
Abstract
Background:
Topoisomerase I (Topo I) poisons (e.g., camptothecin (CPT)), used to treat cancer, cause DNA breaks that are most cytotoxic during S phase. PARP-1 promotes DNA repair and PARP inhibitors (PARPi) sensitise cells to Topo I poisons. We aimed to determine whether chemosensitisation is also S phase specific using rucaparib, a potent PARPi in advanced clinical evaluation.
Methods:
The impact of rucaparib, on CPT-induced cytotoxicity was measured in human colon cancer (LoVo) and leukaemic (K562) cells in asynchronous and cell cycle phase-separated cultures. Topoisomerase I and PARP levels and activity and the effect of rucaparib on DNA single-strand breaks (SSBs), double-strand breaks (DSBs) and collapsed replication fork induction and repair were determined in cell cycle phase-separated cells.
Results:
The cytotoxicity of CPT was greatest during S phase, partially attributable to high Topo I activity, and rucaparib preferentially sensitised S-phase cells. Rucaparib increased CPT-induced DNA SSBs in all phases of the cell cycle, and increased DSB and γH2AX foci in S and G2, with γH2AX foci being highest in S-phase cells. Repair of SSBs and DSBs was most rapid during S then G2 phases and was substantially hindered by rucaparib.
Conclusions:
Rucaparib preferentially sensitises S-phase cells by increasing the frequency of collapsed replication forks.
Similar content being viewed by others


Main
Topoisomerase I (Topo I) forms a reversible complex with DNA catalysing the formation and re-annealing of single-strand breaks (SSBs) in DNA to relieve torsional stress associated with transcription, replication or repair. Topo I poisons such as camptothecin (CPT) stabilise the complex in the broken conformation leading to persistent SSB. Their cytotoxicity is thought to be primarily due to collision of the replication fork with the cleavable complex forming a stalled replication fork and single-ended DNA double-strand break (DSB; reviewed in Pommier, 2006; Gilbert et al, 2012) and is highest during S phase. The cytotoxicity of the Topo I poisons correlates with the level of Topo I-generated DNA breaks, which is dependent on Topo I activity (Pfister et al, 2009). Topo I activity is higher in malignant cells and correlates with disease progression in colorectal and ovarian cancers, making it an attractive target for anticancer chemotherapy (van der Zee et al, 1991; Tsavaris et al, 2009; Smith et al, 2013). Camptothecin derivatives with improved pharmacological properties, irinotecan (Camptosar) and topotecan (Hycamptin), are used in the treatment of colorectal and ovarian cancers, respectively (Douillard et al, 2000). Targeting the repair of Topo I poison-mediated DNA damage may improve the activity of Topo I poisons.
Camptothecin-induced DNA lesions are repaired by overlapping DNA repair pathways. Topo I-associated SSBs are repaired by the base excision repair (BER) pathway (Caldecott and Jeggo, 1991; Barrows et al, 1998) and stalled replication forks and DSBs are primarily repaired by homologous recombination repair (HRR), including excision of Topo I from the DNA by the MRN exonuclease complex that initiates HRR (reviewed in Pommier, 2006; Gilbert et al, 2012). Base excision repair and HRR defects confer a five-fold and 10-fold sensitivity to CPT, respectively (Smith et al, 2005). Poly(ADP-ribose) polymerase-1 (PARP-1) plays a major role in the sensing and repair of DNA SSBs and DSBs. It is a key component of BER and contributes to the restart of stalled replication forks during HRR (Bryant et al, 2009). PARP-1 is activated by CPT-induced DNA breaks and, via recruitment of XRCC1 (El-Khamisy et al, 2003) it promotes the cleavage of Topo I from the DNA by tyrosyl-DNA phosphodiesterase 1 (TDP1) and subsequent DNA repair (Plo et al, 2003). PARP reduces CPT-induced replication fork reversal and limits DNA strand breakage (Ray Chaudhuri et al, 2012). Genetic inactivation of PARP-1 sensitises cells to Topo I poisons (Chatterjee et al, 1989) and PARP-1 null mice are hypersensitive to Topo I poison toxicity (Burkle et al, 2000). PARP inhibitors (PARPi) enhance the cytotoxicity of Topo I poisons in vitro (Delaney et al, 2000; Bowman et al, 2001; Smith et al, 2005; Miura et al, 2012) and in vivo (Miknyoczki et al, 2003; Calabrese et al, 2004; Tentori et al, 2006). Enhancement of Topo I activity by PARPi has been associated with inhibition of DNA repair (Bowman et al, 2001; Smith et al, 2005). Although Topo I poison-induced DNA breakage and cytotoxicity in replicating and non-replicating cells has been studied, little is known about the potentiation by PARPi during different phases of the cell cycle.
PARPi are undergoing clinical evaluation, including combinations with Topo I poisons and initial reports indicate that the PARPi, ABT-888 (Veliparib) increased topotecan-induced DNA breaks in circulating tumour cells (Kummar et al, 2011). A greater understanding of the interaction of PARPi with Topo I poisons is needed to optimise the combination clinically. Here we demonstrate that the clinically active PARPi, rucaparib (AG-014699) increases total CPT-induced DNA breaks and inhibits their repair in all phases of the cell cycle but increased DSB and γH2AX formation predominantly in S phase.
Materials and Methods
Chemicals
All chemicals and reagents, including tissue culture media, were provided by Sigma (Poole, Dorset, UK) unless otherwise stated. The PARP-1 inhibitor rucaparib(AG0140699: 1-(4 dimethylaminomethylphenyl)-8-9-dihydro-7H-2,7,9a-benzo(cd)azulen-6-one) provided by Agouron/Pfizer Pharmaceuticals GRD, La Jolla, CA, USA) was stored at −20°C at a concentration of 5 mM in water and used at a final concentration of 0.4 M. Camptothecin was stored at −20°C as 10 mM aliquots in anhydrous DMSO.
Cytotoxicity assays
Human colon carcinoma (LoVo) and chronic myelogenous leukaemia (K562) cells were obtained from the ATCC (Manassas VA, USA), maintained at low in RPMI 1640 culture medium supplemented with 10% fetal calf serum passage and authenticated by STR profiling (LGC standards, Teddington, UK). Cell survival was determined by clonogenic survival assay following exposure of exponentially growing cells to CPT±rucaparib as indicated in the Results section, prior to re-seeding for colony formation either directly (LoVo) or in 0.15% low melting point agarose (SeaKem ME Cambrex, Berks, UK) in medium (K562). Colonies were stained with crystal violet (LoVo) or MTT (K562).
Centrifugal elutriation
The use of chemicals, serum starvation or double thymidine block to synchronise cells have been criticised, not only for the cytotoxicity of these methods, but also their inability to genuinely synchronise cells (Cooper, 2003; Cooper et al, 2006, 2008). We therefore used centrifugal elutriation to separate the cells into different phases of the cell cycle. Cells were separated into G1, S and G2/M fractions using a Beckman Avanti J-20 centrifuge equipped with JE-5.0 elutriation rotor and Sanderson elutriation chamber (BeckmanCoulter.com). The apparatus was pre-sterilised with 6% H2O2, rinsed with sterile PBS and filled with sterile culture medium (flow rate 15 ml min−1) and a rotor speed of 2500 rpm prior to injecting 1.5–2.5 × 108 cells in 5 ml medium into the system to equilibrate. Cell fractions were collected into 50 ml tubes by increasing the flow rate by 5 ml min−1 under sterile conditions. An aliquot of each fraction was stained with propidium iodide and analysed by flow cytometry (FASCcan, with CellQuest software, BD Biosciences, Oxford, UK) and processed on ModFit LT software (Verity Software, Topsham, ME, USA). The purest fractions enriched with cells in G1, S and G2 cell cycle phase were selected for subsequent experiments.
PARP activity
PARP activity in digitonin-permeabilised cells was measured by immunological detection of the ADP-ribose polymer product with the 10H antibody (kind gift from Dr A Burkle, University of Konstanz, Konstanz, Baden-Württemberg, Germany) after maximal stimulation of PARP activity with exogenous oligonucleotide in the presence of an excess of NAD+ as previously described (Plummer et al, 2008).
Topo I activity
Topo I activity was measured using the Topo I relaxation activity kit (www.Topogen.com). Nuclear lysates were prepared in ice-cold TEMP buffer (10 mM Tris-HCL, pH=7.5, 1 mM EDTA, 4 mM MgCl2, 0.5 mM PMSF) by centrifugation at 1500 g for 10 min at 4°C followed by suspension in ice-cold TEP buffer (10 mM Tris-HCL, pH=7.5, 1 mM EDTA, 0.5 mM PMSF) and an equal volume of 1 M NaCl for 40 min followed by centrifugation at 15 000 g for 30 min (4°C) to remove final debris. The protein concentration of the supernatant was measured by BCA assay (Fisher Scientific, Loughborough, UK) and reactions were performed according to manufacturer’s instructions using proprietary supercoiled DNA and 1 μl of test extract. Reactions were terminated with loading buffer and loaded on to a 1% agarose gel. Electrophoresis was run at 2.5 V cm−1 in TAE buffer (40 mM Tris, pH=8, 20 mM acetic acid, 1 mM EDTA). The gel was than stained with 0.5 μg ml−1 ethidium bromide for 20 min, images were captured using the GelDoc system (Bio-Rad, Hemel Hempstead, UK) and a trans illuminator and analysed by ImageJ (http://imagej.nih.gov/ij/; gel analysing options).
DNA breakage and stalled replication forks
DNA breaks were determined by single cell gel electrophoresis using the Trevigen Comet Assay Kit (Trevigen, AMS Biotechnologies, Oxford, UK) according to the manufacturer’s protocol. All preparation was done under dimmed light to avoid additional DNA damage. Briefly, 5 × 105 cells, suspended in low melting point agarose were spread on the pre-coated wells of the comet slide and allowed to gel prior to lysis. To measure SSB (‘total’ breaks as a fraction will also be DSB and alkali-labile sites) comets slides were incubated in chilled alkaline solution (300 mM NaOH, 40 mM EDTA, pH>13) for 30 min at 4°C to denature the DNA strands prior to electrophoresis in alkaline solution at a current of 200–300 mA under applied voltage 0.75 V cm−1 for 30 min. To measure DSB electrophoresis was carried out with 1 × TBE, pH=10 buffer, which does not permit DNA denaturation, at 1 V cm−1 for 30 min. Breakage was assessed by determining the Olive tail moment (OTM: expressed as (tail mean−head mean) × % of DNA in the tail/100) in >100 cells for each experimental condition.
Camptothecin-induced DSB and stalled replication forks were also measured by immunofluorescence microscopy of H2AX phosphorylation (γH2AX; Furuta et al, 2003) in K562 cells deposited onto microscope coverslips using a Thermo-Shandon cytospin centrifuge (Fisher Scientific) and fixed in cold methanol at −20°C. Coverslips were incubated with anti-phospho serine 139 H2AX (clone JBW301, mouse monoclonal antibody; Upstate, Millipore Corp, Watford, UK) diluted 1 : 1000 in blocking buffer (10% milk, 0.1% TritonX-100) for 1 h at 37°C, and secondary antibody (goat polyclonal to Mouse IgG antibody Chromeo 546) at a dilution of 1 : 1000 for 1 h at 37°C, mounted onto microscope slides using Vector-shield Mounting Medium (Vector Laboratories, Peterborough, UK). For each sample, two images of DAPI and two corresponding images of γH2AX were recorded on a Leica Epi-fluorescence Microscope using Image Spot Advance software and analysed by ImageJ and a custom macro, PZFociEZ (http://www.pzfociez.com/), to count the number of foci/nucleus in at least 100 nuclei per assay.
Results
We investigated the potentiation of CPT by the PARPi, rucaparib in human colon carcinoma, LoVo cells, because the Topo I poison, irinotecan, is commonly used to treat colorectal cancer. There was a marked time-dependency of the sensitivity of these cells, with 99% of cells killed by a 24-h exposure to 10 nM CPT (Figure 1A) but only 45% killed by a 1-h exposure to the same concentration of CPT (Figure 1B). Indeed, there was a substantial CPT-resistant population as increasing the CPT concentration to 300 nM only resulted in 63% cell kill after 1 h (Figure 1B). There was a 1.5- to two-fold enhancement of CPT cytotoxicity by the PARPi, rucaparib, during the 24 h exposure but only a 1.1 to 1.3-fold enhancement after a 1-h exposure. We postulated that the difference in sensitivity to CPT after a 1-h compared to a 24-h exposure may have been due to the fraction of cells passing through S phase during the exposure period. LoVo cells have a cell cycle time of ∼24 h with ∼40% of cells in S phase (Supplementary Figure 1), suggesting that the cells killed by a 1-h exposure to CPT were the 40–45% of cells that passed through S phase during the exposure period. To investigate the cell cycle-dependency further we examined the cytotoxicity of CPT at different phases of the cell cycle. Rather than using synchronisation of the cells with cytotoxic chemicals or nutrient deprivation we separated the cells into different phases by centrifugal elutriation. Since this requires the cells to be in suspension we determined the cytotoxicity of a 1-h exposure of LoVo cells in suspension to CPT, with and without rucaparib, and the data were not significantly different from those obtained after exposure of adherent LoVo cells (Figure 1B). It proved difficult to obtain pure populations of cells in the different phases due to the high degree of aneuploidy in these cells (Supplementary Figure 1; Drewinko et al, 1976). Nevertheless, using cell cycle phase-enriched populations it was apparent, not only that CPT-induced cytotoxicity was greatest during S phase, but also that PARP inhibition only sensitised S-phase cells to CPT (Table 1).

Effect of the PARP inhibitor, rucaparib, on clonogenic survival of LoVo cells. Exponentially growing LoVo cells were treated with increasing concentrations of CPT in the presence ( ○ ) or absence ( ● ) of rucaparib for (A) 24 h or (B) 1 h. The comparison between incubating LoVo cells either as a monolayer or in suspension is also shown in (B), where cells in suspension were exposed to CPT in the presence (Δ) or absence (▴) of rucaparib.
Cells that have a stable modal chromosome number and are not highly aneuploid, such as the human myeloid leukaemia K562 cells, (Chen, 1985) make the best candidates for cell cycle phase separation by centrifugal elutriation and it was possible to obtain G1, S and G2 fractions that were 85% pure (Supplementary Figure 2). Following the cell cycle progression of the G1 fraction of K562 cells indicated that most cells remained in G1 at 2 h but 90% had progressed to S phase by 4 h (Supplementary Figure 3) and progression to G2 occurred between 8 and 10 h. Centrifugal elutriation did not affect the viability of these cells since the G1, S and G2 fractions grew at the same rate as the asynchronous, non-elutriated cells (Supplementary Figure 4) and it did not induce DNA strand breaks (Supplementary Figure 5). Previous studies have demonstrated that PARP inhibitors sensitise asynchronous K562 cells to CPT (Smith et al, 2005). We now show that, like the LoVo cells, not only are the S-phase K562 cells two-fold more sensitive to CPT alone, as expected, but also sensitisation by PARP inhibition was ⩾two-fold in the S-phase fraction but not significant in G1 or G2 (Figure 2).

Cell survival elutriated K562 cells. Elutriated K562 cells in G1, S and G2 cell cycle phases were exposed to 10 or 100 nM CPT for 1 h in the presence (black bars) or absence (white bars) of 0.4 μM rucaparib. Cytotoxicity was measured by clonogenic assay and survival was expressed as percentage of untreated control. Data are mean of three independent experiments±s.e.m.
A major determinant of sensitivity to CPT is Topo I activity, which we found to be greatest in S phase (Figure 3A and B), suggesting that this may contribute to the greater sensitivity of this fraction. Topo I activity appeared not to be related to Topo I protein levels as these showed a modest increase as the cells progressed from G1 to G2 (Supplementary Figure 6). Differences in Topo I activity did not seem to adequately explain the differential sensitivity of the different phases as Topo I activity was around three-fold higher in G2 compared to G1 (Figure 3A and B), but the sensitivity of G1 and G2 fractions to CPT was similar (Figure 2). Also, changes in PARP activity in different phases of the cell cycle did not appear to be determined by PARP-1 protein levels (Supplementary Figure 6) and did not explain the greater sensitisation of S-phase cells: we found that PARP activity/cell increased throughout the cell cycle and was approximately twice as high in G2 compared to G1 (Figure 3C). This may reflect the increase in DNA content as PARP-1 is reported to be present at the concentration of 1 molecule per kB DNA (D’Amours et al, 1999).

Topo I and PARP activity in G1, S and G2 elutriated K562 cells. (A) Nuclear extracts were prepared from asynchronous or elutriated cells. Approximately, 1 μl of the extracts (containing from 0.01 to 0.15 μg protein) was incubated with 1 μg of supercoiled plasmid and reaction products were separated on agarose gel. ‘Sc’ refers to a negative control supercoiled plasmid, and ‘R’ is a positive control of fully relaxed plasmid. Samples from asynchronous (AS), and G1, S and G2 are shown with increasing amount of protein extract, going from left to right. (B) the bands from A were quantified using ImageJ gel analysis, and Topo I activity is shown as the amount of relaxed DNA as a percentage of total DNA. (C) Exponentially growing K562 cells were elutriated and fractions containing >90% cells in G1, S and G2 cell cycle phases were used to measure PARP-1 activity (data are mean±s.e.m. of three independent experiments).
Since the cytotoxicity of Topo I poisons is related to the number of DNA breaks induced, we determined whether CPT-induced DNA breakage in the presence and absence of rucaparib explained the cell cycle-dependent sensitivity of K562 cells. Measurement of DNA breakage by single cell gel electrophoresis (comet assay) under alkaline conditions measures DNA SSBs and DSBs and alkali-labile sites, however, the vast majority of the breaks will be single stranded. Camptothecin induced a concentration-dependent increase in breaks at all phases of the cell cycle (Figure 4A). DNA breakage was lowest in G1-phase cells with both S and G2-phase cells having substantially higher numbers of DNA breaks. PARP inhibition significantly increased DNA breakage by >two-fold (P=0.08 to 0.01) in all phases of the cell cycle (Figure 4A) such that DNA break levels induced by CPT+rucaparib were similar in S and G2 and both were higher than in G1.

The effect of PARP inhibition on the level of DNA breaks in cell cycle phase-separated K562 cells. (A) Exponentially growing K562 cells were separated into cell cycle phases and exposed to increasing concentrations of CPT in the presence (open symbols) or absence (solid black symbols) of 0.4 μM rucaprib (R) for 30 min, prior to measurement of total DNA breaks by alkaline comet assay. Graphs showing mean Olive tail moment (OTM)±s.e.m. from four independent experiments expressed as percentage of control untreated cells. (B) Similarly, elutriated (G1, S, and G2) K562 cells were exposed to increasing concentrations of CPT±0.4 μ M rucaprib (R) for 30 min, and DNA DSB measured using the neutral comet assay. Data are mean+s.e.m. from single representative experiment expressed as a percentage of control. (C) K562 cells were treated with increasing concentrations of CPT in the presence (black bars) or absence (white bars) of 0.4 μ M rucaparib for 30 min, and then separated into individual cell cycle phases and stained for the presence of γH2AX foci. Foci number per cell was measured using ImageJ and the macro PZFociEZ, as described in methods. Data show mean±95% confidence interval calculated from at least 100 cells per treatment, from one experiment (representative of three individual experiments).
DNA DSBs are more profoundly cytotoxic than SSBs and the cytotoxicity of Topo I poisons is thought to be related to DSB formation at replication. Surprisingly, measurement of DSB formation by CPT indicated that they were only slightly higher in S-phase cells (Figure 4B). Rucaparib did not significantly increase the DSBs in G1-phase cells but increased the DSBs in S and G2-phase cells 1.5 to 1.8-fold (P=0.03). We also measured stalled replication forks together with DSBs by determining γH2AX foci formation following exposure to CPT and rucaparib. Not surprisingly, foci numbers per cell were lowest in G1-phase cells and highest in S-phase cells following exposure to CPT alone (Figure 4C). PARP inhibition barely altered the foci levels in G1-phase cells but caused a 1.7-fold and 2.4-fold increase in foci in S-phase cells exposed to 10 and 1000 nM CPT, respectively (P<0.0001 for both concentrations), with the corresponding increases in G2-phase cells being two-fold and 1.8-fold (P=0.06 and <0.0001, respectively). Cells exposed to the combination during S phase had the highest number of foci.
Although rucaparib increased DNA SSBs and DSBs to a similar extent in S- and G2-phase cells (Figure 4) it caused a much more profound cytotoxic sensitisation of S-phase cells (Figure 2) and our previous studies indicated that the effect of PARP inhibition on DNA repair was an important factor in CPT sensitisation (Smith et al, 2005). We therefore investigated whether DNA repair differed at different phases of the cell cycle and whether there was a cell cycle-dependent differential effect of rucaparib on repair. After a 2-h recovery period in drug-free medium there was substantial repair of the total breaks induced by a 30-min pulse of 100 nM CPT (Figure 5A). Repair was more rapid in S phase, with only 38% of breaks remaining at 2 h compared to 46 and 48% in G1 and G2, respectively. PARP inhibition retarded repair in all phases of the cell cycle but with a greater effect in S- and G2- phase cells. Investigation of DNA DSBs indicated that these too were rapidly repaired in S and G2 phases, such that only 25% of breaks remained after 2 h (Figure 5B). In G1-phase cells fewer DSBs were detected and they were repaired more slowly, with 55–60% of breaks remaining after 2 h incubation in fresh medium. Rucaparib inhibited repair at all phases of the cell cycle such that 75–80% of the breaks remained unrepaired after 2 h.

The effect of PARP-1 inhibition on DNA strand break repair in elutriated K562 cells. (A) K562 cells were separated into G1, S and G2 phases, and treated with 100 nM CPT for 30 min (black bars). After 2 h of incubation in drug-free medium (white bars) or rucaprib (R)-containing medium (grey bars) the break levels were measured by alkaline comet assay. The level of total DNA breaks at the end of 2-h recovery period was evaluated by expressing the breaks remaining as percentage of those at the end of the 30 min CPT exposure (time 0) using the formula: 100 × T/C × √((t/T)2 + (c/C)2) where c is s.d. of time 0 (C) and t is s.d. of post 2 h of recovery (T). Data showed mean+s.e.m. of three independent experiments. (B) Similarly, the effect of rucparib on the repair of double-strand breaks was measured using the neutral comet assay. K562 cells were separated into G1, S and G2 phases, and treated with 100 nM CPT for 30 min (black bars). After 2 h of incubation in drug-free medium (white bars) or rucaparib (R)-containing medium (grey bars) the percentage of DSBs remaining were assessed as described in A.
Discussion
Topo I poisons have been in clinical use for several years, showing efficacy in colorectal tumours (Douillard et al, 2000). One promising new way to improve the effectiveness of Topo I poison therapy is the use of PARPi. PARPi are currently undergoing clinical trial but their use in combination with Topo I poisons has not been optimised (Gilbert et al, 2012). Veliparib has been investigated in phase I clinical trials with both topotecan and irinotecan, and olaparib with topotecan (Kummar et al, 2011; LoRusso et al, 2011; Samol et al, 2012). In the topotecan study, veliparib reduced PARP activity in both tumour and peripheral blood mononuclear cells (PBMC), increased DNA breaks in circulating tumour cells and PBMCs, and importantly resulted in some disease stabilisation. However, in all these studies the toxicities associated with the Topo I poison were exacerbated, indicating that improvements in either dosing or scheduling are needed.
Initially our studies focussed on the colorectal cell line, LoVo. Results revealed that CPT-induced cytotoxicity was both time- and concentration-dependent, with virtually all cells killed by low concentrations (10 nM) of CPT when cells were exposed for a complete cell cycle. However only ∼50% of cells were killed by a short pulse with much higher concentrations of CPT, where the plateau in survival suggested a resistant subpopulation. On the basis of published literature, and our data showing that 40% of Lovo cells are in S phase, we assumed that the S-phase cells represented the sensitive fraction. We estimated that only 40–50% of cells would pass through S phase and be killed during the 1-h exposure period, compared to virtually all cells passing through S phase over 24 h. Inhibition of PARP-1 activity by rucaparib cells resulted in a two-fold sensitisation of CPT-induced cytotoxicity during 24-h exposure, consistent with previous reports using other PARP inhibitors and cell lines (Bowman et al, 2001; Calabrese et al, 2004; Bryant et al, 2009). Potentiation of CPT-induced cytotoxicity during a short pulse exposure appeared to be slightly greater than at 24 h, suggesting that potentiation may be greater for S-phase cells.
We used centrifugal elutriation, to separate the cells into different phases of the cell cycle. This technique did not cause DNA damage and, since the S-phase fraction did not have a higher alkaline comet OTM, Okazaki fragments formed during S phase did not contribute to comet tails. Neither did it affect cell viability. In both Lovo and K562 cells CPT was substantially more cytotoxic to S-phase cells than to G1- or G2-phase cells. Rucaparib did not significantly enhance CPT cytotoxicity to Gl- and G2-phase cells in either of the cell lines but caused an approximately two-fold sensitisation of CPT in S-phase Lovo and K562 cells. The S-phase specificity of enhancement by PARP-1 inhibitors is not exclusive to Topo I poisons: veliparib potentiated the DNA methylating agent, temozolomide to a much greater extent in S phase compared to G1 (Liu et al, 2008).
Increased sensitivity to Topo I poisons has been related to elevated levels of Topo I (Pfister et al, 2009). We found that Topo I activity was highest in S-phase K562 cells, linking the increased sensitivity to CPT during this phase to increased Topo I activity. However, Topo I activity in G2 was greater than in G1 but the cytotoxicity was similar in both phases. The high Topo I activity in G2 was associated with a higher level of DNA SSB but not the much more cytotoxic DSB. Cytotoxicity may be related more to DSB and particularly collapsed replication forks that were highest in S phase. In contrast, PARP activity increased throughout the cell cycle, and since the level in G2 was two-fold higher than in G1, this may be related to DNA content. Cell cycle phase changes in PARP-1 and Topo I activity were not related to the levels of PARP-1 or Topo I expression suggesting that both proteins are regulated by posttranslational modification. The highest activity of PARP occurred in G2 phase, but the effect of rucaparib on Topo I activity was no greater in G2 than in other phases, consistent with our previous observations that PARP inhibition did not increase Topo I activity (Smith et al, 2005).
Investigation of the mechanism underlying the greater potentiation of CPT cytotoxicity in S phase by PARP inhibition focussed on measuring the induction of DNA breaks and their repair. Alkaline comets measure total DNA breakage, but in the context of Topo I poisons, these will be largely SSB. The data indicated that although more SSBs were formed in S and G2 phases (possibly as a function of both the Topo I activity and DNA content), rucaparib increased these CPT-induced breaks to a similar degree in all phases of the cell cycle. Rucaparib also inhibited the repair of these lesions to a similar extent in all phases of the cell cycle. Therefore, it does not appear that the effect of rucaparib on SSB levels is responsible for the S-phase-specific sensitisation.
During S phase, the CPT-stabilised Topo I-DNA complexes can be converted into highly cytotoxic collapsed replication forks and DSBs. The hypothesis that these lesions were more closely associated with chemosensitisation was investigated using two methods; neutral comet assay and γH2AX focus formation. These complementary assays measure subtly different end points: neutral comet assays measure the migration of broken DNA into a gel under an electric current and are analogous to pulse-field gel electrophoresis (Olive, 2009) and the γH2AX focus assay measures the phosphorylation of H2AX by DNA damage-activated kinases (ATM, ATR and DNA-PK) and is therefore a sensitive measure of collapsed replication forks as well as DSBs (Darzynkiewicz et al, 2009). The advantage of both these methods is that analysis of individual cells is made allowing population dynamics to be discovered. Measurement of DSB induction by neutral comet assays revealed that they were indeed higher during S phase and that rucaparib significantly increased the level of DSBs in S phase. Similar studies showed that increased cytotoxicity of temozolomide by the PARP inhibitor ABT-888 in S phase was correlated with the level of DSB (Liu et al, 2008). We found that γH2AX foci were low in G1 and highest in S phase as expected, and very substantially increased by rucaparib in S phase. However, the increase in γH2AX foci in G2 cells was not accompanied by a corresponding increase in the induction of DSB as measured by neutral comets. Recent data implicate the XPF-ERCC1 endonuclease in an alternative pathway to PARP-TDP-1 for the removal of CPT-stabilised Topo I-DNA complexes and the induction of γH2AX foci (Zhang et al, 2011). XPF-ERCC1 is key to nucleotide excision repair of UV damage, which is known to induce γH2AX foci (Revet et al, 2011). It is possible that PARP inhibition shifts repair to this pathway leading to an increase in γH2AX foci and our data would suggest that this occurs in S and G2. Interestingly, the increase in γH2AX foci in G2 by rucaparib was not accompanied by a significant increase in cytotoxicity. However, the role of XPF-ERCC1-mediated repair of CPT-induced DNA damage and cytotoxicity is not entirely clear as although XPF knockdown was shown to reduce γH2AX foci it only caused a very marginal increase in CPT sensitivity, and ABT-888 sensitised cells irrespective of XPF levels (Zhang et al, 2011).
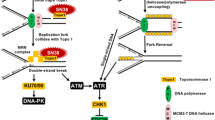
Our data indicate that although PARP inhibition does not cause a greater induction of DSB in G2 it does impede DNA DSB resolution in G2 as well as S phase, but this is only critical to survival in S phase. Recent data suggest that CPT causes replication fork slowing (independently of DSB formation) and that PARP is needed to reverse arrested replication forks after exposure to Topo I poisons, such that PARP inhibition prevented fork reversal and restart leading to a greater accumulation of γH2AX foci (Ray Chaudhuri et al, 2012). Considering our data in the light of the model proposed by (Ray Chaudhuri et al, 2012) and the established role of PARP in the repair of Topo I poison-induced damage by BER, together with the mechanisms described by (Zhang et al, 2011), we propose a modified model (Figure 6). In this model Topo I-associated SSB are repaired by PARP and XRCC1-mediated sequential recruitment of TDP-1 then DNA Polβ and ligase I to execute repair. In the presence of rucaparib, SSB persist in G1 but in G2 XPF-ERCC1 may excise a portion of Topo I-bound DNA, creating NER intermediates that contribute to the measurement of SSB in alkaline comets and also activate H2AX phosphorylation. In S phase the DNA SSB stall replication forks and convert to DSB that activate H2AX phosphorylation. As the increase in γH2AX foci was the most remarkable effect of rucaparib during S phase, we suggest that inhibition of replication fork restart has the most profound implications for Topo I poison cytotoxicity.

A model for repair of Topo I-induced lesions during the cell cycle. Topo I poisons (e.g., camptothecin) stabilise the covalent complex between DNA and Topo I protein. Recruitment of TDP1 (which hydrolyses the 3′-phosphotyrosyl bond that links Topo I to the DNA) is mediated via PARP and XRCC1. Base excision repair (BER) is completed by PARP and XRCC1 in concert with in-filling activity of POLB and ligase1. The presence of a PARP inhibitor results in the persistence of SSB in G1, and during S phase and homologous recombination repair (HRR), the SSB stall replication forks and create DSB that stimulate phosphorylation of H2AX. In G2, PARP inhibition may lead to activation of XPF/ERCC1-mediated nucleotide excision repair (NER) to remove the Topo I-bound DNA, which creates a NER intermediate that activates H2AX phosphorylation.
The data presented here show that PARP inhibition causes S-phase-specific chemosensitisation of Topo I poisons that is related to the impact of PARP inhibition on stalled replication forks. This has clinical implications suggesting that rapidly growing tumours would be most sensitive to the combination, and that scheduling is critical to ensure both drugs are present for long enough for all tumour cells to enter S phase. Cancer cells generally have dysfunctional DNA cell cycle control and/or repair pathways, which underlie their differing vulnerabilities to a spectrum of cytotoxic agents (Curtin, 2012). We anticipate, but have not tested directly, that normal cells would also be most sensitive to Topo I poisons, alone and in combination with PARPi, during S phase. Most normal cells are in G1/G0 and replicating normal cells generally enter S phase in a synchronous fashion (Mormont and Levi, 2003) including cells in the gut mucosa, the site of dose-limiting toxicity by Topo I poisons. In mice (a nocturnal species) the peak in S phase occurs at 1.00 am and Topo I poisons are profoundly toxic when administered at 0200 hours (15% survival) compared to 1400 hours (90% survival; reviewed in Rich et al, 2002). Similarly, in humans the peak in S phase is around midday (Smaaland et al, 2002) and in clinical trials irinotecan toxicity was less toxic if administered at 0500 hours. These data support the hypothesis that normal cells are also more sensitive to Topo I poisons during S phase and we would predict that the combination of a Topo I poison and a PARPi would also be more toxic in this phase. Using a chronotherapy approach it may therefore be possible to schedule the treatment to target the cancers, which are asynchronous but spare replicating normal cells, which generally enter S phase in a synchronous manner.
Change history
23 September 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Barrows LR, Holden JA, Anderson M, D’Arpa P (1998) The CHO XRCC1 mutant, EM9, deficient in DNA ligase III activity, exhibits hypersensitivity to camptothecin independent of DNA replication. Mutat Res 408: 103–110.
Bowman KJ, Newell DR, Calvert AH, Curtin NJ (2001) Differential effects of the poly(ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity. Br J Cancer 84: 106–112.
Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T (2009) PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 28: 2601–2615.
Burkle A, Schreiber V, Dantzer F, Oliver FJ, deMurcia G, Menissier-de Murcia J (2000) Biological significance of poly(ADP-ribosylation) reactions: molecular and genetic approaches. In: From DNA Damage and Stress Signalling to Cell Death: Poly ADP-Ribosylation Reactions, de Murcia G, Shall S (eds.), pp 80–124 O.U.P: Oxford, UK.
Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, Durkacz BW, Hostomsky Z, Kumpf RA, Kyle S, Li J, Maegley K, Newell DR, North M, Notarianni E, Stratford IJ, Skalitzky D, Thomas HD, Wang L-Z, Webber SE, Williams KJ, Curtin NJ (2004) Preclinical evaluation of a novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor, AG14361, with significant anticancer chemo- and radio-sensitization activity. J Natl Cancer Inst 96: 56–67.
Caldecott K, Jeggo P (1991) Cross-sensitivity of gamma-ray-sensitive hamster mutants to cross-linking agents. Mutat Res 255: 111–121.
Chatterjee S, Cheng MF, Trivedi D, Petzold SJ, Berger NA (1989) Camptothecin hypersensitivity in poly(adenosine diphosphate-ribose) polymerase-deficient cell lines. Cancer Commun 1: 389–394.
Chen TR (1985) Modal karyotype of human leukemia cell line, K562 (ATCC CCL 243). Cancer Genet Cytogenet 17: 55–60.
Cooper S, Chen KZ, Ravi S (2008) Thymidine block does not synchronize L1210 mouse leukaemic cells: implications for cell cycle control, cell cycle analysis and whole-culture synchronization. Cell Prolif 41: 156–167.
Cooper S, Iyer G, Tarquini M, Bissett P (2006) Nocodazole does not synchronize cells: implications for cell-cycle control and whole-culture synchronization. Cell Tissue Res 324: 237–242.
Cooper S (2003) Rethinking synchronization of mammalian cells for cell cycle analysis. Cell Mol Life Sci 60: 1099–1106.
Curtin NJ (2012) DNA repair dysregulation, from cancer driver to therapeutic target. Nature Rev Cancer 12: 108–117.
D’Amours D, Desnoyers S, D’Silva I, Poirier GG (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342: 249–268.
Darzynkiewicz Z, Halicka DH, Tanaka T (2009) Cytometric assessment of DNA damage induced by DNA topoisomerase inhibitors. Methods Mol Biol 582: 145–153.
Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, Durkacz BW, Hostomsky Z, Newell DR (2000) Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res 6: 2860–2867.
Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, Gruia G, Awad L, Rougier P (2000) Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 355: 1041–1047.
Drewinko B, Romsdahl MM, Yang LY, Ahearn MJ, Trujillo JM (1976) Establishment of a human carcinoembryonic antigen-producing colon adenocarcinoma cell line. Cancer Res 36: 467–475.
El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW (2003) A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res 31: 5526–5533.
Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, Nussenzweig A, Aladjem MI, Bonner WM, Pommier Y (2003) Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem 278: 20303–20312.
Gilbert DC, Chalmers AJ, El-Khamisy SF (2012) Topoisomerase I inhibition in colorectal cancer: biomarkers and therapeutic targets. Br J Cancer 106: 18–24.
Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, Jia L, Weil M, Speranza G, Murgo AJ, Kinders R, Wang L, Parchment RE, Carter J, Stotler H, Rubinstein L, Holingshead M, Melillo M, Pommier Y, Bonner W, Tomaszewski JE, Doroshow JH (2011) Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumours and lymphomas. Cancer Res 71: 5626–5634.
Liu X, Shi Y, Guan R, Donawho C, Luo Y, Palma J, Zhu GD, Johnson EF, Rodriguez LE, Ghoreishi-Haack N, Jarvis K, Hradil VP, Colon-Lopez M, Cox BF, Klinghofer V, Penning T, Rosenberg SH, Frost D, Giranda VL (2008) Potentiation of temozolomide cytotoxicity by poly(ADP)ribose polymerase inhibitor ABT-888 requires a conversion of single-stranded DNA damages to double-stranded DNA breaks. Mol Cancer Res 6: 621–629.
LoRusso P, Ji JJ, Li J, Heilbrun LK, Shapiro G, Sausville EA (2011) Phase I study of the safety, pharmacokinetics, and pharmacodynamics of the poly(ADP-ribose) polymerase (PARP) inhibitor veliparib (ABT-888; V) in combination with irinotecan (CPT-11) in patients with advanced solid tumors. J Clin Oncol 29 (suppl): abstract 3000.
Miknyoczki SJ, Jones-Bolin S, Pritchard S, Hunter K, Zhao H, Wan W, Ator M, Bihovsky R, Hudkins R, Chatterjee S, Klein-Szanto A, Dionne C, Ruggeri B (2003) Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Cancer Ther 2: 371–382.
Miura K, Sakata K, Someya M, Matsumoto Y, Matsumoto H, Takahashi A, Hareyama M (2012) The combination of olaparib and camptothecin for effective radiosensitization. Radiat Oncol 7: 62.
Mormont MC, Levi F (2003) Cancer chronotherapy: principles, applications, and perspectives. Cancer 97: 155–169.
Olive PL (2009) Impact of the comet assay in radiobiology. Mutat Res 681: 13–23.
Pfister TD, Reinhold WC, Agama K, Gupta S, Khin SA, Kinders RJ, Parchment RE, Tomaszewski JE, Doroshow JH, Pommier Y (2009) Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol Cancer Ther 8: 1878–1884.
Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y (2003) Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair 2: 1087–1100.
Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, Curtin N, Boddy A, McHugh P, Newell D, Harris A, Johnson P, Steinfeldt H, Dewji R, Wang D, Robson L, Calvert H (2008) Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res 14: 7917–7923.
Pommier Y (2006) Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 6: 789–802.
Ray Chaudhuri A, Hashimoto Y, Herradoe R, Neelsen KL, Fachinetti D, Bermejo R, Cocito A, Constanzo V, Lopes M (2012) Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19: 417–424.
Revet I, Feeney L, Bruguera S, Wilson W, Dong TK, Oh DH, Dankort D, Cleaver JE (2011) Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc Natl Acad Sci USA 108: 8663–8667.
Rich TA, Shelton CH III, Kirichenko A, Straume M (2002) Chronomodulated chemotherapy and irradiation: an idea whose time has come? Chronobiol Int 19: 191–205.
Samol J, Ranson M, Scott E, Macpherson E, Carmichael J, Thomas A, Cassidy J (2012) Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs 30: 1493–1500.
Smaaland R, Southern RB, Laerum OD, Abrahamson JF (2002) Rhythms in human bone marrow and blood cells. Chronobiol Int 19: 101–127.
Smith DH, Christensen IJ, Jensen NF, Markussen B, Rømer MU, Nygård SB, Müller S, Nielsen HJ, Brünner N, Nielsen KV (2013) Mechanisms of topoisomerase I (TOP1) gene copy number increase in a stage III colorectal cancer patient cohort. PLoS One 8: e60613.
Smith LM, Willmore E, Austin CA, Curtin NJ (2005) The novel poly (ADP-Ribose) polymerase inhibitor, AG14361, sensitizes cells to topoisomerase I poisons by increasing the persistence of DNA strand breaks. Clin Cancer Res 11: 8449–8457.
Tentori L, Leonetti C, Scarsella M, Muzi A, Mazzon E, Vergati M, Forini O, Lapidus R, Xu W, Dorio AS, Zhang J, Cuzzocrea S, Graziani G (2006) Inhibition of poly(ADP-ribose) polymerase prevents irinotecan-induced intestinal damage and enhances irinotecan/temozolomide efficacy against colon carcinoma. FASEB J 20: 1709–1711.
Tsavaris N, Lazaris A, Kosmas C, Gouveris P, Kavantzas N, Kopterides P, Papathomas T, Agrogiannis G, Zorzos H, Kyriakou V, Patsouris E (2009) Topoisomerase I and IIalpha protein expression in primary colorectal cancer and recurrences following 5-fluorouracil-based adjuvant chemotherapy. Cancer Chemother Pharmacol 64: 391–398.
van der Zee AG, Hollema H, de Jong S, Boonstra H, Gouw A, Willemse PH, Zijlstra JG, de Vries EG (1991) P-glycoprotein expression and DNA topoisomerase I and II activity in benign tumors of the ovary and in malignant tumors of the ovary, before and after platinum/cyclophosphamide chemotherapy. Cancer Res 51: 5915–5920.
Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y (2011) Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res 39: 3607–3620.
Acknowledgements
We are grateful to Dr Zdenek Hostomsky for providing rucaparib and to CR UK for financial support (grant ref C5201/A6710).
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Znojek, P., Willmore, E. & Curtin, N. Preferential potentiation of topoisomerase I poison cytotoxicity by PARP inhibition in S phase. Br J Cancer 111, 1319–1326 (2014). https://doi.org/10.1038/bjc.2014.378
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.378
Keywords
This article is cited by
-
The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: a review
Cell & Bioscience (2020)
-
Insight into DNA substrate specificity of PARP1-catalysed DNA poly(ADP-ribosyl)ation
Scientific Reports (2020)
-
2-Nitroimidazoles induce mitochondrial stress and ferroptosis in glioma stem cells residing in a hypoxic niche
Communications Biology (2020)
-
CHK1 and RAD51 activation after DNA damage is regulated via urokinase receptor/TLR4 signaling
Cell Death & Disease (2016)
