
Abstract
Mutation in the TP53 gene positively correlates with increased incidence of chemoresistance in different cancers. In this study, we investigated the mechanism of chemoresistance and epithelial-to-mesenchymal transition (EMT) in colorectal cancer involving the gain-of-function (GOF) mutant p53/ephrin-B2 signaling axis. Bioinformatic analysis of the NCI-60 data set and subsequent hub prediction identified EFNB2 as a possible GOF mutant p53 target gene, responsible for chemoresistance. We show that the mutant p53-NF-Y complex transcriptionally upregulates EFNB2 expression in response to DNA damage. Moreover, the acetylated form of mutant p53 protein is recruited on the EFNB2 promoter and positively regulates its expression in conjunction with coactivator p300. In vitro cell line and in vivo nude mice data show that EFNB2 silencing restores chemosensitivity in mutant p53-harboring tumors. In addition, we observed high expression of EFNB2 in patients having neoadjuvant non-responder colorectal carcinoma compared with those having responder version of the disease. In the course of deciphering the drug resistance mechanism, we also show that ephrin-B2 reverse signaling induces ABCG2 expression after drug treatment that involves JNK-c-Jun signaling in mutant p53 cells. Moreover, 5-fluorouracil-induced ephrin-B2 reverse signaling promotes tumorigenesis through the Src-ERK pathway, and drives EMT via the Src-FAK pathway. We thus conclude that targeting ephrin-B2 might enhance the therapeutic potential of DNA-damaging chemotherapeutic agents in mutant p53-bearing human tumors.
Similar content being viewed by others


Main
Ephrin-B2, member of the largest receptor tyrosine kinase family, is a bidirectional signaling molecule important in development,1 vascularization,2 nervous system patterning (axon guidance),3, 4 angiogenesis,5, 6 and cancer.5 The presence of the Eph receptor on neighboring cells is a key determinant in activating the signaling cascade in the interacting ephrin-B2 expressing cells.5
The TP53 gene is frequently mutated in human cancers.7 It is evident that many of these mutant forms of the p53 protein not only lose their tumor-suppressive function, but also gain new oncogenic properties independent of the wild-type function.8, 9 Termed the gain-of-function (GOF) hypothesis, this notion first received support when the ectopic expression of different missense p53 mutants (R175H, R273H, D281G, and V143A) in TP53-null cells was shown to promote tumor formation in mice.10 GOF mutant p53 proteins are divided into two main classes based on their structures. Members of the first class have disrupted sequence-specific contacts with DNA (DNA contact mutants; for example, R273H), whereas those belonging to the second have disrupted global conformation (conformational/structural mutants; for example, R175H).7
Tumors carrying mutant p53 do not respond well to chemotherapy.11, 12 Clonogenic survival assays revealed that mutant p53-expressing cells are more refractory to the cytotoxic effects of chemotherapeutics.13 The overexpression of GOF mutant p53 might directly enhance tumor cell resistance to anticancer agents.14, 15 Such selective GOF of mutant p53 might contribute to chemotherapeutic failure.12
In this study, we performed systematic bioinformatic analyses to build a novel prediction model, identifying several potential GOF mutant p53 target genes. We show that one such target gene, EFNB2, is responsible for chemoresistance. Furthermore, silencing of EFNB2 promoted high sensitivity to 5-fluorouracil (5-FU) both in vitro and in vivo. We further explored the mechanism of chemoresistance and epithelial-to-mesenchymal transition (EMT) involving drug-induced ephrin-B2 reverse signaling in mutant p53-harboring tumors.
Results
Predicting gene signatures that confer chemoresistance to mutant p53 tumors
We used a ‘response engineering module’ to connect the inherent gene signature of mutant p53 cancer cells with chemoresistance16 (Supplementary Figure S1A). The NCI-60 cell lines (http://dtp.nci.nih.gov) with known sensitivity against 1400 chemical compounds17 were classified into p53 wild-type and mutant groups (Supplementary Table S1 and S2). Cell lines with normalized log10 (GI50) values at least 0.8 above or below the mean were defined as resistant or sensitive to the respective compounds (Supplementary Figure S1B). We selected 36 compounds that satisfied the above criteria for at least 10 cell lines (five each for resistant and sensitive), and showed significant difference (P<0.05) between the number of sensitive and resistant mutant p53 cell lines (Supplementary Table S3). Interestingly, most of the p53 mutant cell lines showed chemoresistance, whereas wild-type p53 cell lines were sensitive (Figure 1a). 5-FU-resistant and -sensitive cell lines of the NCI-60 cell repertoire are enlisted in Supplementary Table S4. To generate the gene expression signature, we selected four commonly used anticancer drugs: 5-FU, doxorubicin, carboplatin, and camptothecin. We further refined the analysis by classifying the NCI-60 cell lines based on the presence of GOF mutant and wild-type p53 (Supplementary Table S5). The analysis revealed that GOF mutant p53-bearing cell lines were mostly resistant to at least 37 compounds compared with those bearing wild-type p53 (P<0.05; Supplementary Figure S1C). Interestingly, all the drugs except camptothecin appeared in both the analyses (Figure 1a and Supplementary Figure S1C).

Predicting gain-of-function (GOF) mutant p53 regulated gene signature. (a) Classification of sensitive/resistant cell lines on the basis of wild-type and mutant p53 status across 1400 drugs. (b) Heat map showing differential gene expression pattern. (c) 1st level interaction network of 262 genes. Nodes that belonged to 262 gene set are larger in size. Hubs (n=42) are red in color. Only two hubs (ubiquitin and EGFR) do not belong to the original 262 genes. (d) Count of pathways for 42 hubs. Total 76 unique pathways are classified into seven groups according to their overall action. Total number of hub proteins mapped onto a particular group of pathways is marked at the base of each bar. (e) Pearson’s correlation coefficient (PCC) of expression is calculated for 42 hub and their interactors in 11 cancer types. Distribution of PCC for each hub is represented by a box plot. Mean of the distribution are marked by the red square on the boxes. The white stars indicate the PCC of co-expression in colorectal cancer. The number of GO cellular component (CC), GO biological process (BP), and pathways (PTH) are presented by the three colored bar codes at the top of the box plot
Next, to build a drug-specific gene signature, each gene in the NCI-60 gene expression data set (GEO ID: GSE5846) was evaluated based on two criteria: (1) correlation between expression of the gene and GI50 values of resistant versus-sensitive cell lines and (2) differential expression of the gene in resistant versus-sensitive cell lines. The Gene Set Enrichment Analysis tool18 was used to obtain a gene signature that might contribute to the drug-resistant phenotype of mutant p53 cell lines (see Methods for details). We found 884 genes showing differential expression in 5-FU-resistant versus -sensitive cell lines (P<0.003; enrichment score=0.36). Similar analysis was carried out for doxorubicin (921 genes), carboplatin (1345 genes), and camptothecin (105 genes). The heat map showed significant differential expression in the sensitive versus resistant NCI-60 cell lines in response to the individual drugs (Figure 1b, Supplementary Table S6).
Identification of hub proteins from four gene signatures
Altogether, 2975 unique genes were selected from four gene signatures that offer chemoresistance (Supplementary Table S7). Of these, 262, which showed resistance to any two drugs, were extracted (Supplementary Table S7). These 262 genes were then utilized to identify the ‘hubs’ involved in protein–protein interactions of cellular signaling systems (Supplementary Figure S2A). Human-specific interactors with significant confidence scores for these proteins were extracted from the IREF-Index19 and STRING databases.20 Among these 262 proteins, 171 presented 1315 unique interactions with 1144 different proteins from both the databases. We used a statistical method, where proteins possessing more than nine interactions were defined as hubs for the 1st level interaction network (see Methods for details). Forty-two unique hubs were identified of which 40 belonged to the 262 gene signatures (Figure 1c). Twelve of these hubs were common to the 106 hubs (Supplementary Figure S2B) that came from the 2nd level interaction network, comprising of 4844 interactions among the 1135 proteins (see Methods for details). All interactions were mapped to their four original source databases (Supplementary Figure S2C). Enrichment of cellular pathways, as defined by the KEGG (Kyoto Encyclopaedia of Genes and Genomes) database,21 was also analyzed for the hubs and hub clusters (hub+interactors). Key signaling pathways such as JAK-STAT, cytokine-cytokine receptor interaction, MAPK, TGF-beta, and other diverse types of cancer pathways (for example, cancers of the colon, breast, pancreatic and so on), were found to be highly enriched by the hubs alone (Figure 1d) as well as with their interacting proteins (Supplementary Figure S2D). The average Pearson correlation coefficient for all the 42 hub genes and their interacting partners was <0.5, indicating that the identified hubs might act as ‘date hubs’ linking different function-specific modules or complexes within the cellular pathways (Figure 1e). The importance of the 42 hubs was further established by their involvement in multiple cellular localizations, pathways, and biological processes (Supplementary Figure S2D). This was also supported by the observation that their interactors possessed higher connectivity and are involved in multiple cellular processes (Supplementary Figure S2E).
Expression of ephrin-B2 is regulated by mutant p53 in DNA-damaging context
We determined the IC50 values for 5-FU, adriamycin, and etoposide in the human colorectal cancer cell line SW480 and its metastatic counterpart SW620, where the latter was found to be more resistant (Supplementary Figures S3A and B). Comparison of the mRNA expression of 40 hub genes in these cell lines (GEO ID: GSE 1323) showed that EFNB2 is significantly upregulated in SW620 cells (Supplementary Figure S3C). Similar analysis of the NCI-60 data set also revealed that EFNB2 expression is significantly high in GOF mutant p53-harboring cells compared with those harboring wild-type p53 (Supplementary Figure S3D). Thus, we conclude that EFNB2 can potentially impart chemoresistance to GOF mutant p53-harboring cancer cells. Further, we observed both dose- and time-dependent increase in ephrin-B2 protein and mRNA expressions in GOF mutant p53 cells upon 5-FU treatment (Supplementary Figures S4A–D; Supplementary Figures S5A and B). However, 5-FU did not alter ephrin-B2 expression either in wild-type p53 or HCT116p53−/− cells (Supplementary Figures S4A–D; Supplementary Figures S5A and B). Knockdown of mutant p53 in MIAPaCa2 and SW480 cell lines also did not change ephrin-B2 expression upon drug treatment suggesting the requirement of mutant p53 (Figure 2a). Furthermore, ectopically expressed contact-defective mutant (R273H), but not wild-type p53, induced ephrin-B2 expression in 5-FU-treated HCT116p53−/− cells (Figure 2b). We also observed high levels of EFNB2 mRNA in three stable GOF mutant p53-harboring H1299 cells under 5-FU-treated conditions, but not in parental p53-null H1299 cells (Supplementary Figure S5C). In addition, we observed a dose-dependent increase in EFNB2 promoter luciferase activity in four endogenous mutant p53 cells (Supplementary Figure S5D). The 5-FU-induced exogenous EFNB2 promoter luciferase activity in the four mutant p53 cells was abolished when mutant p53 was knocked down (Figure 2c). Furthermore, ectopic expression of mutant p53 in HCT116p53−/− cells induced high luciferase activity while that of wild-type p53 failed to evoke any response in presence of 5-FU (Figure 2d). Moreover, chromatin immunoprecipitation (ChIP) experiments in both endogenous and ectopically expressed GOF mutant p53 cells revealed recruitment of the mutant protein on the EFNB2 promoter (Supplementary Figures S6A and B). However, it is notable that not all mutant forms of p53 are equally proficient in being recruited to the promoter (Supplementary Figures S6A and B). Interestingly, H179Q mutant p53 failed to recruit on the EFNB2 promoter in vivo (Supplementary Figure S6B). Taken together, our findings suggest that some p53 GOF mutants, not the wild-type counterpart, transcriptionally activate EFNB2 expression in response to DNA damage.

Gain-of-function mutant p53 transcriptionally activates ephrin-B2. (a) Ephrin-B2 protein level remains unchanged upon addition of 5-FU in mutant p53 siRNA (80 nM) treated- MIAPaCa-2 and SW480 cells but upregulation was noticed upon drug treatment in scrambled siRNA-treated cells. (b) Transient transfection of gain-of-function mutant p53 (R273H) in HCT116p53−/− cells showed increase of ephrin-B2 expression upon 5-FU addition, whereas wild-type p53 transfected cells served as a control. (c) EFNB2 promoter activity remain constant upon incubation of mutant p53 siRNA (80 nM) and 5-FU in four different endogenous mutant cells, whereas sharp increment of EFNB2 promoter activity has been noticed upon treatment of drug in scrambled siRNA-treated cells. Immunoblot is showing decrease in p53 expression. (d) Increase of EFNB2 promoter activity has been observed by co-transfection of either R273H or R175H mutant p53 cDNA plasmids along with drug treatment. Wild-type p53 transfected cells served as a control. Immunoblot is showing overexpression of p53 protein. All histograms were expressed as means±S.D. of three independent experiments. *, **, ***, and **** indicate P≤0.05, P≤0.01, P≤0.001, and P≤0.0001, respectively. Histograms show densitometric values of band intensity
Transcriptional activation of EFNB2 promoter by mutant p53 requires CCAAT module
Bioinformatic analysis revealed the absence of wild-type p53 consensus binding site and presence of two CCAAT modules in the EFNB2 promoter (Supplementary Figure S7A and B). Indeed, ectopic expression of CCAAT-binding wild-type NF-YA protein elevated the luciferase activity in mutant p53-expressing stable H1299 cell lines, whereas the dominant-negative form of NF-YA22 yielded decreased activity (Figure 3a). We also observed high EFNB2 promoter-driven luciferase activity upon DNA damage in non-silencing controls but not in p53-knocked down SW480 cells (Supplementary Figure S7C). Further, ectopic expression of both the wild-type and dominant-negative forms of NF-YA did not alter the luciferase activity in mutant p53-knocked down cells (Supplementary Figures S7D and E). In addition, co-immunoprecipitation experiments revealed mutant p53 and NF-Y interaction in non-silencing control but not in mutant p53-knocked down cells under both 5-FU-treated and untreated conditions (Figure 3b). Reciprocal co-immunoprecipitation produced similar results in SW480 cells involving mutant p53, NF-YA, and NF-YB (Supplementary Figure S8A). However, the H179Q mutant p53, which did not recruit on EFNB2 promoter earlier, failed to interact with NF-YA (Supplementary Figure S8B). We observed that NF-YA and NF-YB recruitment on the EFNB2 promoter requires mutant p53 (Figure 3c), and occupancy of the NF-Y-mutant p53 complex increased following DNA damage in non-silencing control cells (Figure 3c). Surprisingly, total mutant p53 recruitment on the EFNB2 promoter did not change post drug treatment both in non-silencing control and p53-knocked down cells (Figure 3c). Conversely, mutant p53 recruitment on the EFNB2 promoter was abrogated upon NF-YA knockdown in SW480 cells (Figure 3d). This knockdown, however, did not abolish NF-YB recruitment on the EFNB2 promoter (Figure 3d) as NF-YB and NF-YC heterodimerization is a prerequisite for NF-YA association.22 Wild-type p53 recruitment on the NF-Y target promoter EFNB2 was also not observed in HCT116 cells (Supplementary Figure S8C). On the contrary, NF-YA recruitment was significantly increased in these cells upon 5-FU treatment (Supplementary Figure S8C). Our findings were bolstered by a report that the mutant p53-NF-Y complex binds to Plk2 promoter containing CCAAT boxes.23 The authors also showed that unlike mutant p53, recruitment of wild-type p53 on the Plk2 promoter occurs through the canonical p53 binding sites.23 Therefore, we conclude that the NF-Y-mutant p53 transcription complex recruits on the EFNB2 promoter, thus activating its expression in a DNA damage-dependent manner.

Mutant p53 in association with NF-Y facilitates ephrin-B2 transactivation. (a) Dual Luciferase assay showing ectopic expression of wild-type (WT) NF-YA protein enhances EFNB2 promoter activity. (b) Co-immunoprecipitation experiment showing the presence of mutant p53 and NF-Y complex in scrambled shRNA-transduced SW480 cells. (c) Chromatin immunoprecipitation (ChIP) experiment showing recruitment of NF-YA, NF-YB, and mutant p53 proteins on the endogenous EFNB2 promoter in control shRNA-transduced, but not in p53 shRNA-transduced SW480 cells. (d) Chromatin immunoprecipitation (ChIP) experiment showing recruitment of mutant p53, NF-YA, and NF-YB protein on the endogenous EFNB2 promoter in control siRNA-transfected cells, but not in NF-YA siRNA-transfected SW480 cells. (e) ChIP assay in mutant p53-harboring SW480 cells showing recruitment of acetyl-p53 (Lys 382) and p300 on the EFNB2 promoter in the presence of triply acetylated (wild-type) peptide (380–386; K381Ac/K382Ac/K386Ac) and unacetylated (mutant) peptide (380–386; K381A/K382A/L383A). (f) Triply acetylated peptide (380–386; K381Ac/K382Ac/K386Ac) treatment in mutant p53-harboring SW480 cells inhibit 5-FU-induced enhancement of ephrin-B2 expression. All histograms were expressed as means±S.D. of three independent experiments. *, **, ***, and **** indicate P≤0.05, P≤0.01, P≤0.001, and P≤0.0001, respectively. Histograms show densitometric values of band intensity
DNA damage-induced acetylated mutant p53–p300 interaction facilitates ephrin-B2 transactivation
To understand the mechanism of DNA damage-induced transcription of EFNB2 by the NF-Y-mutant p53 complex, we investigated the post-translational modification of mutant p53. We observed significant dose- and time-dependent increase (Supplementary Figures S9A and B, respectively) in its acetylated form (K382) upon treatment with 5-FU, doxorubicin, and etoposide. We also observed a concomitant increase in ephrin-B2 levels, suggesting that acetylated mutant p53 activates transcription (Supplementary Figures S9A and B). This was further supported by the observation that inhibiting acetylation by anacardic acid did not evoke drug-induced activation of ephrin-B2 (Supplementary Figure S9C). Next, we observed enrichment of acetylated mutant p53 on the endogenous EFNB2 promoter in 5-FU-treated cells versus untreated controls (Figure 3e). In line with this observation, p300 was also selectively recruited on the EFNB2 promoter following drug treatment in SW480 cells (Figure 3e). In a co-immunoprecipitation experiment, we noticed increased association of p300 with mutant p53 upon DNA damage in non-silencing control, but not in p53-knocked down, SW480 cells (Supplementary Figure S9D). Earlier, it was demonstrated that a triply acetylated peptide involving residues 380–386 of wild-type p53 (380–386; K381Ac/K382Ac/K386Ac) inhibits transcription regulatory function of the protein.24 However, the same was not true for a mutant peptide (380–386; K381A/K382A/L383A).24 We employed these two peptides in a ChIP experiment to test whether the triply acetylated (wild-type) peptide could also inhibit the recruitment of acetylated GOF mutant p53 on the EFNB2 promoter (Figure 3e). We observed enhanced p300 and acetylated mutant p53 (K382) recruitment on the EFNB2 promoter upon DNA damage in SW480 cells pretreated with the unacetylated (mutant) peptide (Figure 3e). In contrast, the recruitment was inhibited in presence of the triply acetylated (wild-type) peptide (Figure 3e). Similarly, pretreatment of SW480 cells with the triply acetylated (wild-type), but not unacetylated (mutant), peptide inhibited DNA damage-induced ephrin-B2 protein expression (Figure 3f and Supplementary Figure S9E). Interestingly, peptide treatment had no effect on either the drug-induced acetylation (K382) of mutant p53 or the total endogenous mutant p53 level. We thus conclude that drug-induced upregulation of ephrin-B2 in mutant p53 cells is preceded by p300 recruitment on the EFNB2 promoter following mutant p53 acetylation (Figure 8d).
Ephrin-B2 knockdown sensitizes mutant p53 tumor cells to DNA-damaging chemotherapeutic agents in vitro
Mutant p53 tumors exhibit chemoresistance by virtue of their GOF activities.12 Altered levels of ephrin-B2 lead to a more aggressive tumor phenotype and are associated with shortened survival in patients with colorectal cancer.25 Here, we observed increased ephrin-B2 expression in SW480 cells upon treatment with chemotherapeutic agents (Supplementary Figure S9A and B). In addition, ephrin-B2 knockdown in SW480 cells and R273H mutant p53-expressing H1299 cells resulted in decreased cell viability upon DNA damage and the phenotype is rescued by ephrin-B2 expression in SW480 cells (Figures 4a and b; Supplementary Figure S10A). We observed caspase-3 and PARP-I cleavage upon ephrin-B2 knockdown in SW480 cells treated with chemotherapeutic agents, indicating apoptotic death (Supplementary Figures S10B and C). Ephrin-B2 knockdown also sensitized other GOF mutant p53-harboring cells of diverse tumor origins (Supplementary Figures S11A–D). Furthermore, ectopic expression of ephrin-B2 in chemosensitive HCT116 cells harboring wild-type p53 conferred enhanced drug resistance (Supplementary Figures S11E and F). Next, we generated four ephrin-B2-knocked down stable cell lines, of which three showed effective knockdown (Supplementary Figures S12A and B) and lower IC50 values compared with control cells (Supplementary Figure S12C). Ephrin-B2 stable knockdown cells exhibited significant decrease in both radii and areas of spheroids upon 5-FU treatment in three-dimensional (3D) cell culture system (Figures 4c and d). Moreover, cell viability assays in 3D culture indicated significant killing with lower IC50 values (Figure 4e). In addition, each knocked down cell exhibited time-dependent increase in PARP-I and caspase-3 cleavage upon 5-FU treatment (Figure 4f). Notably, silencing of ephrin-B2 alone did not induce apoptosis, but sensitized cells toward conventional chemotherapeutic agents. We conclude that chemoresistance in mutant p53 cells is due to transcriptional activation of EFNB2 by mutant p53, and perturbations of ephrin-B2 can sensitize mutant p53-bearing cancer cells to DNA-damaging chemotherapeutic agents.

Knockdown of ephrin-B2 in mutant p53 cancer cells showing drug sensitivity. (a) Cell viability assay in endogenous SW480 cells and (b) R273H mutant p53-expressing stable H1299 cell lines showed enhanced sensitivity to 5-FU (10 μg/ml for 48 h). (c) Fluorescence micrograph showing clear spheroids in three-dimensional (3D) cell culture assay. ‘r’ represents radius. Scale bar denotes 200 μm. (d) Dose-dependent decrease of spheroid size (μm) and area (× 10−2 mm2) in 3D cell culture. (e) Dose-dependent decrease of cell viability in 3D cell culture. (f) Immunoblot showing time-dependent increase of PARP-I and Caspase-3 cleavage in ephrin-B2 knocked down cell lines. All histograms, spheroid size, and IC50 data were expressed as means±S.D. of three independent experiments. *, **, ***, and **** indicate P≤0.05, P≤0.01, P≤0.001, and P≤0.0001, respectively
Ephrin-B2-knocked down tumors are sensitive to 5-FU in vivo
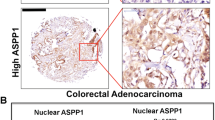
We next examined tumor growth in nude mice inoculated with either non-silencing control or ephrin-B2-knocked down SW480 cells. Further, these mice were either treated with 5-FU (5 mg/kg) or left untreated (see Methods for details). The control mice did not show any significant change in either tumor volume or weight between the PBS- and 5-FU-treated groups (Figure 5a and b; Supplementary Figure S13A). On the contrary, ephrin-B2-knocked down tumors showed significantly decreased volume and weight upon 5-FU treatment (Figures 5a and b; Supplementary Figure S13A). Notably, the control tumors achieved significant volume (>200 mm3) after 16 days of inoculation, whereas the ephrin-B2-knocked down tumors achieved the same only after 32 days. This supports the role of ephrin-B2 in tumorigenesis reported earlier.26 We further observed that both ABCG2 and ABCC1 expression increased markedly upon drug treatment in the control, but not in the knocked down mice group (Figure 5c and d). We confirmed efficient knockdown of ephrin-B2 in the tumors, (Supplementary Figure S13B) whereas mutant p53 expression remained unaltered between the two groups (Supplementary Figure S13C). To ascertain whether our findings in the cell line and mice are reflected in human colon cancer patients, we determined the expression of ABCC1, ABCG2, and EFNB2 in neoadjuvant therapy-resistant (Supplementary Figure S13D) and -sensitive (Supplementary Figure S13E) FFPE tumor tissue sections. Detailed patient history is provided in Supplementary Table S8. Both ABCC1 and EFNB2 expression was increased in patients with no response against neoadjuvant therapy, versus those with response (Figures 5e and f). ABCG2 mRNA was not detectable, suggesting it had low or no expression at all (data not shown). Thus, we conclude that ephrin-B2 is a key determinant in conferring chemoresistance that involves major drug transporter genes.

Silencing of ephrin-B2 inhibits tumor growth in vivo upon 5-FU treatment. (a) Representative graphs indicate tumor growth rate in either non-silencing control or ephrin-B2 knocked down SW480 cells. (b) The weight of the tumors was measured in gms. Histograms are represented as means±S.E.M. (c and d) Total RNA was isolated from SW480 shScrambled (n=5) and SW480 shEphrin-B2#53 (n=5) tumors. Expression of two genes (ABCG2 and ABCC1) was measured. (e and f) Total RNA was isolated from neoadjuvant therapy-resistant human colon carcinoma (n=10) and neoadjuvant therapy-sensitive human colon carcinoma (n=10). mRNA expression of two genes (ABCC1 and EFNB2) was measured
Ephrin-B2 reverse signaling confers drug resistance to mutant p53 cancers via the JNK/c-Jun pathway
To investigate the mode of ephrin-B2 signaling (forward or reverse) that promotes chemoresistance, we treated mutant p53-harboring SW480 cells and its EFNB2 stably knocked down counterpart with both the agonists- EphB4 Fc and ephrin-B2 Fc- that in turn activate ephrin-B2 reverse and EphB4 forward signaling, respectively. We observed an increase in ABCG2/BCRP in control cells in the presence of EphB4 Fc compared with control Fc (Figure 6a). However, the ABCG2 levels were unaltered in ephrin-B2-knocked down cells as well as post ephrin-B2 Fc stimulation (Figure 6a). Thus, ephrin-B2 reverse, and not EphB4 forward, signaling enhances the ABCG2 levels. We also observed increment in the ABCG2 mRNA level in presence of 5-FU, but not under ephrin-B2-knocked down condition (Figure 6b). However, ectopic expression of ephrin-B2 in the knocked down cells restored the ABCG2 levels (Figure 6b). Moreover, the MAPK family member JNK and its downstream member c-Jun showed increased phosphorylation in agonist-treated (EphB4 Fc) control versus knocked down cells (Figure 6c). In addition, we observed reduced phosphorylation of both JNK (T183/Y185) and c-Jun (S63) upon 5-FU treatment in knocked down cells compared with the controls (Figure 6d). We also observed enhanced recruitment of c-Jun and RNA polymerase II on the ABCG2 promoter following 5-FU treatment in control but not in the knocked down cells, suggesting its regulation by c-Jun27 (Figures 6e and f). We observed that knockdown of JNK in SW480 cells resulted in PARP-I cleavage upon 5-FU treatment (Figure 6g). This suggests that JNK is a key determinant of apoptosis, and ephrin-B2 imparts chemoresistance and helps to evade apoptosis through JNK signaling (Figure 8d). Altogether, we conclude that under DNA-damaging conditions, GOF mutant p53 increases ephrin-B2 expression, which in turn induces ABCG2 expression through the c-Jun/JNK signaling pathway (Figure 8d).

Ephrin-B2 reverse signaling exhibits chemoresistance influencing ABCG2 expression. (a) Immunoblot analysis of ABCG2 in agonist-treated SW480 cells (b) mRNA expression of ABCG2 in ephrin-B2 knocked down and overexpressed condition. (c) Immunoblot analysis of phospho-JNK and phospho-c-Jun in agonist-treated SW480 cells. (d) Cells were harvested at 48 h post 5-FU treatment and subjected to immunoblot analysis with α-phospho-JNK and α-phospho-c-Jun. Silencing of ephrin-B2 was ascertained with α-ephrin-B2. (e) ChIP assay showing recruitment of c-Jun and RNA polymerase II on the EFNB2 promoter. (f) ChIP Assay showing recruitment of RNA polymerase II and no recruitment of c-Jun on the GAPDH promoter. (g) Knockdown of JNK protein in SW480 cells increases PARP-I cleavage. Histograms show densitometric values of band intensity and express as means±S.D. of three independent experiments. * indicates P≤0.05
Ephrin-B2 reverse signaling promotes cancer cell proliferation and drives EMT in mutant p53 cancers
Ephrin-B2 reverse signaling transmits extracellular signals to the nucleus through the Src family of kinases.28 Indeed, we too observed increased phosphorylation of both Src (Y418) and p44/p42 MAPK (T202/Y204) in agonist-treated control but not in knocked down cells (Figure 7a). Similarly, ephrin-B2-knocked down cells presented lower phosphorylated Src and p44/p42 MAPK upon 5-FU treatment, clearly implying ephrin-B2 as a stimulant for cell proliferation (Figure 7b). Furthermore, we observed increased apoptosis along with concomitant decrease in survival and proliferation in Src and p44/p42 MAPK (ERK)-knocked down cells, establishing the essentiality of ephrin-B2 reverse signaling in this process (Figures 7c–e). Thus, inhibition of ephrin-B2 expression decreases the survival and proliferation rates and hence, sensitizes cancer cells to apoptotic cell death (Figure 8d).

Ephrin-B2 reverse signaling promotes cell proliferation in mutant p53 cancer. (a) Immunoblot analysis of α-phospho SRC and α-phospho p44/p42 MAPK in agonist-treated cells. (b) Cells were harvested at 48 h post 5-FU treatment and subjected to immunoblot analysis with α-phospho SRC and α-phospho p44/p42 MAPK. (c) Knockdown of Src protein and (d) ERK (p44/p42 MAPK) protein in SW480 cells increase PARP-I cleavage with concomitant decrease in PCNA expression. (e) 5-FU induced decrease of viability in SW480 cells following pretreatment with either Src or ERK siRNA. All histograms were expressed as means±S.D. of three independent experiments. *, **, ***, and **** indicate P≤0.05, P≤0.01, P≤0.001, and P≤0.0001, respectively. Histograms show densitometric values of band intensity

Ephrin-B2 reverse signaling drives EMT in mutant p53 cancer. (a) Agonist-treated cells were subjected to immunoblot analysis with α-phospho FAK, α-E-cadherin, α-SNAI-1, and α-ID-1. (b) 5-FU-treated non-silencing control and ephrin-B2 knocked down cells were subjected to immunoblot analysis with α-phospho FAK, α-E-cadherin, α-Vimentin, α-Snail, and α-Slug. Silencing of ephrin-B2 was ascertained with α-ephrin-B2. (c) Perturbation of FAK in SW480 cells reverses EMT phenotype. (d) Model of our working hypothesis showing ephrin-B2 expression is upregulated in response to DNA damage by NF-Y/mutant p53/p300 complex. Drug induced ephrin-B2 reverse signaling evade apoptosis and impart chemoresistance involving JNK/c-Jun signaling. Ephrin-B2 reverse signaling also drives EMT and promotes cancer cell proliferation involving Src/FAK and Src/ERK signaling, respectively
Cancer cells with EMT phenotypes are shown to be unresponsive to chemotherapy.29, 30 Src and its associated tyrosine kinase, FAK, are at the core of the EMT phenotype.31 Here, we observed abrogation of FAK phosphorylation (Y576/Y577) in ephrin-B2-knocked down cells (Figure 8a), which was reduced even in presence of 5-FU (Figure 8b). Subsequent investigation of EMT markers in both control and knocked down cells revealed substantial enhancement of E-cadherin protein expression in the knocked down cells treated with either agonist (Figure 8a) or 5-FU (Figure 8b). Moreover, we observed significant changes in both SNAI-1 and ID-1 expression levels following agonist treatment (Figure 8a). We also observed reduced expression of vimentin, snail, and slug proteins in EFNB2-knocked down cells upon 5-FU treatment, clearly indicating the involvement of ephrin-B2 in the EMT process (Figure 8b). Furthermore, we noticed high E-cadherin and low vimentin expression in SW480 cells following FAK knockdown (Figure 8c). Based on the above results, we propose that GOF mutant p53-mediated ephrin-B2 activation modulates the EMT process by virtue of Src/FAK signaling, thereby promoting drug resistance (Figure 8d).
Discussion
Half of the human cancers carry p53 mutations.7 Some of these mutant proteins are potentially oncogenic, capable of promoting tumorigenesis through different cellular mechanisms.32, 33, 34 Managing drug-resistant cancer is challenging, and a combinatorial approach is required to enhance drug sensitivity. Based on a previously described predictive methodology to tap genetic contributions to drug sensitivity,16 we designed a new model to identify gene signatures that might contribute to GOF mutant p53-mediated chemoresistance. Our prediction analysis identified a hub gene, EFNB2 that presented differential expression between GOF mutant and wild-type p53-bearing cell lines when treated with chemotherapeutic agents. This observation was supported by a report that mutant p53-harboring ovarian cancer cells exhibit high EFNB2 expression compared with those harboring wild-type p53.35 Hubs are generally associated with greater importance in regulating key cellular pathways, and perturbations of genes encoding hubs are more likely to confer lethality than those of non-hubs.36 Hence, identification of mutant p53-bound hub genes and subsequent perturbation of their expression might be crucial in understanding the mechanism(s) of chemoresistance, and greatly facilitate therapeutic intervention in a large proportion of tumors.
The bidirectional signaling molecule ephrin-B2 becomes clustered upon interaction with its Eph receptor37 and activates key signaling moieties such as Src,28 PDGFRβ,37 VEGFR2,38 STAT3,39 and many more. Until date, only one study has reported the involvement of Eph-ephrin signaling in conferring radioresistance, but the mechanism is poorly understood.40 The ABC transporter family of genes- ABCC1, ABCB1, and ABCG2- are engaged in imparting multidrug resistance to cancer cells. It is known that wild-type p53 represses ABCB1 expression,41 whereas mutant p53 activates it.42 In this study, we report for the first time that ephrin-B2 reverse signaling regulates ABCG2 expression through JNK-c-Jun signaling in mutant p53-expressing cancer cells. Eph receptors induce EMT through oncogenic signaling pathways such as Wnt, Ras/MAPK, Akt-mTOR, and Notch.43 We also observed that the Src/FAK signaling is involved in the EphB4/ephrin-B2–induced EMT process. Perturbation of ephrin-B2 reverses the EMT phenotype by E-cadherin downregulation and hence makes mutant p53-expressing cancer cells more susceptible to chemotherapeutics.
In summary, ephrin-B2 downregulation inhibits colon carcinoma cell growth, promotes apoptosis, reverses the EMT phenotype, and decreases drug resistance in vitro as well as in mouse model. Neo-adjuvant therapy-resistant colorectal tumors also exhibit high EFNB2 and ABC transporter gene expression. Thus, targeting the members of Eph/Ephrin signaling pathways such as ephrin-B2 has the potential to circumvent drug resistance when applied in conjunction with other conventional chemotherapeutic agents. This further suggests that many more genes are yet to be identified whose deregulation is central to some of the key features exhibited by GOF mutant p53 tumors, enhanced chemoresistance being one of them.
Materials and Methods
Cell culture
Human colon cancer cell lines HCT116p53+/+ (CCL-247), and SW480 (CCL-228), human pancreatic cancer cell lines MIAPaCa2 (CRL-1420), and PANC-1 (CRL-1469) and human breast cancer cell lines MDA-MB231 (HTB-26), and SkBr3 (HTB-30) were purchased from American Type Culture Collection. HCT116p53−/− and Huh-7 cell line was a kind gift from Professor Bert Vogelstein (Johns Hopkins University, Baltimore, MD, USA) and Dr. Suvendra Nath Bhattacharyya (CSIR-Indian Institute of Chemical Biology, Kolkata, India), respectively. Mutant p53-expressing stable cell lines (H1299-EV, H1299-R175H, and H1299-R248W) were kind gifts from Dr. Varda Rotter (Weizmann Institute of Science, Israel) and H1299-R273H cell line was generated in our laboratory. All cell lines were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco). All cell lines were routinely screened and tested negative for mycoplasma contamination.
3D cell culture
Cells were seeded onto round-bottom non-tissue culture–treated 96-well plates (BD Biosciences, San Jose, CA, USA) at a concentration of 5000 cells/well in 100 μl DMEM (Gibco), containing 10% FBS (Gibco) and supplemented with 20% methylcellulose stock solution (Sigma-Aldrich, St. Louis, MO, USA). The methylcellulose stock solution was prepared as described previously.44 Spheroids were grown under standard culture conditions (5% CO2 at 37 °C) and drug treatment was carried out for 72 h.
Plasmids and peptides
1106-bp (−929 nt to +176 nt respective to the transcription start site) long region of the human EFNB2 promoter was a kind gift from Dr. Christopher CW Hughes (University of California, Irvine, CA, USA) and it has been thoroughly characterized in another study.45 The Renilla luciferase construct (pRL-TK) was purchased from Promega Corp (Madison, WI, USA). The wild-type p53 (pCMV-p53) and the mutant p53 (pCMV-R175H and pCMV-R273H) expression plasmids were a kind gift from Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD, USA) and full-length EFNB2 cDNA clone was purchased from Origene Technologies (Rockville, MD, USA). Δ4YA13 vector (NF-YA) and Δ4YA13m29 dominant-negative vector (DN-NF-YA) were a kind gift from Dr. Giovanni Blandino (Regina Elena Cancer Institute, Rome, Italy). Triply acetylated wild-type peptide (380–386; K381Ac/K382Ac/K386Ac) and mutant peptide (380–386; K381A/K382A/L383A) were synthesized in our laboratory24 and were used at different concentrations for 48 h. The concentration of peptide was measured by standard BCA (Thermo Scientific, Rockford, IL, USA) method.
Drugs and agonists treatment
For induction of DNA damage, four cytotoxic drugs such as 5-FU (Sigma-Aldrich), doxorubicin (Sigma-Aldrich), etoposide (Sigma-Aldrich) and cisplatin (Sigma-Aldrich) were used at different doses for indicated times. Two agonists, EphB4 Fc, and ephrin-B2 Fc (R&D Systems, Minneapolis, MN, USA), at a concentration of 2 μg/ml were used to induce ephrin-B2–mediated reverse and forward signaling, respectively.
siRNA treatment
Cells were seeded in 6-cm dishes and cultured for 18–24 h. The next day, cells were washed with PBS, added Opti-MEM reduced-serum medium (Gibco) and transfected with either 80 nmol/l human TP53 siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or 80 nmol/l human EFNB2 siRNA (Santa Cruz Biotechnology and Dharmacon, Lafayette, CO, USA) or 100 nmol/l human SRC siRNA (Dharmacon) or 100 nmol/l human JNK siRNA (Dharmacon) or 100 nmol/l human ERK siRNA (Dharmacon) or 100 nmol/l human FAK siRNA (Dharmacon) or 80 nmol/l human NF-YA siRNA (Santa Cruz Biotechnology) or Negative Control siRNA (Ambion Inc., Austin, TX, USA) using DharmaFECT 4 (Dharmacon). After 5 h, antibiotic-containing DMEM supplemented with 10% FBS was added and cells were incubated for a total of 72 h before further processing.
Short hairpin RNA transfection
pGIPZ lentiviral short hairpin RNA (shRNA) vectors for human EFNB2, TP53, and non-silencing control were purchased from Open Biosystems (Thermo Scientific) and were prepared as previously described.46 SW480 cells were infected with lentiviral shRNA; after puromycin (Gibco) selection, stable selected cells were used for the experiments.
Xenograft tumor model
Six- to 8-week-old female nude mice (BALB/C; nu/nu) were obtained from the National Institutes of Health and housed in the institutional animal facilities. All animal work was performed under protocols approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Patient samples
Neo-adjuvant therapy-resistant (n=10) and -sensitive (n=10) colon carcinomas were obtained from the archive samples of the hospital section at Mayo Clinic in Rochester, MN, USA. Before sample collection, written informed consent was taken from each patient and the study was approved by the Mayo Clinic Institutional Review Board.
In-vivo antitumor activity
To establish tumor growth in nude mice, either 5 × 106 non-silencing control cells or stable ephrin-B2 knockdown SW480 cells, resuspended in 100 μl of PBS, were injected subcutaneously into the left flank. Tumors were allowed to grow for 16–30 days until they reached significant volume (>200 mm3), and mice were then randomized into two groups (five animals per group). Group 1 was treated with PBS alone, whereas group two was treated with 5-FU. Drug at doses of 5 mg/kg was administered three times in a week intra-peritoneally. After 10 successive treatments, all tumor-bearing mice were killed by asphyxiation with CO2; tumors were removed, weighed, and measured. A part of each tumor was homogenized to obtain total cellular RNA that was used for real-time PCR-mediated expression analysis.
Transfection and luciferase assays
For the dual-luciferase reporter assay, promoter luciferase constructs (0.5 μg), along with pRL-TK plasmids (0.1 μg), were co-transfected into different cells using Polyfect (Qiagen, Hilden, Germany). The cells were incubated in Opti-MEM reduced-serum medium (Gibco) for 5 h and then switched to antibiotic-containing DMEM containing 10% FBS. After 48 h of incubation, the cells were harvested and luminescence was measured according to manufacturer’s protocol (Promega Corp). For transient overexpression assay, cells were seeded in 6-cm dishes and cultured for 18–24 h. The next day, cells were washed with PBS (Gibco), Opti-MEM reduced-serum medium was added and cells were transfected with cDNA expression plasmids at indicative doses. After 5 h, antibiotic-containing DMEM supplemented with 10% FBS was added and cells were incubated for a total of 48 h before further processing.
Quantitative real-time PCR
Total RNA from different cell lines was isolated using either TRIZOL (Invitrogen, Carlsbad, CA, USA) or RNeasy Plus (Qiagen) according to manufacturer’s protocol. Isolated RNA (5 μg) was treated with DNase-I (Promega Corp) and 1 μg of the DNase-treated RNA was used for cDNA preparation with random hexamer (Invitrogen) and MMLV-RT (Promega Corp). Real-time PCR was performed in the 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with power SYBR Green PCR Master Mix (Applied Biosystems). The comparative threshold cycle method (ΔΔCt) was used to quantify relative amounts of product transcripts with GAPDH as endogenous reference controls. Primer sets for various genes are enlisted in Supplementary Table S9.
All the human patients’ samples were available on slides as paraffin sections, and total cellular RNA was extracted using RNeasy FFPE kit (Qiagen). Of the isolated total RNA, a 100-ng amount was used for cDNA preparation using iScript (Biorad, Hercules, CA, USA) according to the manufacturer’s protocol. Real-time PCR was performed in the 7500 Real-Time PCR System (Applied Biosystems) using power SYBR Green PCR Master Mix (Applied Biosystems). The comparative threshold cycle method (ΔCt) was used to quantify relative amounts of product transcripts with GAPDH as endogenous reference controls. Primer sets for various genes are listed in Supplementary Table S9.
ChIP assays
ChIP assay was done with EZ-ChIP kit from Millipore (Billerica, MA, USA) according to the manufacturer’s protocol. PCR amplification from the immunoprecipitated chromatin was done with different promoter primers (Supplementary Table S9). In the amount of 10 μg each, anti-p53 antibody (Santa Cruz), anti–RNA polymerase II antibody (Santa Cruz), anti-p300 antibody (Santa Cruz), anti–acetyl-p53 antibody (Upstate, Billerica, MA, USA), anti–NF-YA antibody (Santa Cruz), anti–NF-YB antibody (Santa Cruz) and anti–c-Jun antibody (Cell Signaling Technology, Danvers, MA, USA) were used for immunoprecipitation. Normal rabbit antiserum (Cell Signaling Technology) was used as negative control and 5% of the total lysate was used as an input control. The GAPDH promoter primers were used as a negative control for c-Jun.
Western blot analysis
Western blot analysis was performed using antibodies against ephrin-B2 (Abcam, Cambridge, UK; ab96264), p53 (Santa Cruz; DO-1), PLC-γ1 (Santa Cruz; 1249), acetyl-p53 (Upstate), NF-YA (Santa Cruz; C-18) NF-YB (Santa Cruz; FL-207) β-actin (Sigma, St. Louis, MO, USA), PARP-I (Cell Signaling Technology; 9542), caspase-3 (Cell Signaling Technology; 9662), cleaved PAPR-I (Cell Signaling Technology; 9541), cleaved caspase-3 (Cell Signaling Technology; 9661), p300 (Santa Cruz; C-20), ABCG2 (Cell Signaling Technology; 4477), phospho-Src (Cell Signaling Technology; 2101), total Src (Cell Signaling Technology; 2123), phospho p44/p42 MAPK (Cell Signaling Technology; 9101), total p44/p42 MAPK (Cell Signaling Technology; 9102), phospho-JNK (Cell Signaling Technology; 4668), total JNK (Santa Cruz, C-17), phospho-c-Jun (Cell Signaling Technology; 2361), total c-Jun (Cell Signaling Technology; 9165), phospho FAK (Cell Signaling Technology; 3281), total FAK (Cell Signaling Technology; 3285), E-cadherin (Santa Cruz, G-10), SNAI-1 (Santa Cruz, H-130), ID-1 (Santa Cruz, C-20), Snail (Cell Signaling Technology; 3879), Vimentin (Cell Signaling Technology; 3390), Slug (Cell Signaling Technology; 9585) in different cell lysates. Cells were treated with different doses of cytotoxic drugs and incubated for indicated times. Whole-cell lysates in radioimmunoprecipitation assay buffer supplemented with protease inhibitor cocktail (Sigma) and with or without phosphatase inhibitor (Cell Signaling Technology) were then prepared. Supernatant was collected by centrifugation at 13 000 rpm for 10 min. Subsequently, samples were subjected to SDS-PAGE and then transferred to polyvinyl difluoride membranes (Millipore) and immunoblotted for the proteins of interest. Antibody-reactive bands were detected by enzyme-linked chemiluminescence (Thermo Scientific). Densitometric values of bands by ImageJ software are plotted. Proteins were normalized to either with β-actin levels or with PLC-γ1 levels. In case of phospho protein, normalization was done with their corresponding total protein level.
Apoptotic assays
To measure PARP-I cleavage, 1 × 105 SW480 cells were seeded and transiently transfected as mentioned earlier. At indicated time points after drug treatment, cells were fixed with 4% paraformaldehyde (Sigma) and permeabilized with fluorescence-activated cell sorter permeabilizing solution (BD Biosciences). Before staining, permeabilized cells were treated with heat-inactivated 2% normal bovine serum to block nonspecific staining. Cells were then stained with normal mouse sera or mouse anti–human-cleaved PARP-I antibody (Cell Signaling Technology). After they were washed, cells were incubated with multiple adsorbed Alexa Fluor 647-conjugated secondary antibody (Molecular Probes, Invitrogen), washed, and analyzed with BD FACSAria (Becton-Dickinson, San Jose, CA, USA).
Cell survival assays
Cells were treated with drugs at indicated concentration in 96-well flat-bottomed microtitre plates and cell proliferation was assessed with WST-1 cell proliferation reagent (Roche, Basel, Switzerland) after 72 h of incubation. Absorbance of samples was measured at 450 nm. From the absorbance values, the viability index was calculated.
For 3D cell culture, cell viability was measured using CellTiter-Glo Luminescent assay kit (Promega Corp) according to the manufacturer’s protocol. Luminescence was measured and viability index has been calculated from this value. Confocal microscopy of these spheroids has been performed using a Zeiss LSM 780 confocal laser scan microscope.
Co-immunoprecipitation and western blotting
SW480 cells were lysed with cold immunoprecipitation buffer (50 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS), which contains 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 0.5% aprotinin, and 2 μg/ml pepstatin A. Lysate protein (500 μg) was incubated with 2 μg of indicated antibodies at 4 °C overnight and then with 50 μl of protein G-conjugated agarose beads (Sigma-Aldrich) at 4 °C for 3–5 h. After the beads were washed with the same buffer, immunoprecipitates were resuspended in 2 × Laemmeli sample buffer for western blot analysis.
Immunohistochemistry
For immunohistochemistry, 5-μm paraffin-embedded sections of cancer tissues were deparaffinized and then subjected to hematoxylin and eosin immunochemistry according to the manufacturer’s instructions (DAB 150; Millipore).
Identification of gene expression signature
We used NCI-60 gene expression data set (GEO ID: GSE5846) for identification of gene expression signature as described previously.16 Genes, that are differentially expressed (GEO ID: GSE5846) in resistant versus-sensitive group for 5-FU, were identified (student's t-test; P<0.05). In addition, correlation was checked between expressions of genes and GI50 values across cell lines (correlation P<0.05). Genes showed significant expression change between resistant and sensitive as well as show significant correlations with GI50 values were selected as drug-specific gene signature. The human genes were rank-ordered based on differential expression for resistant versus-sensitive cell lines. Gene Set Enrichment Analysis scores the relative position of each gene in the resistant signature. A heat map was generated using the Gene Set Enrichment Analysis tool showing significant expression difference in the sensitive versus resistant NCI-60 cell lines to individual drugs (see Supplementary Table S6 for the list of all differentially expressed genes). We labeled each signature as VSR-Carbo, VSR-camptothecin, VSR- Dox and VSR-5-FU (see Figure 1b). Such signatures provide information on biological processes, pathways, or genes that operate in chemoresistance tumors; and scope for targeted therapy against such modules.
Identification of hub genes
Human-specific protein–protein interactions were extracted from the IREF-Index19 and STRING20 database. From both the databases only experimentally established interactions were taken based on their respective confidence scores. For IREF-Index, the confidence was measured by NP score ≥2 and for STRING database the confidence was measured by experimental score ≥0.7 and combined score ≥0.7. Two interaction levels were considered. In the first level only the interactions mediated by 262 gene signatures (Supplementary Table S7) were taken and in the 2nd level all interactions among their interactors were also taken. In-house Perl scripts were used to identify the hubs. ‘Hubs’ were defined as proteins, which showed higher connectivity than other nodes in the network. In this study the degree threshold for defining hub was calculated by z-score of degree. Z-score 1 was set as threshold, which corresponds to degree 9 and 20 in 1st and 2nd level networks, respectively. Construction of second level network was important for two reasons. First, the first level network is comprised of the direct neighbors of those 262 genes hence the network was radial and isolated in small modules. These isolated modules were connected to each other by addition of second level neighbors to investigate the impact of these 262 genes in a larger context. Second, it was crucial to investigate whether the neighbors of hubs in first level were also important when investigated in a larger network context, which asserts that the hub proteins not only have high number of connection but also connected to highly connected proteins also. Identification of hubs was crucial for analysis of protein–protein interaction network because hubs were found to have special biological properties: they tend to be more essential than non-hub proteins,47, 48 they are found to have a central role in modular organization of the protein interaction network,36, 49 and some studies indicate that hub proteins may also be evolutionarily conserved to a larger extent than non-hubs.50 Hubs can be divided into party hubs, which comprise a specific cellular process or protein complex; and date hubs, which link together and convey information between different function-specific modules or -complexes. Date hubs generally display low co-expression correlation with their interacting partners, whereas party hubs have high co-expression correlation. Average pearson’s correlation coefficient of mRNA expression of 42 hub genes and their interacting partners were calculated. Expression data were collected from different experiments performed on different cell lines available at Oncomine database.51 Cellular pathway information for the hubs and their interactors were collected from the KEGG (Kyoto Encyclopedia of Genes and Genomes) database,21 which is a collection of biological pathways. An in-house Perl script was used to calculate the extent of co-occurrence of biological pathways within the hub clusters. The sub cellular localization and biological process were mapped as per gene ontology (GO) of the hubs. GO for cellular component and biological processes were extracted from gene association file (GOC validation date 7 October 2014) from GO database.52 mRNA expression data were collected from different experiments performed on different cancer cell lines available at Oncomine database.51 Expression data were collected for 11 different types of cancer from the following data sets; Beroukhim multicancer data set (leukemia, breast cancer, sarcoma, lung cancer, medulloblastoma, melanoma, meningioma, myelodysplasia, overian cancer, prostate cancer, and thyroid cancer) and Jaiswal multicancer data set (colorectal cancer, glioma, lung cancer). Date and party hubs were calculated for each of these cancer types separately. The average Pearson correlation coefficient of co-expression for each hub in each cancer type was plotted as a box-whisker distribution (see Figure 1e).
Statistical analysis
Statistical significant differences between groups were determined by the Student's t-test using GraphPad QuickCalcs online software (http://www.graphpad.com/quickcalcs/contingency1.cfm). Two tailed P-value were defined as P≤0.05 (*), P≤0.01 (**), P≤0.001 (***), and P≤0.0001 (****).
Abbreviations
- 5-FU:
-
5-Fluorouracil
- GOF:
-
gain-of-function
- NCI:
-
national cancer institute
- GEO:
-
gene expression omnibus
- EMT:
-
epithelial-to-mesenchymal transition
References
Cayuso J, Xu Q, Wilkinson DG . Mechanisms of boundary formation by Eph receptor and ephrin signaling. Dev Biol 2015; 401: 122–131.
Tallquist MD, Soriano P, Klinghoffer RA . Growth factor signaling pathways in vascular development. Oncogene 1999; 18: 7917–7932.
Pasquale EB . Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol 2005; 6: 462–475.
Senturk A, Pfennig S, Weiss A, Burk K, Acker-Palmer A . Ephrin Bs are essential components of the Reelin pathway to regulate neuronal migration. Nature 2011; 472: 356–360.
Pasquale EB . Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 2010; 10: 165–180.
Brantley-Sieders DM, Chen J . Eph receptor tyrosine kinases in angiogenesis: from development to disease. Angiogenesis 2004; 7: 17–28.
Muller PA, Vousden KH . Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014; 25: 304–317.
Brosh R, Rotter V . When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9: 701–713.
Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA 2003; 100: 8424–8429.
Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M et al. Gain of function mutations in p53. Nat Genet 1993; 4: 42–46.
Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I et al. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994; 84: 3148–3157.
Blandino G, Levine AJ, Oren M . Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999; 18: 477–485.
Peled A, Zipori D, Rotter V . Cooperation between p53-dependent and p53-independent apoptotic pathways in myeloid cells. Cancer Res 1996; 56: 2148–2156.
Shetzer Y, Solomon H, Koifman G, Molchadsky A, Horesh S, Rotter V . The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis 2014; 35: 1196–1208.
Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev 2012; 26: 830–845.
Yadav VK, Kumar A, Mann A, Aggarwal S, Kumar M, Roy SD et al. Engineered reversal of drug resistance in cancer cells—metastases suppressor factors as change agents. Nucleic Acids Res 2014; 42: 764–773.
Stinson SF, Alley MC, Kopp WC, Fiebig HH, Mullendore LA, Pittman AF et al. Morphological and immunocytochemical characteristics of human tumor cell lines for use in a disease-oriented anticancer drug screen. Anticancer Res 1992; 12: 1035–1053.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550.
Razick S, Magklaras G, Donaldson IM . iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics 2008; 9: 405.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011; 39: D561–D568.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M . KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 2010; 38 (Database issue): D355–D360.
Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006; 10: 191–202.
Valenti F, Fausti F, Biagioni F, Shay T, Fontemaggi G, Domany E et al. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle 2011; 10: 4330–4340.
Debnath S, Chatterjee S, Arif M, Kundu TK, Roy S . Peptide-protein interactions suggest that acetylation of lysines 381 and 382 of p53 is important for positive coactivator 4-p53 interaction. J Biol Chem 2011; 286: 25076–25087.
Liu W, Ahmad SA, Jung YD, Reinmuth N, Fan F, Bucana CD et al. Coexpression of ephrin-Bs and their receptors in colon carcinoma. Cancer 2002; 94: 934–939.
Abengozar MA, de Frutos S, Ferreiro S, Soriano J, Perez-Martinez M, Olmeda D et al. Blocking ephrinB2 with highly specific antibodies inhibits angiogenesis, lymphangiogenesis, and tumor growth. Blood 2012; 119: 4565–4576.
Zhu MM, Tong JL, Xu Q, Nie F, Xu XT, Xiao SD et al. Increased JNK1 signaling pathway is responsible for ABCG2-mediated multidrug resistance in human colon cancer. PLoS One 2012; 7: e41763.
Wu HC, Chang CH, Peng HY, Chen GD, Lai CY, Hsieh MC et al. EphrinB2 induces pelvic-urethra reflex potentiation via Src kinase-dependent tyrosine phosphorylation of NR2B. Am J Physiol Renal Physiol 2011; 300: F403–F411.
Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 2009; 69: 5820–5828.
Zhang W, Feng M, Zheng G, Chen Y, Wang X, Pen B et al. Chemoresistance to 5-fluorouracil induces epithelial-mesenchymal transition via up-regulation of Snail in MCF7 human breast cancer cells. Biochem Biophys Res Commun 2012; 417: 679–685.
Avizienyte E, Frame MC . Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol 2005; 17: 542–547.
Freed-Pastor WA, Prives C . Mutant p53: one name, many proteins. Genes Dev 2012; 26: 1268–1286.
Muller PA, Vousden KH . p53 mutations in cancer. Nat Cell Biol 2013; 15: 2–8.
Peled A, Schwartz D, Elkind NB, Wolkowicz R, Li R, Rotter V . The role of p53 in the induction of polyploidity of myelomonocytic leukemic M1/2 cells. Oncogene 1996; 13: 1677–1685.
Davidson BA, Rubatt JM, Corcoran DL, Teoh DK, Bernardini MQ, Grace LA et al. Differential angiogenic gene expression in TP53 wild-type and mutant ovarian cancer cell lines. Front Oncol 2014; 4: 163.
Han JD, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 2004; 430: 88–93.
Nakayama A, Nakayama M, Turner CJ, Hoing S, Lepore JJ, Adams RH . Ephrin-B2 controls PDGFRbeta internalization and signaling. Genes Dev 2013; 27: 2576–2589.
Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 2010; 465: 487–491.
Bong YS, Lee HS, Carim-Todd L, Mood K, Nishanian TG, Tessarollo L et al. ephrinB1 signals from the cell surface to the nucleus by recruitment of STAT3. Proc Natl Acad Sci USA 2007; 104: 17305–17310.
Stahl S, Kaminskyy VO, Efazat G, Hyrslova Vaculova A, Rodriguez-Nieto S, Moshfegh A et al. Inhibition of Ephrin B3-mediated survival signaling contributes to increased cell death response of non-small cell lung carcinoma cells after combined treatment with ionizing radiation and PKC 412. Cell Death Dis 2013; 4: e454.
Bush JA, Li G . Cancer chemoresistance: the relationship between p53 and multidrug transporters. Int J Cancer 2002; 98: 323–330.
Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem 2001; 276: 39359–39367.
Li RX, Chen ZH, Chen ZK . The role of the EPH receptors in the cancer-related epithelial-mesenchymal transition Running title: Eph receptors and EMT. Chin J Cancer 2013; 33: 231–240.
Longati P, Jia X, Eimer J, Wagman A, Witt MR, Rehnmark S et al. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013; 13: 95.
Kim JH, Peacock MR, George SC, Hughes CC . BMP9 induces EphrinB2 expression in endothelial cells through an Alk1-BMPRII/ActRII-ID1/ID3-dependent pathway: implications for hereditary hemorrhagic telangiectasia type II. Angiogenesis 2012; 15: 497–509.
Cao Y, Wang L, Nandy D, Zhang Y, Basu A, Radisky D et al. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res 2008; 68: 8667–8672.
Jeong H, Mason SP, Barabasi AL, Oltvai ZN . Lethality and centrality in protein networks. Nature 2001; 411: 41–42.
He X, Zhang J . Why do hubs tend to be essential in protein networks? PLoS Genet 2006; 2: e88.
Albert R, Jeong H, Barabasi AL . Error and attack tolerance of complex networks. Nature 2000; 406: 378–382.
Wuchty S, Almaas E . Peeling the yeast protein network. Proteomics 2005; 5: 444–449.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 2004; 6: 1–6.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25: 25–29.
Acknowledgements
We thank Dr Santu Bandyopadhyay (CSIR-Indian Institute of Chemical Biology, India) for his valuable suggestion during FACS analysis. We are also grateful to Sneha Vivekanandan (Mayo Clinic, USA) and Julie S Lau (Mayo Clinic, USA) for their valuable help in 3D cell culture assay and nude mice experiment, respectively. We are grateful to Dr Sumana Bhattacharjya for her assistance in manuscript editing. We also acknowledge Mayo Clinic Histology Core Facility for their assistance in this work. This work was supported by CSIR-Mayo Clinic Collaboration for Innovation and Translational Research Grant CMPP-08 to S Roychoudhury and MLP-1203 to S Chowdhury; Council of Scientific and Industrial Research (CSIR) Grant IAP-001 to S Roychoudhury, and BSC-0121 to S Chakrabarti; Department of Biotechnology (DBT) Grant BT/01/COE/05/04 to S Roychoudhury. This work was also partly supported by NIH Grant CA150190 to D Mukhopadhyay and Mayo Clinic Foundation. SKA, AD, and MB are supported by predoctoral fellowship from the Council of Scientific and Industrial Research (New Delhi, India). S Chowdhury is a Senior Research Fellows of the Wellcome Trust/Department of Biotechnology India Alliance (500127/Z/09/Z). VKY acknowledges postdoctoral fellowship from Wellcome Trust/Department of Biotechnology India Alliance (500127/Z/09/Z).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Oren
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Alam, S., Yadav, V., Bajaj, S. et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ 23, 707–722 (2016). https://doi.org/10.1038/cdd.2015.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.133
This article is cited by
-
Eph receptors and ephrins in cancer progression
Nature Reviews Cancer (2024)
-
IKKα promotes lung adenocarcinoma growth through ERK signaling activation via DARPP-32-mediated inhibition of PP1 activity
npj Precision Oncology (2023)
-
The crosstalk between ubiquitin-conjugating enzyme E2Q1 and p53 in colorectal cancer: An in vitro analysis
Medical Oncology (2023)
-
Mutant p53 in cancer: from molecular mechanism to therapeutic modulation
Cell Death & Disease (2022)
-
ASCL1-regulated DARPP-32 and t-DARPP stimulate small cell lung cancer growth and neuroendocrine tumour cell proliferation
British Journal of Cancer (2020)
