
Abstract
Recent epigenomic studies have predicted thousands of potential enhancers in the human genome. However, there has not been systematic characterization of target promoters for these potential enhancers. Using H3K4me2 as a mark for active enhancers, we identified genome-wide EP interactions in human CD4+ T cells. Among the 6 520 long-distance chromatin interactions, we identify 2 067 enhancers that interact with 1 619 promoters and enhance their expression. These enhancers exist in accessible chromatin regions and are associated with various histone modifications and polymerase II binding. The promoters with interacting enhancers are expressed at higher levels than those without interacting enhancers, and their expression levels are positively correlated with the number of interacting enhancers. Interestingly, interacting promoters are co-expressed in a tissue-specific manner. We also find that chromosomes are organized into multiple levels of interacting domains. Our results define a global view of EP interactions and provide a data set to further understand mechanisms of enhancer targeting and long-range chromatin organization. The Gene Expression Omnibus accession number for the raw and analyzed chromatin interaction data is GSE32677.
Similar content being viewed by others


Introduction
Gene transcription is controlled by functional interactions between promoters and enhancers. The combination of sequence analysis, RNA mapping and epigenetic modification patterns has allowed for the accurate annotation of most gene promoters. Although it is more difficult to predict enhancer elements, recent studies suggested that functional enhancers are often associated with certain histone modifications. In fact, elevated histone acetylation signals are reliable indicators for the existence of functional enhancers 1, 2, 3. Consistently, the histone acetyltransferases (HATs), p300 and other HATs are associated with active enhancers in different tissues 4, 5. Histone methylation, particularly the methylation of H3 lysine 4, was associated with enhancer elements 6, 7, 8, 9. Using a combination of these histone modification patterns, HAT-binding profiles and chromatin accessibility to DNase digestion, thousands of potential enhancers have been predicted in various human cell types 10, 11. Comprehensive analysis of histone modification states and gene expression profiles in multiple cell types linked several groups of tissue-specific enhancers to the expression of proximal genes that have important functions in the respective lineages 12. However, since enhancers may activate their target promoters over long distances, sometimes even on a different chromosome 13, a systematic analysis of genome-wide direct interactions between enhancers and promoters is needed to assign the predicted enhancers to their specific target promoters.
An enhancer regulates its target promoter via direct physical interaction, which can be revealed by chromatin conformation capture (3C) analysis 14. For example, the spatial proximity mediated by GATA1 and FOG-1 is required for activation of the β-major globin promoter by the locus control region 15. Thus, the 3C-based direct interaction assays would be powerful tools to identify target promoters for potential enhancers. A strategy, termed chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) that combined 3C, ChIP and next-generation sequencing, has been designed to identify genome-wide potential enhancers mediated by a specific transcription factor 16. Since a sequence-specific transcription factor may recognize only a subset of all potential enhancers in a cell, we decided to use H3K4me2, which is a general mark for active enhancers 8, 17, as a bait to unbiasedly identify enhancer-promoter (EP) interactions in human CD4+ T cells using the ChIA-PET strategy. We identified 6 520 long-distance chromatin interactions, among which are 2 067 potential enhancers that interact with 1 619 target promoters. Our data revealed complex networks of EP, enhancer-enhancer, and promoter-promoter interactions. We found that these enhancers increase the expression of their target promoters and the interacting genes are co-regulated in a tissue-specific manner, likely mediated by tissue-specific EP interactions.
Results
Analysis of genome-wide EP interactions using ChIA-PET and the H3K4me2 antibody
To capture genome-wide EP interactions, human primary resting CD4+ T cells were crosslinked with formaldehyde to covalently stabilize these interactions to keep the interacting enhancer and promoter in spatial proximity. Since enhancers and promoters are marked by H3K4me2 17, 18, we enriched the enhancer- and promoter-containing chromatin fractions by immunoprecipitation using antibodies against H3K4me2. A biotinylated DNA half linker was ligated to the ends of enriched chromatin fragments and then the two chromatin fragments with half linkers, which are in spatial proximity, were ligated to form the full linker, as described 16. The ligation products were digested with MmeI, which recognizes two sites in the full linker, to release a 19-21 bp sequence tag from each end of the chromatin fragments that were ligated to the linker. The resulting tag-linker-tag fragments are further enriched by binding to streptavidin beads and ligated to specific adaptors, followed by PCR amplification and next-generation paired-end sequencing (summarized in Figure 1A). To identify long-range chromatin interactions, we represented paired-end tags by points in the 2-dimensional space Genome × Genome, and identified regions with significantly enriched points (see Materials and Methods). Since neighboring regions in the genome tend to exhibit a higher background of random interactions that may be more frequently captured by crosslinking and adaptor ligation, we only considered interactions that occurred by > 20 kb apart. Our estimates showed that the false discovery rate (FDR) of inter-chromosomal interaction detection is quite high due to intra-bead ligations of random fragment (see Materials and Methods). Therefore, we restricted our analyses to the intra-chromosomal interactions only. From two sequencing runs (Seq 1 and Seq 2) of the same library, we identified 2 934 and 3 171 long-range interactions, respectively. Comparison of these two technical repeats revealed 2 350 shared interactions (80% of the Seq 1) (Figure 1B, left panel), indicating that the sequencing of the libraries has not reached saturation. In an independent biological replicate experiment, we identified 665 long-range (≥ 20 kb) interactions, which showed a significant 60% overlap with those from Seq 1 + 2 (Figure 1B, right panel). Because neither of these two libraries has been sequenced to saturation, we pooled the sequencing tags from these two experiments and identified a total of 6 520 long-range intra-chromosomal interactions (Supplementary information, Table S1).

Identification of genome-wide enhancer-promoter interactions. (A) Experimental scheme. Chromatin fractions prepared from formaldehyde-crosslinked human primary resting CD4+ T cells by sonication were immunoprecipitated using H3K4me2 antibodies. The ChIP DNA was processed following the ChIA-PET protocol. The library was sequenced by paired-end sequencing on HiSeq 2000. (B) Technical and biological replicates indicate significant overlap of detected interactions. (C) Summary of types of interactions. (D) Distance of enhancers to their target promoters. Each column shows the number of enhancer-promoter interactions at a certain distance between the enhancer and target promoter.
The 6 520 long-distance interactions involved 5 939 promoters and 2 499 non-promoter regions (hereafter termed as potential enhancers). Among all the interactions, 2 373 were EP interactions involving 2 067 enhancers and 1 619 promoters. In addition, we also detected 3 669 promoter-promoter interactions and 478 enhancer-enhancer interactions (Figure 1C). Among all the EP interactions, 1 324 (56%) were within 50 kb and 1 049 (44%) were > 50 kb (Figure 1D).
To validate the long-range interaction results, we tested 11 randomly selected interacting regions using 3C-PCR assays and found that nine were positive for interaction (Figure 2A). One specific example of an interaction was detected between the promoters (denoted as P1 and P3) of ANTXR1 and AGAP7 (Figure 2B), which are separated by > 4 million base pairs on chromosome 10. The 3C-qPCR assays indicated a significantly higher interaction between the detected interacting pair (P1 and P3) than the neighboring regions (P1-P2 and P1-P4, respectively). Another interaction was identified between the P1 and P4 regions of PRDM12 and ASS1 genes, separated by 250 kb on chromosome 9. The 3C-qPCR confirmed that the interaction between P1 and P4 were highly specific, in comparison to the neighboring P2, P3 and P5 regions (Figure 2C).

Validation of the genome-wide interaction data. (A) 3C-PCR validation of randomly selected interactions. T cells were crosslinked using formaldehyde, digested with EcoRI and re-ligated following 3C protocols. The purified DNA was analyzed using specific primers for the genomic regions as indicated. β-actin promoter was used as an input control. (B) The specific interaction between the ANTXRL and AGAP7 genes were confirmed by 3C-quantitative PCR assays. The left panel shows the two genomic regions that interact, indicated by two filled boxes linked by a line. The H3K4me2 peaks are shown. P1 to P4 indicate the specific PCR primers designed to test the 3C interactions. The right panel shows the quantitative PCR signals of re-ligation products surrounding EcoRI sites using different combination of PCR primers, as indicated below the panel. (C) The specific interaction between the ASS1 and PRDM12 genes were confirmed as described in panel B.
Identification of EP interaction networks
Enhancer may activate transcription of multiple genes. Whether this phenomenon is a universal mechanism by which enhancers function in promoter activation is not clear. Our data indicated that 9% of all potential enhancers interacted with multiple promoters, while 91% interacted with only a single promoter (Figure 3A, upper panel). The enhancer located upstream of the BCL9L gene on chromosome 11 was found to interact with four distinct promoters. On the other hand, one gene promoter may interact with multiple enhancers. We found that 25% of promoters interacted with two or more enhancers (Figure 3A, lower panel). In particular, we found that the RUNX1 gene on chromosome 21 interacted with six enhancers. This promiscuity of promoters with multiple enhancers may serve as a mode of functional redundancy to ensure stable gene activation.

Identification of enhancer-promoter interaction networks. (A) Different modes of enhancer-promoter interactions as illustrated on the left. The upper panel indicates the number of promoters (under the columns) that interact with each enhancer; the lower panel indicates the number of enhancers (under the column) that interact with one promoter. The numbers on Y axes correspond to the numbers of enhancers (upper panel) and promoters (lower panel) in each category. (B) One cluster of enhancer-promoter interactions on chromosome 19 was shown. Red: enhancers; blue: promoters. (C) The interaction network at the VAV1 locus. (D) The interaction network, gene expression and other features at the VAV1 locus.
Our data revealed complex networks of EP interactions. For example, an interacting network was identified on chromosome 19, which consisted of multiple enhancers and promoters (Figure 3B). VAV1 is critically involved in multiple steps of lymphocyte development 19. Our data revealed extensive long-distance chromatin interaction at the VAV1 locus (Figure 3C and 3D), which harbors several genes, including GPR108, TRIP10, SH2D3A and VAV1 (Figure 3D). We identified four potential enhancers (E1-4) in this region that interacted with their gene promoters (Figure 3D). E3 and E4 were associated with hypersensitivity to DNase digestion and peaks of H3K4me2, H3K4me3, p300, and CTCF binding (Figure 3D). While E1 and E2 interacted with the divergent VAV1 and SH2D3A promoters, E3 skipped the intervening VAV1 and SH2D3A promoters, and instead interacted with the more distant GPR108 and TRIP10 promoters. E4 interacted with all of these promoters in this genomic locus. In addition, the promoters of TRIP10 and VAV1 were also linked by long-distance interaction (Figure 3C and 3D). These results suggest that these four genes may be co-expressed in T cells. Indeed, examination of mRNA profiles indicated that all these four genes were expressed. In contrast, the neighboring gene EMR1 that did not interact with the other four genes was not expressed at a detectable level (Figure 3D).
A subset of chromatin regions interacting with promoters exhibit enhancer activities
Since H3K4me2 marks active potential enhancers 17, the long-distance physical interactions between the enhancers and promoters that we detected may enhance the expression of underlying genes. We first tested whether the chromatin regions interacting with promoters have enhancer activity, using an enhancer reporter assay. Among the seven regions tested, we found that four significantly activated a heat shock reporter promoter (Figure 4A). These results suggest that the identified enhancers may enhance the expression of their target genes. To test whether the enhancers globally increase the expression of their target promoters, we compared the expression levels of the promoters with interacting enhancers to those without enhancers. This analysis indeed revealed that the former group of promoters exhibited significantly higher levels of expression than the latter group (Figure 4B). To test whether the expression of a gene correlates with the number of enhancers interacting with its promoter, we grouped genes according to the number of detected enhancers and compared their expression levels. Interestingly, the expression positively correlated with the number of enhancers in CD4+ T cells but not in HEK293 cells (Figure 4C).

Increased expression is associated with the detected enhancer-promoter interactions. (A) The identified enhancers activate transcription from a heat shock reporter promoter. The potential enhancer was cloned upstream of the basal heat shock promoter driving a luciferase reporter gene and tested in Jurkat T cells. (B) Significantly higher expression is detected for the genes with enhancer-promoter interactions. Box plots of the RPKM values of the genes showing EP interactions (EP genes) and the controls genes that do not have interacting enhancers but have similar levels of H3K4me2 in promoter regions (left panel). The right panel shows the similar levels of H3K4me2 at the enhancer target genes (EP genes) and the control genes. (C) Box plots showing that the expression of a gene is positively correlated with the number of enhancers that interact with its promoter. The expression of genes interacting with different numbers of enhancers (indicated below the panel) is shown for CD4+ T cells (left panel) and HEK293 cells (right panel). (D) Genes interacting with the same enhancer are co-expressed in CD4+ T cells (red: genes linked by the same enhancer; blue: randomized gene list). Y axis indicates the average higher order correlation characteristic of genes linked by a shared enhancer (see Materials and methods for detail) and X axis indicates the normalized expression level. (E) The same analysis as in Figure 4D but using gene expression data in HEK293 cells.
Due to the finding that one enhancer may interact with more than one gene (Figure 3A), we tested whether the genes that interact with the same enhancer tend to be co-expressed. Examination of the four genes that interacted with the BCL9L enhancer indicated that three are highly expressed in CD4+ T cell. In contrast, these genes had low expression levels in HEK293 cells. To test this globally, we identified all enhancers that each interacted with at least two target genes, and examined whether these target genes are co-expressed using a higher order correlation characteristic (see Materials and Methods for details). The analysis revealed that the genes interacting with the same enhancer tended to be co-expressed in CD4+ T cells as revealed by the significant difference between the identified genes that interacted with the enhancers and the randomized gene list (Figure 4D). Interestingly, the significant difference disappeared in HEK293 cells (Figure 4E). These results collectively indicate that the predicted enhancers possess enhancer activity and contribute to the expression of their target genes.
CTCF and p300 binding collectively controls genes involved in lymphocyte function
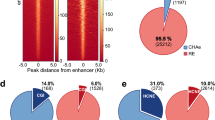
Among the factors involved in chromatin organization and enhancer activities are CTCF and p300. While p300, a histone acetyltransferase, is bound to tissue-specific enhancers 4, CTCF is a ubiquitously expressed chromatin-binding protein 7, 20, 21 and demarcates chromatin domains 21. Recent studies implicated a critical role for CTCF in mediating long-range chromatin interactions 22, 23. Our data revealed that 23% of the predicted enhancers were bound by CTCF, while 14% were bound by p300 and/or CBP (Figure 5A). In addition, 4% were bound by both CTCF and p300 or CBP. At the gene level, 417 genes interacted with enhancers bound by CTCF and 182 genes interacted with enhancers bound by p300 and/or CBP. Notably, only 24 genes interacted with both CTCF-bound enhancers and p300/CBP-bound enhancers (Figure 5B). Gene ontology analysis revealed that the group of genes interacting with either the CTCF-bound or p300-bound enhancers were highly enriched in lymphocyte-related pathways (Figure 5C), which is consistent with the critical involvement of CTCF in the expression of γ-interferon in T help 1 cells 22.

CTCF-bound and p300-bound enhancers jointly control T cell-specific gene expression. (A) Pie diagram showing the fractions of enhancers bound by CTCF or p300 and/or CBP. (B) Venn diagram showing the number of genes interacting with CTCF-bound enhancers, p300-bound enhancers, and both classes of enhancers. (C) The genes interacting with either the CTCF-bound enhancers or p300-bound enhancers are highly enriched in T cell and immune functions as indicated by Gene Ontology analysis. (D) Highly enriched motifs identified in the 2 067 enhancers. (E) Stronger enhancer-promoter interaction was detected at T cell-specific genes. Human CD4+ T cells and IMR90 cells were crosslinked with formaldehyde, digested with EcoRI and the DNA ends were re-ligated. The products derived from long-range chromatin interaction were analyzed by PCR using specific primers.
To seek potential sequence-specific transcription factors that may be involved in these long-range chromatin interactions, we searched for known factor-binding sequence motifs that may be overrepresented in the 2 067 potential enhancers. Among the top motifs were RUNX, GATA, ETS, and STAT6 motifs (Figure 5D), which are recognized by known critical regulators of T cell development and function. Using a published binding data set of GABP1 24, an ETS family member, we found that GABP1-binding sites from Jurkat T cells overlapped with 110 of the 2 067 enhancers (data not shown), suggesting that GABP1 may be involved in the function of a subset of enhancers.
The above results suggest that the EP interactions involved in the T cell-specific genes may be T cell specific. To test this hypothesis, we compared the EP interactions at the GATA3 and CD3g genes, which were expressed in T cells but not in IMR90 cells, between CD4+ T cells and IMR90 cells. Indeed, we detected a stronger interaction between the GATA3 promoter and enhancer in CD4+ T cells than in IMR90 cells, while the interaction at the CD3g gene was similar between the two cell types (Figure 5E).
Interacting promoters are co-expressed
We found that about 56% of the long-distance interacting pairs are promoter-promoter interactions (Figure 1C). To test whether there is any functional significance of these interactions, we plotted the fraction of intra-chromosomal interacting pairs (separated by > 50 kb) versus the expression levels of the two interacting promoters measured by RNA-seq (Figure 6A). Interestingly, a significant difference (P < 10−5) was detected between the interacting pairs and randomized control pairs, indicating that the interacting promoters may be co-expressed. To test whether the co-expression of these interacting genes are conserved in other cell types, we performed a similar analysis using mRNA expression data obtained in HEK293 cells, and found that there was no detectable difference in expression between the interacting pairs and randomized pairs (Figure 6B). These results indicate that the interacting promoters are potentially significantly co-expressed in a tissue-specific manner.

The expression of interacting promoters is correlated. (A) Intra-chromosomal interacting promoters are co-expressed. The pairwise gene expression correlation characteristic (Y axis) versus the gene expression level (X axis) measured by RKPM values in human primary CD4+ T cells is plotted (see Materials and methods). (B) Same as in (A) except that the gene expression was measured in HEK293 cells.
Multiple levels of chromatin organization mediated by long-distance chromatin interaction
Interphase chromatin may take multiple configurations from extended 10 nm to 30 nm fibers and even higher order structure. However, very little is known regarding the folding of chromatin beyond the 30-nm fiber. To examine the global picture of long-distance chromatin interaction and higher order chromatin organization in human T cells, we plotted all interactions between regions > 20 kb distant for chromosome 19 (Figure 7A). The results revealed that several regions of the chromosome are enriched with “local” chromatin interactions (from 20 kb to hundreds of kb), as indicated by the highlighted domains (labeled as a, b, c, d, and e). Interestingly, these highly interactive chromosome domains are also linked by “super” long-distance chromatin interactions. To visualize these interactions, we plotted the density of long-distance interaction using a heat map for chromosome 19 (Figure 7B, red indicates high density; blue indicates low density). The plot revealed five domains along the diagonal, which correspond to the five domains rich of “local” interactions highlighted in Figure 7A. In addition, each of these domains also exhibited interactions with other domains (the red signals outside of the diagonal region). These results indicate that multiple levels of chromatin organization exist in the cells, ranging from “local” interactions to super long-distance “domain” interactions, as summarized in the cartoon (Figure 7C).

Multiple levels of chromatin organization mediated by long-distance chromatin interactions. (A) All interactions between regions > 20 kb distant for chromosome 19 are plotted. The five regions that show dense “local” interactions are highlighted and labeled as domains a-e. (B) The global chromatin interaction heatmap for chromosome 19 is shown. Red color indicates dense interaction and blue color indicates sparse interaction. The five domains (a-e) showing dense interaction are indicated on the chromosome. (C) Cartoon showing the multiple levels of chromatin organization mediated by long-distance chromatin interactions beyond the 30 nm fiber.
Discussion
Using ChIA-PET with antibodies recognizing the H3K4me2 signal, which marks active enhancers in the genome, we identified 6 520 long-distance chromatin interactions in human primary CD4+ T cells. In particular, we identified 2 067 enhancers that interacted with 1 619 specific promoters. We found that 57% of the tested enhancer elements showed enhancer activity on the heat shock reporter promoter, and these enhancers globally increased the expression of their target genes in CD4+ T cells but not in HEK293 cells, consistent with our observation that selected enhancers exhibited T cell-specific interactions with their target promoters. Our results indicate that interacting promoters are co-expressed. Our data also revealed multiple levels of chromatin organization comprising subdomains and domains rich of “local” interactions.
Previous genome-wide studies predicted thousands of potential enhancers based on sequence features, various epigenetic signatures, and regulatory factors 10, 11. Correlation analysis of histone modifications on the predicted enhancers with expression of genes in proximity indicated that a subset of enhancers may regulate the expression of their nearby target genes 12. However, an enhancer can regulate target genes over long distance, sometimes skipping intervening genes or even from different chromosomes. In these cases, it is difficult to predict targets for enhancers based on proximity. Since enhancers must interact with target promoters to activate their expression, direct interaction assays provide a powerful tool to identify target genes for enhancers. In this study, we were able to assign 2 067 potential enhancers to 1 619 specific target promoters in human primary CD4+ T cells using direct interaction assays. Our data indicated that the genes that interact with the identified enhancers exhibited higher expression than the control group of genes, suggesting that the enhancer regions have enhancer activity as indicated by the reporter assays. However, we found that 23% of these enhancer elements were bound by CTCF, an insulator-binding protein. Therefore, the CTCF-bound regions may not function directly to activate transcription of target genes, but instead may act to bring other enhancers in close proximity to target promoters, as suggested by a recent study 23. Nonetheless, our data suggested that CTCF-bound enhancers and p300-bound enhancers collectively control the expression of genes involved in T cell differentiation and function.
Subnuclear organization critically influences gene expression. Transcription of active genes is believed to take place at transcription factories that are foci of hyperphosphorylated RNA polymerase II and are associated with active transcription 25, 26, suggesting that the co-localized genes may be co-regulated. However, there is only sparse evidence on co-localization of genes and their co-regulation in the genome 27, although relocation of genes in the nucleus is observed upon repression or activation 28, 29. Recently, Schoenfelder et al.30 presented an elegant data showing that active globin genes associate with hundreds of other active genes located both on the same and different chromosomes, highlighting the coordinated regulation of genes by co-localization. Consistently, the INS gene promoter physically interacts with the SYT8 gene, located > 300 kb away, which is critical for the regulated expression of the SYT8 gene depending on insulin signals 31. In our study, we comprehensively identified promoter-promoter interactions and showed that interacting promoter pairs are co-expressed. Furthermore, the genes interacting with the same enhancer were significantly co-expressed. Importantly, the co-expression of the interacting genes appeared to be tissue specific, as the expression of linked genes, which were identified from CD4+ T cells, was only significantly correlated in CD4+ T cells and not in HEK293 cells, thus providing genome-wide support for co-localization and co-expression of functionally important genes.
Interphase chromosomes adopt preferred conformations 32, which bring different functional elements into proximity by compartmentalization of the nucleus 33, 34, 35, 36. However, our knowledge about the folding of chromatin beyond the 30-nm fiber is very limited. A recent unbiased mapping of long-range chromatin interactions confirmed the presence of chromosome territories and the segregation of open and closed chromatin compartments 37. The genome-wide interaction data support the fractal globule model of chromatin organization through multiple rounds of crumpling an unentangled chromatin fiber into a series of small globules in a beads-on-a-string configuration. Our data support a model of multiple levels of chromatin organization (Figure 7C) such that chromosomes contain active domains rich in relatively local interactions, and these domains are connected by super long-distance interactions. These super long-distance interactions may be responsible for bringing different chromatin domains to spatial proximity required for the reciprocal translocation observed in various human diseases. What is the force that drives the chromatin organization in the nucleus? One of the best-studied factors of chromatin organization is CTCF, a chromatin insulator-binding protein. CTCF is associated with tens of thousands of sites on chromatin 7, 20, 21 and mediates the functional interaction of several well-studied genomic loci 38, 39. A recent study has unveiled the genome-wide chromatin interactions mediated by CTCF 23. One critical partner for the function of CTCF in forming long-distance chromatin loop is cohesin. CTCF and cohesin are broadly co-localized on mammalian chromosomes 40, 41, 42. However, our data indicated that only a minor fraction of the EP interactions was associated with CTCF binding, suggesting that other factors may be involved. Indeed, we found that motifs for several transcription factor families such as ETS, RUNX, and GATA, which represent key factors for T cell development and function, were identified in the enhancer regions. Future studies are needed to address the mechanisms that regulate the long-distance EP and promoter-promoter interactions.
Materials and Methods
Purification of human CD4+ T-cell, chromatin preparation, H3K4me2 ChIP
1 × 108 CD4+ T cells were isolated from human blood as previously described 7. Cells were then crosslinked with 1% formaldehyde for 10 min at room temperature, followed by sonication of the cells to obtain chromatin fragments ranging in size from 150 bp to 300 bp. The ChIP assay was carried out using the antibody against H3K4me2 (Millipore, 07-030) and the immunoprecipitated chromatin was stored in TE buffer.
ChIA-PET library construction and paired-end sequencing
ChIA-PET libraries were prepared as described 16 and pair-end sequencing was carried out using Illumina Hi-seq sequencer.
3C-PCR assays
Crosslinked CD4+ T cells and IMR90 cells were resuspended in lysis buffer (10 mM Tris-HCl, pH 8.0, 10 mM NaCl, 0.2% NP40, and proteinase inhibitors) and lysed with a Dounce homogenizer. Following EcoRI digestion overnight, 3C-ligated DNA was prepared as previously described 37. Regular PCR products were loaded on 2% agarose gel. For quantitative 3C analysis, real-time PCR was performed as described 31. The PCR primers used for 3C-PCR analyses are listed in the Supplementary information, Table S2.
Reporter assays
Enhancer regions were amplified by PCR and cloned into pGL3-HS vector. Jurkat cells were transfected with the reporter construct using the method as described 1. The PCR primers used for reporter assays are listed in the Supplementary information, Table S3.
Data preprocessing
We performed three sequencing experiments: the biological replicate Exp 1 that consists of two technical replicates Seq 1 and Seq 2, and the biological replicate Exp 2.
The raw sequences were trimmed and the first 36 bp were kept. We modified the linker-filtering code from 16 to output base quality values in addition to the sequence, and used the modified code to remove the linker sequences from the 36-bp reads.
The sequences of the two ends of PETs were separately mapped to the hg18 human genome using the bowtie algorithm 43 with the option “-n 0 -l 20 -m 1 — best” and taking into account the base-quality values. This bowtie option ensured that only uniquely mapped reads with zero mismatches to the reference genome were retained. The mapped locations of PETs were then represented as ordered quadruples (chrom1, pos1, chrom2, and pos2) of chromosome names and positions of the two ends of each PET, with the ordering relation: chrom1 ≤ chrom2 and pos1 ≤ pos2. From the resulting set of PETs, we removed redundant ones. In other words, if the multiple copies of a quadruple (chrom1, pos1, chrom2, and pos2) were present, we kept only one. There are 53 million, 55 million, 73 million, 20 million, and 92 million uniquely mapped non-redundant PETs in Seq 1, Seq 2, Exp 1, Exp 2, and Exp 1 + Exp 2 libraries, respectively.
Identification of chromatin interactions
Each PET may be represented as a point in the two-dimensional space formed by the Cartesian product Genome × Genome, where the Genome is a concatenation of chromosomes separated by 1 Mbp spacers. The problem of identification of chromatin interactions is thus reduced to the detection of clusters of points in the two-dimensional space. In order to efficiently cluster tens of millions of data points, we implemented in C++ the density-based clustering algorithm DBSCAN (Density-Based Spatial Clustering of Applications with Noise) described in 44. We set the DBSCAN parameters: the size of the ɛ-neighborhood of a point, Eps = 3 000 bp, and the minimum number of points required to form a cluster, MinPts = 3. This choice of parameters is optimal as suggested by Supplementary information, Figure S1A, where we plotted the number of statistically significant (FDR < 5%) intra-chromosomal interactions detected for each combination of Eps and MinPts parameters. The choice Eps = 5 Kbp, MinPts = 3 gave slightly more interactions, but the interacting regions became broader and hence less specific. We removed intra-chromosomal PETs of length < 20 kbp from the clustering analysis because such PETs may originate from self-ligation of chromatin fragments in the ChIA-PET procedure. After excluding the intra-chromosomal PETs of length < 20 kb, 68 million PETs remained in Exp 1 + Exp 2 library.
We observed that single nucleotide polymorphisms could cause misalignment of tags and lead to false-positive chromatin interactions. A sequence tag that originates from a genomic locus that has a nucleotide mismatch with the reference genome may fortuitously map to a different locus perfectly without a mismatch. Therefore, we realigned the PETs in the identified clusters to the hg18 human genome using the option “-n 1 -l 20 -m 10 -k 10 –best”, allowing up to one mismatch with the reference genome and up to ten reportable alignments. The aligned reads were then examined for the presence of known single nucleotide polymorphisms (dbSNP build 130) at mismatch locations with the reference genome. A read was discarded from further analysis if a known SNP was present in at least one of its mapped locations. The resulting SNP-filtered PETs were then clustered using DBSCAN algorithm as described above and the interaction PET clusters were identified. The statistical significance of the resulting PET clusters was assessed using the hyper-geometric distribution (as described in 16). The FDR of chromatin interaction detection was estimated by “cutting” and randomly “rejoining” PETs in silico, simulating random ligation events on magnetic beads. We observed that the FDR for the inter-chromosomal interactions is very high. Therefore, only the intra-chromosomal chromatin interactions with FDR < 5% were used for all downstream analyses. A chromatin interaction as determined by the above method is a pair of regions (R1, R2) from the human genome and was displayed as a pair of rectangular boxes connected by a thin line in UCSC genome browser.
As a global test of our data, we estimated the relative (observed/expected) intra-chromosomal interaction frequency of two genomic loci as a function of the linear distance separating the loci. As expected from the flexibility of the chromatin fiber, we found that the frequency decays with increasing linear distance (Supplementary information, Figure S1B).
Annotation of chromatin interactions
We retrieved the hg18 genomic coordinates of transcription start sites of Refseq, UCSC, and Ensembl genes from the UCSC genome table browser. If the center of a region R is less than 5 kb upstream or downstream of the nearest TSS, the region is deemed a “promoter”. Otherwise, it is regarded as an “enhancer”. The overlapping “enhancer” regions were merged to obtain a non-redundant set of enhancers. All “promoter” regions located within 5 kb of the same TSS were regarded as one single promoter. The resulting non-redundant sets of “enhancers” and “promoters” were treated as nodes in our chromatin interaction network. The Cytoscape program 45 was used to display the network.
Enhancer-promoter (EP) genes are more expressed than genes in the control group
EP-promoters are the promoters that interact with the putative enhancers. The H3K4me2 scores of promoters were computed as the maximums of SICER 46 scores of H3K4me2 islands that overlap ± 2 kb region around TSS. In order to have an unbiased comparison, we selected a control group of non-EP promoters that match the EP promoters in terms of H3K4me2 score distribution as follows: we computed the quantiles q0, q10, ..., q100 of the H3K4me2 score distribution of EP promoters at 0%, 10%, 20%,...,100% percentiles of the ranked score list. A total of 150 non-EP promoters were then randomly sampled from each score interval (qi, qi+ 10). The resulting 1 500 non-EP promoters were used as the random controls. The one-sided Kolmogorov-Smirnov test was used to show that EP genes are significantly more expressed than the random control non-EP genes (P < 10−12). The results of the above analyses are displayed in Figure 4B.
The global chromatin interaction heatmap for the chromosome 19
The intra-chromosomal PETs of length at least 20 kbp on the chromosome 19 were represented as points in the two-dimensional space Chrom19 × Chrom19, and the density of points was computed using bivariate Gaussian kernel density estimator. We, thus, have a density value ρ(x,y) for each point (x, y) ∈ Chrom19 × Chrom19. We then computed the average density over all PETs of length L as ρave(L) = (1/N) ∑abs(x-y)=L ρ(x,y), where N =∑abs(x-y)=L1. The average density ρave(L) represents the expected frequency of interaction of two loci separated by the linear distance L bp on the chromosome 19. The negative, zero, and positive values of the difference between observed and expected densities, ξ(x,y) = ρ(x,y) − ρave(abs(x− y)), are color-coded by blue, white, and red colors, respectively (Figure 7B).
Co-expression of the interacting genes
The interacting gene pairs are co-regulated and their expression levels may be correlated. We devised a sensitive statistical test for this purpose. Let {Imn} be the connectivity matrix of the intra-chromosomal promoter-promoter interaction network and let NE be the total number of edges in the network. It is assumed that the genomic distance between interacting promoters is >50 kbp. Thus, Imn = 1 if the promoter m and the promoter n interact, and Imn = 0 otherwise. Let xm be the expression (RPKM) value of the gene whose promoter is m. For each RPKM value of x, let λ(x) be the fraction of interacting promoter pairs in the network {Imn} such that the expression levels of both genes of the pair are at least x. Namely, λ(x) = (1/NE) ∑m < n Θ (xm ≥ x AND xn ≥ x) Imn, where Θ is the function: Θ(true) = 1 and Θ(false) = 0. The function λ(x) is a pairwise gene expression correlation characteristic of the promoter-promoter interaction network. The definite integral ∫λ(x)dx in the interval (0,∞) yields the area under the curve (AUC). The higher the AUC value is, the more correlated the expression values of the gene pairs in the network are.
To show the statistical significance of pairwise correlations, we generated 1 000 random promoter-promoter interaction networks using the same set of promoters and the expression levels of the associated genes as in the network {Imn}, and computed AUC for each random network. From the resulting distribution of AUC values, we estimated the P-value for the statistical significance of AUC of the real network. Since the promoters (nodes) of randomized networks were chosen from the real network, the levels of H3K4me2 at the promoters of real and random networks are similar, thus making the analysis unbiased.
In order to demonstrate the tissue specificity of the correlations, the above analysis was repeated in HEK293 cells using the chromatin interaction network in CD4+T cells, but with the gene expression values in the HEK293 cells obtained from 47.
The results of the above correlation analyses are displayed in Figure 6.
Higher order correlation of genes controlled by the same enhancer
Our data show that multiple genes can interact with the same enhancer (see Figure 3A). Such co-regulated genes are expected to have correlated expression levels. To test this, we devised a sensitive statistical test.
Let S be the set of genes that interact with the same enhancer. For each gene g in the set, we computed a normalized gene expression value yg as the percentage of g's expression value compared to the maximum expression value in S. Namely, yg= 100 (xg/xmax)%, where xg is RPKM value of g and xmax is the maximum of RPKM values in S. For each value, 0 ≤ y ≤ 100, we computed the fraction Φ(y) of genes in the S with normalized expression value of at least y.
Namely, Φ(y) = (1/NS) ∑g∈SΘ(yg ≥ y), where NS is the number of genes in S and the function Θ was defined above. The function Φ(y) is a higher order correlation characteristic of the set S.
Let S1, S2, ..., SM be the sets of genes that interact with enhancers E1, E2, ..., EM, respectively. The number of genes Nk in each set Sk is assumed to be at least two. From the higher order correlation characteristics Φ1, Φ2, ..., ΦM corresponding to these sets, we computed the average characteristic Φave (red curve in Figure 4D). We then computed the AUC = ∫ Φ(y)dy for each set of genes controlled by an enhancer and obtained a distribution of AUC values.
The randomized controls were obtained by randomly sampling M sets of genes of the same sizes as in the real data, i.e. of the sizes N1, N2, ..., NM, from the network of chromatin interactions in CD4+T cells. Using these gene sets, we computed the average higher order correlation characteristic for randomized data (blue dashed curve in Figure 4D).
To obtain the corresponding curves and P-value in HEK293 cells (Figure 4E), we used the same chromatin interaction network from CD4+ T cells as above, but the gene expression values were from the HEK293 cells 47.
Motifs at enhancers
We detected H3K4me2 peaks in CD4+ T cells using model-based analysis of ChIP-Seq algorithm 48, extracted “summit” locations from the peaks, and kept the summits located within enhancers that interact with promoters according to our data. We used HOMER algorithm 49 to find enriched motifs of known factors at ± 300 bp regions surrounding the summits.
References
Roh TY, Wei G, Farrell CM, Zhao K . Genome-wide prediction of conserved and nonconserved enhancers by histone acetylation patterns. Genome Res 2007; 17:74–81.
Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 2011; 107:21931–21936.
Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J . A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011; 470:279–283.
Visel A, Blow MJ, Li Z, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009; 457:854–858.
Wang Z, Zang C, Cui K, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009; 138:1019–1031.
Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007; 39:311–318.
Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823–837.
Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008; 40:897–903.
Pekowska A, Benoukraf T, Zacarias-Cabeza J, et al. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J 2011; 30:4198–4210.
Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108–112.
Boyle AP, Davis S, Shulha HP, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008; 132:311–322.
Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43–49.
Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA . Interchromosomal associations between alternatively expressed loci. Nature 2005; 435:637–645.
Dekker J, Rippe K, Dekker M, Kleckner N . Capturing chromosome conformation. Science 2002; 295:1306–1311.
Vakoc CR, Letting DL, Gheldof N, et al. Proximity among distant regulatory elements at the beta-globin locus requires GATA-1 and FOG-1. Mol Cell 2005; 17:453–462.
Fullwood MJ, Liu MH, Pan YF, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009; 462:58–64.
He HH, Meyer CA, Shin H, et al. Nucleosome dynamics define transcriptional enhancers. Nat Genet 2010; 42:343–347.
Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008; 40:897–903.
Tarakhovsky A, Turner M, Schaal S, et al. Defective antigen receptor-mediated proliferation of B and T cells in the absence of Vav. Nature 1995; 374:467–470.
Kim TH, Abdullaev ZK, Smith AD, et al. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 2007; 128:1231–1245.
Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K . Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res 2009; 19:24–32.
Sekimata M, Perez-Melgosa M, Miller SA, et al. CCCTC-binding factor and the transcription factor T-bet orchestrate T helper 1 cell-specific structure and function at the interferon-gamma locus. Immunity 2009; 31:551–564.
Handoko L, Xu H, Li G, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 2011; 43:630–638.
Valouev A, Johnson DS, Sundquist A, et al. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 2008; 5:829–834.
Jackson DA, Hassan AB, Errington RJ, Cook PR . Visualization of focal sites of transcription within human nuclei. EMBO J 1993; 12:1059–1065.
Wansink DG, Schul W, van der Kraan I, van Steensel B, van Driel R, de Jong L . Fluorescent labeling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J Cell Biol 1993; 122:283–293.
Osborne CS, Chakalova L, Brown KE, et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet 2004; 36:1065–1071.
Ragoczy T, Bender MA, Telling A, Byron R, Groudine M . The locus control region is required for association of the murine beta-globin locus with engaged transcription factories during erythroid maturation. Genes Dev 2006; 20:1447–1457.
Takizawa T, Gudla PR, Guo L, Lockett S, Misteli T . Allele-specific nuclear positioning of the monoallelically expressed astrocyte marker GFAP. Genes Dev 2008; 22:489–498.
Schoenfelder S, Sexton T, Chakalova L, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet 2011; 42:53–61.
Xu Z, Wei G, Chepelev I, Zhao K, Felsenfeld G . Mapping of INS promoter interactions reveals its role in long-range regulation of SYT8 transcription. Nat Struct Mol Biol 2011; 18:372–378.
Cremer T, Cremer C . Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2001; 2:292–301.
Misteli T . Beyond the sequence: cellular organization of genome function. Cell 2007; 128:787–800.
Kosak ST, Groudine M . Gene order and dynamic domains. Science 2004; 306:644–647.
Sexton T, Schober H, Fraser P, Gasser SM . Gene regulation through nuclear organization. Nat Struct Mol Biol 2007; 14:1049–1055.
Dekker J . Gene regulation in the third dimension. Science 2008; 319:1793–1794.
Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009; 326:289–293.
Bell AC, Felsenfeld G . Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000; 405:482–485.
Ling JQ, Li T, Hu JF, et al. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science 2006; 312:269–272.
Wendt KS, Yoshida K, Itoh T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008; 451:796–801.
Parelho V, Hadjur S, Spivakov M, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008; 132:422–433.
Seitan VC, Hao B, Tachibana-Konwalski K, et al. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature 2011; 476:467–471.
Langmead B, Trapnell C, Pop M, Salzberg SL . Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25.
Ester M, Kriegel HP, Sander J, Xu X . A density-based algorithm for discovering clusters in large spatial databased with noise. In: Simoudis E, Han J, Fayyad U, eds. Proceedings of 2nd International Conference on Knowledge Discovery and Data Mining. AAAI Press: Palo Alto. 1996; 226–231.
Cline MS, Smoot M, Cerami E, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2007; 2:2366–2382.
Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W . A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009; 25:1952–1958.
Sultan M, Schulz MH, Richard H, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008; 321:956–960.
Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9:R137.
Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010; 38:576–589.
Acknowledgements
We thank our colleagues Brian Abraham, Alika Maunakea and Daniel Kraushaar for critical reading of the manuscript and Thomas Ried for suggestions. The paired-end sequencing was performed by the DNA Sequencing Core Facility of National Heart, Lung and Blood Institute (NHLBI). This work was supported by the Division of Intramural Research, NHLBI, National Institutes of Health, USA.
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Table S1
The list of 6520 intra-chromosomal interactions (XLSX 532 kb)
Supplementary information, Table S2
PCR primer sequences for 3C-PCR (PDF 55 kb)
Supplementary information, Table S3
PCR primer sequences for the reporter assays (PDF 52 kb)
Supplementary information, Figure S1
Miscellaneous analyses. (PDF 207 kb)
Rights and permissions
About this article
Cite this article
Chepelev, I., Wei, G., Wangsa, D. et al. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res 22, 490–503 (2012). https://doi.org/10.1038/cr.2012.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2012.15
Keywords
This article is cited by
-
Widespread contribution of transposable elements to the rewiring of mammalian 3D genomes
Nature Communications (2023)
-
Characterizing chromatin interactions of regulatory elements and nucleosome positions, using Hi-C, Micro-C, and promoter capture Micro-C
Epigenetics & Chromatin (2022)
-
Enhancer-promoter interaction maps provide insights into skeletal muscle-related traits in pig genome
BMC Biology (2022)
-
Molecular and computational approaches to map regulatory elements in 3D chromatin structure
Epigenetics & Chromatin (2021)
-
Self-organizing maps with variable neighborhoods facilitate learning of chromatin accessibility signal shapes associated with regulatory elements
BMC Bioinformatics (2021)
