
Abstract
Heterozygous mutations in the LMX1B gene cause nail patella syndrome (NPS) that is associated with nail and skeletal malformations, nephropathy, and glaucoma. Previous phenotype studies of Lmx1b null mice revealed dorsal limb and renal anomalies similar to human NPS, which contributed to the identification of heterozygous mutations in this LIM-homeodomain protein LMX1B as the genetic defect responsible for NPS. Despite advanced insight into the role of the Lmx1b transcription factor in a broad range of animal developmental programs, the pathogenic mechanism underlying dominant inheritance of NPS in man remained unclear. Here, we describe for the first time the detection of two entire LMX1B gene deletions and one smaller exonic LMX1B deletion by multiplex ligation-dependent probe amplification (MLPA) in a series of eight unrelated families with classical features of NPS in whom no pathogenic LMX1B mutation was found by sequence analysis. The identification of entire LMX1B deletions strongly confirms that haploinsufficiency is the principal pathogenetic mechanism of NPS and suggests a difference in dosage sensitivity for this gene between mice and man.
Similar content being viewed by others

Introduction
Nail patella syndrome (NPS; OMIM 161200) is an autosomal dominant disorder characterized by nail malformations, patellar aplasia or hypoplasia, additional skeletal abnormalities encompassing iliac horns and elbow dysplasia, glomerulopathy, and glaucoma. Since the identification of the LMX1B gene, encoding an LIM-homeobox transcription factor as being the causative gene of NPS, mutations within the coding sequence have been detected in approximately 85% of families with NPS, including missense, nonsense, frameshift, splice-site mutations, and small intragenic insertions/deletions.1, 2, 3, 4, 5, 6, 7, 8, 9 These pathogenic LMX1B mutations are concentrated in the LIM domains encoded by exons 2–3 that facilitate the interaction with other transcription factors and in the homeodomain encoded by exons 4–6, which is involved in DNA binding. Dunston et al (2004) identified the first mutations in exon 1, upstream of the LIM domains and in exon 6, downstream of the homeodomain.10 As yet, no mutations have been described in exons 7 and 8 encoding the transcriptional activation domain at the carboxy terminus suggesting that mutations in the latter domain not cause the NPS phenotype. In addition, a 17 bp intronic deletion was detected in one NPS family and balanced translocations, presumably disrupting LMX1B have been reported.11, 12 Linkage studies in NPS families did not reveal evidence for locus heterogeneity thus far.
Molecular studies in Lmx1b null mice modelling the NPS phenotype have proven to be pivotal to our understanding of the role of Lmx1b in the normal and disrupted development of dorsal limb structures, the kidney, the eye, and the central nervous system, and particularly focussed on the pathogenesis underlying glomerulopathy in the past decade.13, 14, 15, 16, 17, 18, 19, 20 It is as yet unclear why heterozygous LMX1B mutations cause NPS in humans, whereas Lmx1b heterozygous (+/−) mice are completely normal. The hypothesis that haploinsufficiency is the main pathogenic mechanism underlying dominant inheritance in human NPS is based on the fact that the same phenotypic variability is observed in individuals with LMX1B missense, nonsense, frameshift or splice-site mutations and that the range and severity of symptoms varies both within and between NPS families.1, 2, 3, 9 This assumption is supported by the lack of any dominant-negative effect observed by in vitro experiments studying missense and truncation LMX1B mutations.21, 22 A recent study on Lmx1b+/− mice in which renal damage was induced by unilateral nephrectomy has shown diminished compensatory renal growth with a reduced glomerular volume and a higher glomerulosclerosis index of the remaining kidney compared to the kidneys of Lmx1b+/+ mice.13 The latter experimental animal model further supports the assumption that a minimum dosage of Lmx1b is critical for normal kidney development.13, 15, 20
To investigate whether a reduced dosage of LMX1B causes NPS in man, we searched for deletions of the entire LMX1B gene in families with the classical NPS phenotype. By multiplex ligation-dependent probe amplification (MLPA) analysis with specific probes for the exons 1–8 of LMX1B, we found a deletion of the entire gene in two unrelated individuals and a deletion of exons 3–8 in another patient from a series of eight classic NPS families in whom the genetic defect was not established by sequence analysis.
Methods
Patients
The series of patients included 10 individuals from eight unrelated families with classical features of NPS in whom no pathogenetic LMX1B mutation could be detected by sequencing the exons and exon–intron boundaries. Among these families, four families were incorporated in an earlier genotype−photype study (families no. 29−32,9), the other four families have not been reported previously. Informed consent was obtained from all patients or their parents. Phenotype studies of NPS patients in whom an LMX1B deletion was identified was accomplished as previously described.6
MLPA analysis
A specifically designed set of synthetic oligonucleotide probes located in all exons 1–8 of the LMX1B gene was used for MLPA analysis to identify intragenic and entire LMX1B deletions. The synthetic MLPA probes were created in accordance with the protocol provided by MRC-Holland (http://www.mlpa.com) (Table 1). The probes contain gene-specific sequences; in one case, a stuffer and an extension for amplification. To further establish the size of the genomic deletion identified, we subsequently developed a second set of probes for MPLA analysis of the flanking regions of LMX1B (Table 1). This additional set of probes was located in the FAM125B and ZNF297B genes flanking the 5′ and 3′ end of LMX1B, respectively, and in the next positioned genes PBX3 and ANGPTL2 located ∼0.5 Mb centromerically and telomerically from LMX1B, respectively (UCSC Genome Browser on Human, freeze 2004, http://genome.ucsc.edu) (Figure 1). MLPA was essentially performed according to standard procedures.23, 24 Briefly, 200–400 ng of genomic DNA in a final volume of 8 μl was heated at 98°C for 5 min. After addition of 1.5 μl probe mix per sample, which was mixed 1:1 with a salt solution, samples were heated for 1 min at 95°C and incubated overnight at 60°C. The Ligation-65 was inactivated by heating at 98°C for 1 min. Subsequently, the ligation products were amplified by PCR using the SALSA primers (Table 1), labeled with FAM on the forward primer. Product identification and quantification was performed by capillary electrophoresis on an ABI 3100 genetic analyser (Applied Biosystems).

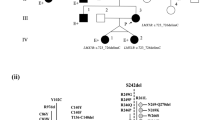
Schematic genomic structure of the LMX1B gene (exons 1–8) localized at chromosome 9q34.1, the size of the three unique deletions, and the localization of MLPA probes in the different genes flanking LMX1B (FAM125B, ZNF297B, PBX3, and ANGPTL2). The horizontal line represents the genomic region at chromosome 9q34.1 encompassing LMX1B, the vertical lines denote the exons of LMX1B, and blackened bars indicate the size of the deletion. In one sporadic case (family A), a de novo deletion of the entire gene was found; in family B, a deletion of the entire gene and flanking genes of both the 5′ and 3′ end of LMX1B was detected; and in family C, a deletion of exons 3–8 of the LMX1B gene was identified.
Results
Deletions of all exons 1−8 of the LMX1B gene were identified by initial MLPA analysis in two unrelated individuals with NPS (family A and B) and a deletion of exons 3–8 was found in another patient (family C) from a series of eight classic NPS families (3/8; (38%) in whom no mutation was found by sequencing LMX1B (Figures 1 and 2). In family A, this entire LMX1B gene deletion was found de novo. Further determination of the size of the genomic microdeletions revealed a deletion of the whole LMX1B gene only in family A, whereas a deletion of the entire LMX1B and flanking FAM125B and ZNF297B genes was shown in family B (Figure 1). In family C, no evidence for the deletion of genes at the 3′ boundary of the LMX1B gene was found. Comprehensive clinical studies of these families revealed renal and extrarenal symptoms comparable with our previously reported families with LMX1B mutations resulting in premature termination codons (PTCs) as well as missense mutations (Supplementary Table 1).9

Pedigrees of three families with nail patella syndrome in whom the entire or partial gene LMX1B deletions were identified. Asterisks indicate individuals in whom MLPA experiments were performed.
Discussion
To the best of our knowledge, this is the first study demonstrating entire LMX1B deletions in human classic NPS, which confirms the hypothesis that haploinsufficiency is the principal pathogenic mechanism of NPS in man. By MLPA analysis of eight unrelated NPS patients in whom no pathogenic LMX1B mutation had been detected by direct sequencing, two unique whole gene and one smaller exonic LMX1B deletion were identified in three kindreds. The present eight families are part of our series of 36 NPS families among which an LMX1B mutation was previously found by sequencing in 78% (28/36) of the families and in which the current entire/smaller exonic LMX1B deletions were shown in 8% (3/36). The smaller exonic LMX1B deletion (exons 3–8) was found in one of the four previously reported families in whom fluorescent in situ hybridization (FISH) analysis failed to reveal this genetic defect (family C, corresponding to family no. 29 in Bongers et al, 2005).9 This finding underscores that FISH analysis lacks the resolution of MLPA in detecting smaller exonic LMX1B deletions. The genetic defect remains to be elucidated in 14% (5/36) of our series of NPS families. In these unresolved families, mutations in the introns, the promoter region, or the 3′ untranslated region of LMX1B as well as complex genomic rearrangements could be causing NPS. The identification of the genetic cause in other families may be instrumental to investigate whether other mechanisms than loss-of-function, such as gain-of-function, may underlie NPS.
Comparison of the NPS phenotype between families with entire/partial gene deletions, nonsense mutations, and missense mutations revealed the same range and severity of symptoms. The observation that entire/partial gene LMX1B deletions and nonsense LMX1B mutations give similar NPS phenotypes supports the assumption that mutations resulting in premature termination codons (PTCs) cause NPS by degradation of mutant LMX1B transcripts via the nonsense-mediated mRNA decay pathway to prevent the synthesis of truncated proteins.25 The comparable phenotype of NPS patients with entire deletions and missense mutations of the LMX1B gene suggests that missense alterations also result in loss-of-function. Previous functional studies further support that human missense mutations in the homeodomain diminish or abolish DNA binding, and ultimately result in decreased transcriptional activity.21, 22 A possible dominant-negative effect of these LMX1B mutations was excluded by mixed transfection studies demonstrating that wild-type LMX1B function was not altered.21, 22 The present finding of entire gene deletions in families with classic NPS confirms beyond any doubt that a reduced dosage of the LMX1B protein causes NPS and emphasizes that loss-of-function is the predominant mechanism underlying NPS pathogenesis.
Recently, a 3.07 Mb deletion on chromosome 9q33.3-34.11 encompassing six genes, including LMX1B and NR5A1, has been identified by array-based comparative genome hybridization (aCGH) in a phenotypic female with 46,XY sex reversal and clinical features of NPS and genitopatellar syndrome.26 In addition to some characteristic manifestations of NPS, including hypoplastic ridged nails, limited elbow extension with dislocated radial heads, and small patellae, she was known with mental retardation, craniofacial dysmorphisms, and genital anomalies. Although this microdeletion encompassing LMX1B might suggest that the NPS features in this patient are the result of haploinsufficiency for LMX1B, the absence of pathognomonic NPS characteristics in this patient in combination with the deletion of five additional genes does not unequivocally demonstrate that haploinsufficiency is underlying dominant inheritance in human NPS.
The fact that the kidney disease observed in Lmx1b null mice resembles a milder variant of nephropathy in individuals with heterozygous deficiency of LMX1B, whereas Lmx1b+/− mice show no ultrastructural anomalies of the glomerular basement membrane and podocytes, suggests a difference in dosage sensitivity for this gene between different species.15, 20, 27 In contrast to the previous observation that Lmx1b+/− mice reveal no ultrastructural anomalies of the kidney, induction of renal damage in this animal model appears to result in functional and structural anomalies of the kidney.13 These studies emphasize that a critical dosage of LMX1B is essential for normal postnatal glomerular function and structure. The role of LMX1B and the effect of a reduced level of LMX1B transcript on the development of other tissues affected in NPS is limited and needs further investigation.
In summary, we identified partial and whole LMX1B deletions by MLPA analysis in 38% (3/8) of patients in whom LMX1B mutations had not been detected; this represents 10% (3/31) of all LMX1B alterations found in our series of 36 families displaying a classical NPS phenotype. This finding provides strong evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance of NPS in man. The extreme variability of the NPS phenotype observed among all different LMX1B mutations challenges the identification of genetic, endogenous, and environmental modifier factors involved in the pathogenesis of NPS.
References
Dreyer SD, Zhou G, Baldini A et al: Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 1998; 19: 47–50.
Vollrath D, Jaramillo-Babb VL, Clough MV et al: Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet 1998; 7: 1091–1098.
Clough MV, Hamlington JD, McIntosh I : Restricted distribution of loss-of-function mutations within the LMX1B genes of nail-patella syndrome patients. Hum Mutat 1999; 14: 459–465.
McIntosh I, Dreyer SD, Clough MV et al: Mutation analysis of LMX1B gene in nail-patella syndrome patients. Am J Hum Genet 1998; 63: 1651–1658.
Seri M, Melchionda S, Dreyer S et al: Identification of LMX1B gene point mutations in italian patients affected with Nail-Patella syndrome. Int J Mol Med 1999; 4: 285–290.
Knoers NV, Bongers EM, van Beersum SE, Lommen EJ, van Bokhoven H, Hol FA : Nail-patella syndrome: identification of mutations in the LMX1B gene in Dutch families. J Am Soc Nephrol 2000; 11: 1762–1766.
Hamlington JD, Jones C, McIntosh I : Twenty-two novel LMX1B mutations identified in nail patella syndrome (NPS) patients. Hum Mutat 2001; 18: 458.
Bongers EM, Gubler MC, Knoers NV : Nail-patella syndrome. Overview on clinical and molecular findings. Pediatr Nephrol 2002; 17: 703–712.
Bongers EM, Huysmans FT, Levtchenko E et al: Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur J Hum Genet 2005; 13: 935–946.
Dunston JA, Hamlington JD, Zaveri J et al: The human LMX1B gene: transcription unit, promoter, and pathogenic mutations. Genomics 2004; 84: 565–576.
Hamlington JD, Clough MV, Dunston JA, McIntosh I : Deletion of a branch-point consensus sequence in the LMX1B gene causes exon skipping in a family with nail patella syndrome. Eur J Hum Genet 2000; 8: 311–314.
Silahtaroglu A, Hol FA, Jensen PK et al: Molecular cytogenetic detection of 9q34 breakpoints associated with nail patella syndrome. Eur J Hum Genet 1999; 7: 68–76.
Endele S, Klein S, Richter S et al: Renal phenotype in heterozygous Lmx1b knockout mice (Lmx1b (+/−)) after unilateral nephrectomy. Transgenic Res 2007; 16: 723–729.
Miner JH, Morello R, Andrews KL et al: Transcriptional induction of slit diaphragm genes by Lmx1b is required in podocyte differentiation. J Clin Invest 2002; 109: 1065–1072.
Morello R, Zhou G, Dreyer SD et al: Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nat Genet 2001; 27: 205–208.
Rohr C, Prestel J, Heidet L et al: The LIM-homeodomain transcription factor Lmx1b plays a crucial role in podocytes. J Clin Invest 2002; 109: 1073–1082.
Suleiman H, Heudobler D, Raschta AS et al: The podocyte-specific inactivation of Lmx1b, Ldb1 and E2a yields new insight into a transcriptional network in podocytes. Dev Biol 2007; 304: 701–712.
Pressman CL, Chen H, Johnson RL : LMX1B, a LIM homeodomain class transcription factor, is necessary for normal development of multiple tissues in the anterior segment of the murine eye. Genesis 2000; 26: 15–25.
Smidt MP, Asbreuk CH, Cox JJ, Chen H, Johnson RL, Burbach JP : A second independent pathway for development of mesencephalic dopaminergic neurons requires Lmx1b. Nat Neurosci 2000; 3: 337–341.
Chen H, Lun Y, Ovchinnikov D et al: Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet 1998; 19: 51–55.
Sato U, Kitanaka S, Sekine T, Takahashi S, Ashida A, Igarashi T : Functional characterization of LMX1B mutations associated with nail-patella syndrome. Pediatr Res 2005; 57: 783–788.
Dreyer SD, Morello R, German MS et al: LMX1B transactivation and expression in nail-patella syndrome. Hum Mol Genet 2000; 9: 1067–1074.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G : Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification1. Nucleic Acids Res 2002; 30: e57.
White SJ, Vink GR, Kriek M et al: Two-color multiplex ligation-dependent probe amplification: detecting genomic rearrangements in hereditary multiple exostoses. Hum Mutat 2004; 24: 86–92.
Behm-Ansmant I, Kashima I, Rehwinkel J, Sauliere J, Wittkopp N, Izaurralde E : mRNA quality control: an ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett 2007; 581: 2845–2853.
Schlaubitz S, Yatsenko SA, Smith LD et al: Ovotestes and XY sex reversal in a female with an interstitial 9q33.3-q34.1 deletion encompassing NR5A1 and LMX1B causing features of Genitopatellar syndrome. Am J Med Genet A 2007; 143: 1071–1081.
Morello R, Dreyer S, Oberg KC, Lee B : Localization of Lmx1b during kidney and limb development and pathogenesis of nail patella syndrome. Am J Hum Genet 1999; 65S: A77.
Acknowledgements
We thank the individuals with NPS and their family for their participation in this study. This work was supported by the Council for Medical and Health Research of the Netherlands Organization for Scientific Research (ZON-MW) through Grant 920-03-131.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Bongers, E., de Wijs, I., Marcelis, C. et al. Identification of entire LMX1B gene deletions in nail patella syndrome: evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance in man. Eur J Hum Genet 16, 1240–1244 (2008). https://doi.org/10.1038/ejhg.2008.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.83
Keywords
This article is cited by
-
Update on CD164 and LMX1A genes to strengthen their causative role in autosomal dominant hearing loss
Human Genetics (2022)
-
Clinical and genetic characterization of nephropathy in patients with nail-patella syndrome
European Journal of Human Genetics (2020)
-
Heterozygous missense variants of LMX1A lead to nonsyndromic hearing impairment and vestibular dysfunction
Human Genetics (2018)
-
Nail-patella syndrome
Pflügers Archiv - European Journal of Physiology (2017)
-
Spectrum of LMX1B mutations: from nail–patella syndrome to isolated nephropathy
Pediatric Nephrology (2017)
