pressure, sodium intake, insulin resistance, and urinary nitrate excretion. Hypertension 1999; 33: 1008–12.
Fujiwara N, Osanai T, Kamada T, Katoh T, Takahashi K, Okumura K . Study on the relationship between plasma nitrite and nitrate level and salt sensitivity in human hypertension: modulation of nitric oxide synthesis by salt intake. Circulation 2000; 101: 856–61.
Miyoshi A, Suzuki H, Fujiwara M, Masai M, Iwasaki T . Impairment of endothelial function in salt-sensitive hypertension in humans. Am J Hypertens 1997; 10: 1083–90.
Rodríguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M, et al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 2001; 59: 2222–32.
Oberleithner H, Riethmüller C, Ludwig T, Shahin V, Stock C, Schwab A, et al. Differential action of steroid hormones on human endothelium. J Cell Sci 2006; 119: 1926–32.
Wang S, Meng F, Mohan S, Champaneri B, Gu Y . Functional ENaC channels expressed in endothelial cells: a new candidate for mediating shear force. Microcirculation 2009; 16: 276–87.
Kusche-Vihrog K, Callies C, Fels J, Oberleithner H . The epithelial sodium channel (ENaC): Mediator of the aldosterone response in the vascular endothelium. Steroids 2010; 75: 544–9.
Wei SP, Li XQ, Chou CF, Liang YY, Peng JB, Warnock DG, et al. Membrane tension modulates the effects of apical cholesterol on the renal epithelial sodium channel. J Membr Biol 2007; 220: 21–31.
Drummond HA, Welsh MJ, Abboud FM . ENaC subunits are molecular components of the arterial baroreceptor complex. Ann N Y Acad Sci 2001; 940: 42–7.
Pérez FR, Venegas F, González M, Andrés S, Vallejos C, Riquelme G, et al. Endothelial epithelial sodium channel inhibition activates endothelial nitric oxide synthase via phosphoinositide 3-kinase/Akt in small-diameter mesenteric arteries. Hypertension 2009; 53: 1000–7.
Fang Z, Carlson SH, Peng N, Wyss JM . Circadian rhythm of plasma sodium is disrupted in spontaneously hypertensive rats fed a high-NaCl diet. Am J Physiol Regul Integr Comp Physiol 2000; 278: R1490–5.
He FJ, Markandu ND, Sagnella GA, de Wardener HE, MacGregor GA . Plasma sodium: ignored and underestimated. Hypertension 2005; 45: 98–102.
Huang BS, Van Vliet BN, Leenen FH . Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am J Physiol Heart Circ Physiol 2004; 287: H1160–6.
Kawano Y, Yoshida K, Kawamura M, Yoshimi H, Ashida T, Abe H, et al. Sodium and noradrenaline in cerebrospinal fluid and blood in salt-sensitive and non-salt-sensitive essential hypertension. Clin Exp Pharmacol Physiol 1992; 19: 235–41.
Gordon FJ, Mark AL . Mechanism of impaired baroreflex control in prehypertensive Dahl salt-sensitive rats. Circ Res 1984; 54: 378–87.
Osborn JL, Roman RJ, Ewens JD . Renal nerves and the development of Dahl salt-sensitive hypertension. Hypertension 1988; 11: 523–8.
Hayakawa H, Coffee K, Raij L . Endothelial dysfunction and cardiorenal injury in experimental salt-sensitive hypertension: effects of antihypertensive therapy. Circulation 1997; 96: 2407–13.
Zhou MS, Kosaka H, Tian RX, Abe Y, Chen QH, Yoneyama H, et al. L-Arginine improves endothelial function in renal artery of hypertensive Dahl rats. J Hypertens 2001; 19: 421–9.
Fujita T, Henry WL, Bartter FC, Lake CR, Delea CS . Factors influencing blood pressure in salt-sensitive patients with hypertension. Am J Med 1980; 69: 334–44.
Ono A, Kuwaki T, Kumada M, Fujita T . Differential central modulation of the baroreflex by salt loading in normotensive and spontaneously hypertensive rats. Hypertension 1997; 29: 808–14.
Huang BS, Leenen FH . Both brain angiotensin II and “ouabain” contribute to sympathoexcitation and hypertension in Dahl S rats on high salt intake. Hypertension 1998; 32: 1028–33.
Huang BS, Wang H, Leenen FH . Enhanced sympathoexcitatory and pressor responses to central Na+ in Dahl salt-sensitive vs -resistant rats. Am J Physiol Heart Circ Physiol 2001; 281: H1881–9.
Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA . Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 1999; 104: R19–23.
Amin MS, Wang HW, Reza E, Whitman SC, Tuana BS, Leenen FH . Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol 2005; 289: R1787–97.
Ergonul Z, Frindt G, Palmer LG . Regulation of maturation and processing of ENaC subunits in the rat kidney. Am J Physiol Renal Physiol 2006; 291: F683–93.
Bunag RD, Miyajima E . Sympathetic hyperactivity elevates blood pressure during acute cerebroventricular infusions of hypertonic salt in rats. J Cardiovasc Pharmacol 1984; 6: 844–51.
Drummond HA, Abboud FM, Welsh MJ . Localization of beta and gamma subunits of ENaC in sensory nerve endings in the rat foot pad. Brain Res 2000; 884: 1–12.
Fricke B, Lints R, Stewart G, Drummond H, Dodt G, Driscoll M, et al. Epithelial Na+ channels and stomatin are expressed in rat trigeminal mechanosensory neurons. Cell Tissue Res 2000; 299: 327–34.
Simchon S, Manger W, Golanov E, Kamen J, Sommer G, Marshall CH . Handling 22NaCl by the blood-brain barrier and kidney: its relevance to salt-induced hypertension in Dahl rats. Hypertension 1999; 33: 517–23.
Wang H, Leenen FH . Brain sodium channels mediate increases in brain “ouabain” and blood pressure in Dahl S rats. Hypertension 2002; 40: 96–100.
Wang H, Huang BS, Leenen FH . Brain sodium channels and ouabainlike compounds mediate central aldosterone-induced hypertension. Am J Physiol Heart Circ Physiol 2003; 285: H2516–23.
Wang H, Leenen FH . Brain sodium channels and central sodium-induced increases in brain ouabain-like compound and blood pressure. J Hypertens 2003; 21: 1519–24.
Hannila-Handelberg T, Kontula K, Tikkanen I, Tikkanen T, Fyhrquist F, Helin K, et al. Common variants of the beta and gamma subunits of the epithelial sodium channel and their relation to plasma renin and aldosterone levels in essential hypertension. BMC Med Genet 2005; 6: 4.
Su YR, Rutkowski MP, Klanke CA, Wu X, Cui Y, Pun RY, et al. A novel variant of the beta-subunit of the amiloride-sensitive sodium channel in African Americans. J Am Soc Nephrol 1996; 7: 2543–9.
Persu A, Coscoy S, Houot AM, Corvol P, Barbry P, Jeunemaitre X . Polymorphisms of the gamma subunit of the epithelial Na+ channel in essential hypertension. J Hypertens 1999; 17: 639–45.
Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J . Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 2008; 117: 1161–71.
AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res 2009; 104: 860–9.
Acknowledgements
The current study was supported by the National Natural Science Foundation of China (No 81070217 and 30871007), the Natural Science Foundation of Heilongjiang Province (No ZD200807-01 and QC2010097), the Overseas Talent Foundation of the Department of Education, Heilongjiang Province (No 1154HZ11), the Key Research Program of the 2nd Affiliated Hospital of Harbin Medical University (No ZD2008-08), and the Postdoctoral Foundation of the 2nd Affiliated Hospital of Harbin Medical University (No ZD2010-01).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sun, Y., Zhang, Jn., Zhao, D. et al. Role of the epithelial sodium channel in salt-sensitive hypertension. Acta Pharmacol Sin 32, 789–797 (2011). https://doi.org/10.1038/aps.2011.72
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.72
Keywords
This article is cited by
-
A Systems Level Analysis of Vasopressin-mediated Signaling Networks in Kidney Distal Convoluted Tubule Cells
Scientific Reports (2015)
-
Proteases, cystic fibrosis and the epithelial sodium channel (ENaC)
Cell and Tissue Research (2013)
-
Direct Activation of ENaC by Angiotensin II: Recent Advances and New Insights
Current Hypertension Reports (2013)
-
Channelopathies and drug discovery in the postgenomic era
Acta Pharmacologica Sinica (2011)
