
Abstract
The human leukocyte antigen (HLA) super-locus is a genomic region in the chromosomal position 6p21 that encodes the six classical transplantation HLA genes and at least 132 protein coding genes that have important roles in the regulation of the immune system as well as some other fundamental molecular and cellular processes. This small segment of the human genome has been associated with more than 100 different diseases, including common diseases, such as diabetes, rheumatoid arthritis, psoriasis, asthma and various other autoimmune disorders. The first complete and continuous HLA 3.6 Mb genomic sequence was reported in 1999 with the annotation of 224 gene loci, including coding and non-coding genes that were reviewed extensively in 2004. In this review, we present (1) an updated list of all the HLA gene symbols, gene names, expression status, Online Mendelian Inheritance in Man (OMIM) numbers, including new genes, and latest changes to gene names and symbols, (2) a regional analysis of the extended class I, class I, class III, class II and extended class II subregions, (3) a summary of the interspersed repeats (retrotransposons and transposons), (4) examples of the sequence diversity between different HLA haplotypes, (5) intra- and extra-HLA gene interactions and (6) some of the HLA gene expression profiles and HLA genes associated with autoimmune and infectious diseases. Overall, the degrees and types of HLA super-locus coordinated gene expression profiles and gene variations have yet to be fully elucidated, integrated and defined for the processes involved with normal cellular and tissue physiology, inflammatory and immune responses, and autoimmune and infectious diseases.
Similar content being viewed by others


Introduction
It is a decade since the first completely annotated and continuous human major histocompatibility complex (MHC) genomic sequence map was published.1 The main purpose of the initial genomic sequences was to produce gene and genomic feature maps incorporating known and predicted gene loci. Since then, the MHC genomic sequence template has been used extensively to investigate single nucleotide polymorphism (SNP) and haplotype variation, gene expression, sequence diversity between and within species, and the evolution of the MHC structural organization.2, 3, 4, 5, 6, 7, 8 The continuing strong interest in the MHC genomic sequence stems from its well-established role in regulating inflammation, the complement cascade and the innate and adaptive (acquired) immune responses using the natural killer (NK) and T-cell systems. The MHC locus contributes to restricted cellular interactions and tissue histocompatibility owing to the cellular discrimination of ‘self’ and ‘non-self’ that requires an essential knowledge of the effects of MHC-matched and -mismatched donors in transplantation medicine9 and transfusion therapy.10 Similarly, a fully annotated MHC genomic and diversity map is useful for understanding autoimmunity11 and for charting the host response to infectious agents.12, 13 Apart from regulating immunity, the MHC genes may have a role in reproduction and social behavior, such as pregnancy maintenance, mate selection and kin recognition.14, 15 The MHC genomic region also appears to influence central nervous system (CNS) development and plasticity,16, 17, 18, 19, 20 neurological cell interactions,21, 22 synaptic function and behavior,23, 24 cerebral hemispheric specialization,25 and neurological and psychiatric disorders.26, 27, 28, 29, 30
The MHC region at ∼4 Mb occupies 0.13% of the human genome (3 × 109 bp), but contains ∼0.5% (>150) of the ∼32 000 known protein coding genes. Many of the MHC gene products are ligands, receptors, interacting proteins, signaling factors and transcription regulators involved in the inflammatory response, antigen processing and presentation as part of the adaptive immune response, and interactions with NK cells and cytokines as part of the innate immune responses. The MHC genomic landscape is composed mainly of genes, retrotransposons, transposons, regulatory elements, pseudogenes and a few remaining undefined sequences. The MHC genomic region is one of the most gene-dense and best-defined regions within the human genome, and the undefined sequences contribute to only a low percentage of the MHC region.
The human leukocyte antigen (HLA) is the name for the human MHC and we will use both names interchangeably in this overview, which outlines the HLA genomic loci, SNP and haplotype diversity, gene interactions and expression, and disease associations. This presentation complements other recent reviews on the human MHC architecture, duplications, diversity, disease and evolution.5, 6, 14, 31, 32, 33
Definition and annotation of gene classifications
Table 1 is a summary of the latest (16 September 2008) locus information gathered on the genomic sequence of the HLA region providing the official gene and locus symbols, geneIDs, gene type, isoforms, mRNA and protein sequence accession numbers, and Online Mendelian Inheritance in Man (OMIM) identification numbers. The genomic sequence of the HLA region used for the present annotations is the PGF haplotype sequence34 that was derived from a consanguineous HLA-homozygous cell line carrying the HLA-A3, -B7, -Cw7, -DR15(DR2) combination of alleles. This sequence is different from the original HLA virtual genomic sequence that was first reported1 and reviewed31 as a continuous, but mixed genomic sequence obtained from different haplotypes. The locus information in Table 1 is divided into five subregions from the telomeric to the centromeric end, the extended class I (GABBR1 to ZFP57), class I (HLA-F to MICB), class III (PPIAP9 to BTNL2), class II (HLA-DRA to HLA-DPA3) and the extended class II (COL11A2 to KIFC1) regions. The definition of the extended class I and II regions is ambiguous, and we have included only four well-analyzed loci in the extended class I and 19 in the extended class II regions as shown in Table 1.
Locus information was assembled by using the Entrez Gene database (http://www.ncbi.nlm.nih.gov/sites/entrez) of the National Center for Biotechnology Information (NCBI) and previously published reports and papers.1, 35 The Homo sapiens official gene symbols and gene names of the MHC genomic region can be accessed by way of the ‘GeneID’ using Entrez Gene at NCBI.36 Of the 224 loci mapped and reported by The MHC Sequencing Consortium in 1999,1 more than half of them (124 loci per 224 loci) were replaced within 5 years with a new and official gene symbol and name approved by the HUGO Gene Nomenclature Committee (HGNC).31 Since then, another 21 gene symbols and names have been changed. We have provided only one ‘old symbol in 2004 and 2008’ in Table 1, but many of the official gene symbols and names have alternate symbols and aliases. For example, the alternative symbols for HLA-F (GeneID 143110) are DADB-68M4.2, CDA12, HLA-5.4, HLA-CDA12 and HLAF. There are 11 alternative names for the gene DDR1 (GeneID 780). The old or alternative gene/locus names and symbols can also be accessed through the GeneID (Table 1) at NCBI.
The assembled loci in Table 1 were classified into four categories of gene status: ‘protein coding,’ ‘gene candidate (candidate),’ ‘non-coding RNA (NC gene)’ and ‘pseudogene (pseudo).’ The descriptor ‘protein coding’ means a gene that is transcribed to mRNA and also has a reliable open reading frame (ORF) and/or a known protein product, with the accession numbers for the mRNA and protein sequences provided. The ‘gene candidate’ is transcribed to mRNA (an mRNA sequence accession number is provided), but has an unknown or uncertain ORF. It may or may not have an accession number for a protein sequence listed. The ‘NC gene’ is transcribed to mRNA (accession number is provided), but does not have any ORF or a known protein or peptide product. The ‘pseudo’ is generally not transcribed to mRNA, and it may be a fragmented gene structure or a retrotransposed and unprocessed cDNA structure. Some of the pseudogenes, such as the P5-1 family in Table 1, are known to be the remnants or hybrids of ancient endoretroviral sequences.37 Interestingly, SNP variants for one of the members of the P5-1 family, the gene locus HCP5 located near HLA-B, have been strongly associated with the progression of HIV infection,13, 38 psoriasis vulgaris and psoriatic arthritis.39
Gene numbers in the HLA region
A total of 253 loci have now been identified and/or reclassified in the 3.78 Mb HLA region of the PGF haplotype34 from BABBR1 located on the most telomeric side of the extended class I region to KIFC1 (past name: HSET) located on the most centromeric side of the extended class II region (Figure 1 and Table 1). There are an additional 29 loci since the 224 loci were first identified in the HLA region and reported in 1999.1 The locus numbers of HLA-DRB and RP-C4-CYP21-TNX subregions generated by gene duplication vary in number and reflect HLA haplotypic differences, as reported earlier.1 When all the loci of the HLA complex were grouped into four categories of gene status, 133, 19, 22 and 79 loci were classified as protein coding, gene candidates, non-coding RNAs and pseudogenes, respectively. It is clear from Table 1 that the non-HLA genes greatly outnumber the HLA-like genes (HLA-class I, MIC and HLA-class II genes). Of the 45 HLA-like genes, 20 were identified as protein coding genes, 4 were NC genes and 21 were pseudogenes. Of the 208 non-HLA genes, 112 were identified as protein coding genes, 20 candidate genes, 18 NC genes and 58 pseudogenes.

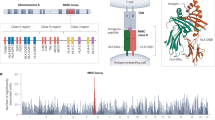
Gene map of the human leukocyte antigen (HLA) region. The major histocompatibility complex (MHC) gene map corresponds to the genomic coordinates of 29 677 984 (GABBR1) to 33 485 635 (KIFC1) in the human genome build 36.3 of the National Center for Biotechnology Information (NCBI) map viewer. The regions separated by arrows show the HLA subregions such as extended class I, classical class I, class III, classical class II and extended class II regions from telomere (left and top side) to centromere (right and bottom side). White, gray, striped and black boxes show expressed genes, gene candidates, non-coding genes and pseudogenes, respectively. The location of the alpha, beta and kappa blocks containing the cluster of duplicated HLA class I genes in the class I region are indicated.
Of the total number of 113 non-HLA protein coding genes, 9 (SFTA2, MUC21, PSORS1C3, MCCD1, SLC44A4, ZBTB12, PRRT1, WDR46 and PFDN6) were newly identified to be functional loci (Tables 1 and 2). Of them, PSORS1C3 is one of the associating genes of psoriasis vulgaris.40 MCCD1 encodes mitochondrial coiled-coil domain 1 and is highly polymorphic, containing approximately one SNP in every 99 basepairs.41 PFDN6 encodes prefoldin subunit 6, and the gene was reported to be overexpressed in certain cancers compared with normal counterparts in a tissue microarray study.42
Thirty-three of the non-HLA expressed genes (GABBR1, MOG, ZNRD1, RNF39, TRIM10, TRIM39, PRR3, ABCF1, DDR1, CCHCR1, TCF19, POU5F1, BAT1, ATP6V1G2, LTB, LST1, AIF1, BAT3, MSH5, EHMT2, STK19, CYP21A2, TNXB, PPT2, AGPAT1, AGER, TAP2, PSMB8, PSMB9, BRD2, COL11A2, SLC39A7 and TAPBP) and HLA-F appear to express spliced variants with an overall average of 2.6 different kinds of spliced variants per gene. One of the recently identified expressed genes with a relatively large number of spliced variants is C6orf25 that is located between LY6G6C and DDAH2 within the class III region. This gene has at least seven spliced variants, and it is a member of the immunoglobulin (Ig) superfamily that encodes a glycosylated, plasma membrane-bound cell surface receptor as well as soluble isoforms. Some of the membrane-bound and soluble products encoded by the C6orf25 splice variants contain two immunoreceptor tyrosine-based inhibitory motifs (ITIMs) that were found to interact by phosphorylation with the SH2-containing protein tyrosine phosphatases SHP-1 and SHP-2.43
Regional analysis of the HLA super-locus
The HLA super-locus can be separated into the traditional five HLA regions with 4, 128, 75, 27 and 19 loci within the extended class I, class I, class III, class II and extended class II regions, respectively (Figure 1 and Table 2).
Extended class I region
In this version of the HLA loci, only four genes (BABBR1, SUMO2P, MOG and ZNP57) have been included in the extended class I region. However, numerous duplicated genes encoding the olfactory receptor, histone, tRNA and zinc-finger protein are located on the telomeric segment of the extended class I region. The hemochromatosis gene (HFE) that is similar in structure to an HLA class I gene is located outside the HLA super-locus ∼3.6 Mb away on the telomeric side of HLA-F and the extended class I region.44
Class I region
The class I region contains the six classical and non-classical HLA class I genes. The non-classical HLA class I genes are differentiated from the classical class I genes on the basis that they have limited polymorphism; the tissue distribution of gene expression is restricted and they appear to play a less well-defined role in transplantation medicine.45 There are 19 HLA class I gene loci, where 3 are classical (HLA-A, -B and -C), 3 non-classical (HLA-E, -F and -G) and 12 non-coding genes or pseudogenes (HLA-S/17, -X, -N/30, -L/92, -J/59, -W/80, -U/21, -K/70, -16, -H/54, -90 and -75), clustered within three separate duplication blocks, designated as the alpha, beta and kappa blocks46 (Figure 1). Of the HLA pseudogenes, HLA-H/54 appears to encode two mRNA sequences (AK090500 and AK308374), whereas the transcript AK127349 and hypothetical protein FLJ45422 sequence were mapped to a part of overlapping exons of HLA-L/92. The FLJ45422 gene is composed of five exons and contains an Ig domain constant region (IGc) and transmembrane domain, but its polymorphisms and function are unknown.
There are seven MIC genes, which are HLA class I-like genes, distributed across the three duplication blocks; two are expressed within the beta block, whereas the remainder are non-expressed pseudogenes within the kappa and alpha blocks.46, 47, 48 These MIC genes have been generated with HLA class I genes by several rounds of segmental duplication events.35 There are 34 non-HLA class I protein coding genes distributed between the duplication blocks that from an evolutionary perspective are termed anchor or framework genes.48, 49
Overall, there are 128 loci within the 1.8 Mb class I region from HCP5P15 to MICB, with 42 expressed genes, 12 gene candidates, 10 non-coding genes and 64 (50%) pseudogenes (Table 2). Of the 54 protein coding genes and gene candidates, 7 non-HLA genes (LOC100133214, FLJ45422, LOC100133303, LOC100129065, LOC729792, HCG22 and PSORS1C3) were identified in the region after the previous locus information report.31 Of the 42 protein coding genes, 4 (SFTA2, MUC21, CCHCR1 and PSORS1C3) were previously unknown to be functional loci, and TUBB received a new official symbol and name (Table 1).
Class III region
The class III region, located between the class I and II regions, contains 75 loci within 0.9 Mb of DNA from PPIAP9 to BTNL2 (Table 1), with 55 protein coding genes and 5 (6.7%) pseudogenes (Table 2). Most of the protein coding genes and gene candidates were described earlier in the locus information report of 2004,31 but three genes (LY6G6F, C6orf26 and LOC100128067) were identified more recently. LY6G6F belongs to a cluster of leukocyte antigen-6 (LY6) genes in the class III region and it encodes a type I transmembrane protein belonging to the Ig superfamily,43 which may have a role in signal transduction in response to platelet activation.50 Of the 55 protein coding genes, 5 (MCCD1, SLC44A4, EHMT2, ZBTB12 and PRRT1) were previously unknown to be functional loci, and three (VARS, LSM2 and CFB) had a symbol and name change (Table 1). In addition, five small nuclear RNA sequences (SNORD84, SNORD117, SNORA38, SNORD48 and SNORD52) were identified in the vicinity of the BAT1, BAT2 and C6orf48 genes, respectively.51, 52, 53 The class III region has no known HLA class I- and class II-like genes, but contains the complement factor genes, C2, C4, CFB, the cytokine genes TNF, LTA and LTB, and many genes with no obvious relationship to immune function or inflammation. The gene combination of RP-C4-CYP21-TNX is modular in structure and varies in copy number and has haplotypic variability. Many of the gene products expressed in the class III region have fundamental roles in cellular processes, such as transcription regulation (BAT1, VARS, RDBP, STK19, SKIV2L, CREBL1 and PBX2), housekeeping (DOM3Z, NEU1, AGPAT1, CL1C1 and CSNK2B), biosynthesis, electron transport and hydrolase activity (PPT2, DDAH2 and ATP6V1G2) and protein–protein interactions for either intracellular or intercellular interactions, chaperone function and signaling (C6orf46, HSPA1A, HSPA1B, BAT3, BAT8, AGAR, RNF5, FKRPL, TNXB, NOTCH4).
Class II region
The class II region spans 0.7 Mb of DNA and contains the classical class II alpha and beta chain genes, HLA-DP, -DQ and -DR that are expressed on the surface of antigen-presenting cells to present peptides to T-helper cells. There are 27 loci identified within the class II region from HLA-DRA to HLA-DPA3 (Table 1), with 17 protein coding genes, seven gene candidates and five pseudogenes (Table 2). In total, 19 of the loci are HLA class II-like sequences, including the 15 classical HLA class II loci and the four non-classical HLA class II loci (HLA-DM and -DO). The HLA-DRB loci are variable in number and MHC haplotype-dependent. The HLA-DRB locus in the PGF haplotype (Table 1) contains four copies of the HLA-DRB gene, HLA-DRB1 (coding), -DRB5 (coding), -DRB6 (non-coding) and -DRB9 (non-coding), whereas the HLA-DRB copy numbers vary for other haplotypes.5 All of the 17 protein coding genes were previously known to be functional genes. Of all the protein coding genes in this region, BRD2 (alias RING3) is the only gene without an established immune function. It is a transcription factor with widespread specificity, possibly remodeling chromatin complexes through interactions with histone acetyltransferase complexes, and its activity is high in myeloid leukemias.54 Although BRD2 may have a homologous sequence in yeast and Drosophila, it is strongly linked with the MHC of most vertebrates in the evolutionary path from sharks to man.48
Extended class II region
The extended class II region spans 0.2 Mb of DNA from COL11A2 to KIFC1 (Table 1), with 19 loci; that is, 15 protein coding genes, 1 gene candidate, 1 non-coding gene and 2 pseudogenes (Table 2). There was only one newly identified gene candidate (LOC646720) since the locus information report of 2004.31 However, of the protein coding genes, two (WDR46 and PFDN6) were previously unknown to be functional genes.
Interspersed repeats
Apart from the gene loci, 49.5% of the HLA genomic sequence is composed of interspersed repeat elements, such as SINE (Alu, MIR), LINE (LINE1 and 2, L3/CR1), LTR elements (ERVL, ERV class I and class II) and DNA elements (hAI-Charlie, TeMar-Tigger). Table 3 presents a summary of the repeat elements as detected by RepeatMasker (http://www.repeatmasker.org/). A comparable analysis with slightly different results and annotations (data not shown) was obtained with the repeat analysis program CENSOR.55
Genomic diversity
HLA genes
A total of 3201 HLA allele sequences (2215 in class I and 986 in class II) were released by the IMmunoGeneTics HLA (IMGT/HLA) database release 2.22 in July 2008 (http://www.ebi.ac.uk/imgt/hla/). The IMGT/HLA Database is a specialist database for HLA sequences. Ten years ago, the allele numbers were only 964, but since then the numbers have increased by ∼200–300 allele sequences each year. Of the 2176 HLA class I alleles, 673, 1077, 360, 9, 21 and 36 alleles were counted in HLA-A, -B, -C, -E, -F and -G genes, respectively (Table 4); 2110 and 66 alleles were counted in the classical and non-classical HLA class I genes, respectively. Of 986 HLA class II alleles, 3, 669, 34, 93, 27, 128, 4, 7, 12 and 9 alleles were counted in HLA-DRA, -DRB, -DQA1, -DQB1, -DPA1, -DPB1, -DMA, -DMB, -DOA and -DOB genes, respectively (Table 4), with 954 and 32 alleles in the classical and non-classical HLA class II genes, respectively. In addition, 64 and 30 alleles were detected for the MHC class I-like gene, MICA and MICB, respectively.
Microsatellites
A total of 1527 microsatellite loci (846 in class I, 295 in class III and 386 in class II) were detected in the COX-MHC sequence (accession number NT_113891) by the Sputnik program (http://espressosoftware.com/pages/sputnik.jsp). Of them, 268 microsatellites (146 in class I, 61 in class III and 61 in the II) were developed as genetic markers.56 These polymorphic microsatellite markers have been useful for precise mapping of disease-related genes within the HLA region in linkage analysis and disease association studies.57, 58 Moreover, they provide a powerful tool to study recombination events in this region, which contributes to haplotypic diversification. Detailed microsatellite marker information is provided by the dbMHC database of the NCBI (http://www.ncbi.nlm.nih.gov/gv/mhc/main.fcgi?cmd=init).
SNPs
A total of 60 928 to 71 569 SNPs were detected in a pairwise analysis of five different genomic sequence assemblies (PGF, Celera, HuRef, C6_COX and C6_QBL), ranging from GABBR1 to KIFC1, by dbSNP (http://www.ncbi.nlm.nih.gov/SNP/). SNP markers are useful for constructing HLA haplotypes and for precise mapping of disease-related genes within the HLA region.59, 60, 61, 62 Figure 2 shows the marked peaks and troughs of the SNP distributions for the pairwise analysis of the five assemblies. The main peak diversities were observed not only in genomic segments harboring the highly polymorphic HLA-A, -B, -C, -DR, -DQ and -DP loci but also within some non-HLA loci such as those telomeric of HLA-C. Therefore, the HLA diversity is not limited to the antigen/T-cell receptor)-interacting sites of the HLA molecules,63 but spreads to the surrounding loci as hitchhiking diversity owing to the accumulated effect of overdominant selection acting on HLA loci.3 Interestingly, several disease-related genes, such as diffuse panbronchiolitis, psoriasis vulgaris, rheumatoid arthritis and sarcoidosis, were identified in the hitchhiking diversity-affected segments.57, 58, 64, 65 It was hypothesized by Shiina et al.3 that some non-HLA disease alleles co-evolved with the positively selected HLA loci that were in linkage with harmful polymorphisms within the negative or neutrally selected non-HLA loci in response to various selection, population, genetic and environmental factors.

Single nucleotide polymorphism (SNP) distribution within the human leukocyte antigen (HLA) region. Diversity plots (a–e) drawn by comparing the released SNPs in dbSNP database against the reference assembly sequence determined in 19991 (accession no. NT_007592) (a), Celera alternate assembly sequence (accession no. NW_923073) (b), HuRef alternate assembly sequence based on HuRef SCAF_1103279188254 (accession no. NW_001838980) (c), c6_COX sequence (accession no. NT_113891) (d) and c6_QBL sequence (accession no. NT_113893 to NT_113897) (e). Gray backgrounds show significantly higher SNP regions that may have been generated by hitchhiking diversity.3
Genomic variation
The HLA genomic variations generated by HLA-DRB gene copy number in class II and/or the copy number variations (CNVs) of the RP-C4-CYP21-TNX gene combination in class III were previously associated with a number of different autoimmune diseases well before the complete, continuous HLA super-locus sequence was available.46 The HLA-DR haplotypes consist of a number of copies of coding and non-coding HLA-DR genes. The expressed DRB sequences have been assigned to four different loci, DRB1, 3, 4 and 5. The highly polymorphic DRB1 alleles (Table 4) are present in all haplotypes, whereas DRB3, 4 and 5 are present only in some haplotypes, as are the HLA-DRB2 and HLA-DRB6 to -DRB9 pseudogenes. The HLA-DRB2 pseudogene lacks exon 2 and contains a 20-nt deletion in exon 3, which has interrupted the correct translational reading frame.66 The common HLA-DR alleles, major allotypes and their association with disease have been reviewed by Marsh.67 The low and high copy numbers of the C4 gene in the class III region have been recently associated as risk and protective genes, respectively, for systemic lupus erythematosus (SLE) susceptibility in European Americans.68
Genomic variations, such as insertion or deletion (InDel), inversion and other CNV, have been detected in recent genome-wide studies by comparative genomic hybridization (CGH) array mapping, fosmid end mapping, Mendelian inconsistencies, paired-end mapping of 454 sequencing reads, SNP chips and computational mapping of re-sequencing traces.69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 From the Database of Genomic Variants (http://projects.tcag.ca/variation/; 26 June 2008), 181 variations (50 InDels, 1 inversion and 130 CNVs) were detected at 49 genomic positions of the HLA region, especially within the HLA class I and II gene regions and a part of the class III region (Table 5). Some InDels are repetitive elements, such as Alu, HERV, L1 and SVA, or were generated by the influence of repetitive elements.7, 34, 80, 81, 82, 83
Intra- and extra-MHC gene interactions
MHC genes do not function in isolation from other genes in the human genome, but they may interact with other genes inside (local or intra-MHC gene interaction) or outside the MHC region (global or extra-MHC gene interactions). The MHC gene interactions may be viewed as quantitative interactions between alleles at different loci that affect fitness or contribute to complex disease phenotypes (epistasis),84, 85 as simple statistical interactions between alleles at different loci (linkage disequilibrium or LD) as a consequence of functional selection or a hitchhiking effect,86, 87 as functional protein-binding interactions detected by two-hybrid, affinity capture or phage display methods,88 or as protein–DNA interactions such as those between transcription factors and gene promoter and enhancer regions89, 90 or between replication protein factors and DNA replication sites and elements.91, 92 The study of genetic interactions can reveal gene function, the nature of the mutations, functional redundancy, transcription regulation and protein interactions in normal and disease processes.
Table 6 provides an example of some protein interactions encoded by genes located inside and outside the MHC genomic region. Of the interactions between different genes within the MHC, the most definitively studied examples are those involved in protein dimer formation and peptide presentation in the adaptive immune response. In the former case, the interaction of the HLA class II alpha and beta proteins encoded by the classical class II A and B gene loci, respectively, have long been known to form the alpha and beta heterodimer chains and consequently have received extensive investigations at various levels, including X-ray structural analysis.93, 94 The interaction of proteins involved in antigen presentation, such as HLA class I proteins, TAP1, TAP2, HLA-DM and TAPBP, have also been extensively studied.95, 96 The interactions between the alleles of the HLA-DR haplotypes, which are in strong LD, were found to affect the immune response levels and disease susceptibility. For example, the results obtained for two multiple sclerosis-associated HLA-DR alleles at separate loci of the HLA-DR2 haplotype in a humanized mice functional assay imply that the LD between these two alleles is due to a functional epistatic interaction.97 Accordingly, one allele modifies the T-cell response activated by the second allele through activation-induced cell death resulting in a milder form of multiple sclerosis. Other protein interactions encoded by genes within the MHC genomic region include those between RFP5 and BAT5, C4B and C2, CFB and C4B, LTA and LTB, IER3 and BAT3, and between MRPS18B and NFKBIL1.
Examples of protein interactants encoded by genes inside and outside the MHC are more numerous than those encoded by genes within the MHC genomic region. Recent research has focused strongly on the HLA class I interactions with the killer Ig receptor (KIR) genes and the leukocyte Ig-like receptor (LIR) gene family encoded in the leukocyte receptor complex (LRC) on chromosome 19q13.98, 99 Combinations of HLA class I and KIR variants have been associated with autoimmunity, viral infections, pregnancy-related disorders and cancer.100, 101 Similarly, the proteins encoded by the MICA and MICB genes (Table 6) are known to interact with KLRC4 and KLRK1 that are encoded by the genes on chr 12, to regulate innate immunity by way of the NK cell systems.47 The proteins encoded by the C4, CFB and C2 genes in the HLA class III region are involved in complement activation and consequently interact with proteins encoded by genes from outside the MHC (Table 6). Allelic variations between the MHC complement genes and non-MHC gene sequences have been associated with macular degeneration and SLE.102 Recently, Lester et al.103 reported finding an epistasis between the MHC C4 gene region and the RCAa block in primary Sjögren syndrome. The RCAa block (regulators of complement activation, 1q32) contains critical complement regulatory genes such as CR1 and MCP, and the epistasis was attributed to an interaction between C4 and its receptor, CR1, encoded within the RCAa block. Furthermore, the IFN-regulator factor 5 (IRF5) gene variants located on chr 7q32 were found to interact with the class I MHC locus in people with psoriasis104 and possibly other autoimmune diseases.105
Most proteins encoded by the 132 protein coding genes within the MHC interact with proteins encoded by genes outside the MHC region. The protein and genetic interactions of the MHC genes listed in Table 1 can be accessed and viewed by way of the GeneID number. For example, the interaction data and online links for the MDCI gene (GeneID: 9656), mediator of DNA damage checkpoint 1, which is required to activate the intra-S phase and G2/M phase cell cycle checkpoints in response to DNA damage, includes information on the peptide or protein interactants, the interacting genes, the source databases (Human Protein Reference Database (HPRD) or BioGRID) and published references (PubMed). The 13 genes found to interact with MDC1 and listed at Entrez Gene are ATM, BRCA1, CHEK2, H2AFX, NBN, SMC1A, TP53, TP53BP1, CENPC1, CHEK2, GATA4, H2AFX and HDAC10. In another example, the protein expressed by the CCHCR1 gene (ID:54535), which has at least three splice variants, was identified to promote steroidogenesis by interacting with STAR, the steroidogenesis acute regulatory protein106 encoded by a gene on chr 8p (Table 6), which may be downregulated in psoriatic keratinocytes.107 A public online service for protein interaction datasets is also provided by BioGRID at http://www.thebiogrid.org/index.php and the HPRD at http://www.hprd.org/index_html. The knowledge extracted from protein interaction databases might assist in a more efficient organization and analysis of genome-wide studies by revealing which gene interactions warrant epistatic investigation.
MHC and genome-wide gene expression profiling
Most knowledge on MHC gene expression at the transcript and protein levels has depended on individual gene studies (Table 1). However, in recent years, the development of genome-wide gene expression assays, including some or many of the MHC genes, has provided a more global perspective of different expression patterns in immune- and disease-related pathways. Gene expression profiling of normal and diseased cells and/or tissues using oligonucleotides, cDNA or genomic arrays has been a particularly successful by-product of genome sequence research. Global transcriptome studies are performed using various descriptive, experimental and disease conditions, and the data are often deposited into public databases, such as Gene Expression Omnibus (GEO), that can be accessed online for review and/or reanalysis (http://www.ncbi.nlm.nih.gov/geo/).
Genome-wide gene expression data have permitted an examination and comparison of the mRNA profiles expressed by genes both inside and outside the MHC region. For example, in our study of the gene transcription patterns in the skin lesions of four Japanese patients with psoriasis vulgaris and three normal controls, we found that only seven MHC genes (LY6G6C, CDSN, TAP1, HLA-G, HLA-F, TUBB and CFB) from a total of approximately 90 MHC protein coding and non-coding genes represented on the HUG95A Affymetrix oligonucleotide array of 12 000 human genes were significantly upregulated in the affected skin compared with normal skin; no significant statistical changes occurred in the expression of the classical HLA class I and II genes.108 The only MHC gene that was significantly downregulated in the psoriatic lesions was GABBR1. Most of the 263 significantly upregulated changes in the psoriatic-affected skin occurred for genes located outside the MHC region that were involved with interferon mediation, inflammation immunity, cell adhesion, cytoskeleton restructuring, protein trafficking and degradation, RNA regulation and degradation, signaling transduction, apoptosis and atypical epidermal cellular proliferation and differentiation. Bioinformatics analysis of the significantly upregulated genes in psoriatic skin compared with normal skin, using a commercially available computer network program (MetaCore) in Figure 3, shows that inflammation and cell cycle regulation were the two most significant molecular pathways involved in psoriasis by way of the STAT and Myc gene regulatory systems as well as by the MHC genes, HLA-G (interacting with KIR2DL4 and ILT2 on chr 19), DDR1 and TNF (MetaCore Applications (2007) http://www.genego.com/pdf/PsoriasisCS.pdf). The HLA-G locus was recently found to also interact with the IRF5, encoded by gene variants on chr 7q32 in Swedes with psoriasis.104

The involvement of major histocompatibility complex (MHC) genes, HLA-G, DDR1 and tumor necrosis factor (TNF)-alpha, in the molecular pathways of psoriasis. The whole-genome microarray data of Kulski et al.108 were evaluated using the MetaCore software package to identify the molecular character and pathways involved in psoriasis. The MHC genes are highlighted by black squares. Red rectangles and orange ovals represent the genes involved in the inflammation and cell cycle regulation pathways (thick blue lines), respectively, and red circles represent overexpressed key transcription regulators. The figure was produced by MetaCore from GeneGo Inc. (St Joseph, MI, USA). The color reproduction of this figure is available on the html full text version of the manuscript.
Other investigators have used similar gene microarray assays to identify the patterns of MHC and non-MHC gene transcription in skin lesions of patients with psoriasis,109, 110 atopic dermatitis111 and porokeratosis, a skin disorder of keratinization.112 Gene expression profiling of peripheral blood mononuclear leukocytes has been performed on psoriasis patients for disease stage prediction113, 114 and treatments with therapeutic TNF and IFN-gamma antibodies.115 Leukocytes and/or lymphocytes express more than 75% of the human genome and provide an alternative to tissue biopsies for studies of the association between HLA gene activity and autoimmune diseases, such as psoriasis, asthma, rheumatoid arthritis (RA) and SLE. A number of different MHC-related diseases, including SLE,114, 116 RA117, 118 and OA,119 have been investigated by gene expression profiling. For example, van der Pouw Kraan et al.120 used cDNA microarray technology to subclassify RA patients and to disclose different disease pathways in rheumatoid synovium. They found that among the 121 genes overexpressed in one of the main tissue groups (RA-I) identified by a hierarchical clustering of gene expression data, 9 genes from the MHC region were indicative of an adaptive immune response, whereas another group (RA-II) expressed genes suggestive of fibroblast dedifferentiation. Microarray analyses of peripheral blood cells from patients with psoriatic arthritis identified downregulation of innate and acquired immune responses as well as the MHC genes from the PSORS1 and PSORS2 susceptibility loci.121
Peripheral arterial occlusive disease (PAOD: OMIM 606787) is commonly found in elderly patients as a result of atherosclerosis of large and medium peripheral arteries, or aorta, and often coexists with coronary artery disease and cerebrovascular disease. Recently, Fu et al.122 analysed 30 femoral arteries (11 with intermediate and 14 with advanced atherosclerotic lesions and 5 normal femoral arteries) by genome-wide gene expression profiling using the Affymetrix microarray platform and found that most of the MHC class II and complement molecules were significantly upregulated in the intermediate lesions, but not in the advanced lesions. They concluded from the results of their expression study that different immune and inflammatory responses occur at different stages of PAOD and development of artherosclerotic lesions. The MHC class II and complement gene activity was related in different ways to the Toll-like receptor signaling and NK cell-mediated cytotoxicity enrichment found to take place in the intermediate and advanced atherosclerotic lesions.
HLA-wide gene expression profiling using the Affymetrix microarray platform also allows researchers an opportunity to determine the degree of positive and negative coordination between HLA and non-HLA gene expression in controlled experiments, cell and tissue types, and in population and disease studies. For example, Figure 4 shows the microarray expression profiles for some non-HLA class I genes relative to the expression of the non-classical HLA class I genes, HLA-E, -F and -G, in established cell lines derived from different cancers, with data provided by The Cancer Genome Anatomy Project (http://cgap.nci.nih.gov/Genes). It can be seen in Figure 4 that the FLOT1 gene was expressed at highest levels in cancer cells derived from the CNS, whereas DDR1 and TRIM15 (alias Hs.591789) were expressed most strongly in the colonic cancer cell lines. In comparison, the non-classical HLA class I genes were expressed most consistently at moderate to high levels in the cell lines derived from renal carcinomas. The variable expression of TRIM15 among the different cancer cell types is notable given its possible antiviral role in innate immunity.123, 124

The relative expression of some human major histocompatibility complex (MHC) class I genes in different cancer cell lines. The gene examples from the class I region are non-human leukocyte antigen (HLA) genes (DDR1, IER3, HCG18, PPP1R11, RPP21, DHX16, GTF2H4, GNL1, RNF39, TRIM31, Hs.591789 (TRIM15), FLOT1 and PP1R10) and the non-classical HLA class I genes, HLA-E, -F and -G. The data are taken from The Cancer Genome Anatomy Project at the National Cancer Institute (USA) using the batch gene finder to find the expression data for the selected genes of interest (query) in the gene list of NC160_U133 (Affymetrix platform). The present image for the transcriptome analysis was produced online at http://cgap.nci.nih.gov/Genes/BatchGeneFinder using only the selected gene list shown in the image. The level of transcriptional activity in the cells ranged from the highest (red squares) to the lowest (blue squares) according to the color scale indicated at the top left-handed side of the figure. The rectangular blocks labeled (a–e) within the matrix of the figure highlight the detection probes with relatively high expression levels of FLOT1 in central nervous system (CNS) cancer cells (a), DDR1 (b) and Hs.59178 (TRIM15) (c) in colon cancer cells, IER3 and HLA-E in melanoma (d) and the non-classical HLA class I genes (e) in the renal cancer cells. Of the list of cancerous tissue at the bottom of the matrix, ‘Leuk’ is leukemia and ‘P’ is prostate. The color reproduction of this figure is available on the html full text version of the manuscript.
Although an HLA and global picture of gene expression in tissues and cells can be obtained by using a full set of Affymetrix GeneChips, CGH for SNP analysis in combination with gene expression is still a relatively new and demanding approach for the study of complex diseases. CGH, in an attempt to improve functional genome research and disease associations, is particularly useful for detecting genomic sequence alterations or gene CNVs125, 126 that might be associated with disease. For example, CNVs of defensin genes on chr 8 were found to be strongly associated with Crohn's disease and the skin disease, psoriasis.127, 128 Similar studies on the effects of genomic alteration or CNVs on the expression of MHC genes are still limited, but a few recent reports suggest that this approach might yield important new insights into the interaction between the genes of the MHC and other genomic regions in disease studies. For example, the study by Jiang et al.129 using cDNA microarrays to detect the simultaneous genomic and expression alterations in prostate cancer, has implicated the dysregulation of exogenous antigen presentation through MHC class II and protein ubiquitination during protein-dependent protein catabolism in the tumorigenic process. They found that the expressions of the MHC genes ABCF1, HLA-DRB1 and HLA-A, located on the chromosome 6p21, and of the MHC class II chaperone gene, CD74, located on 5q32 were both significantly downregulated, probably as a consequence of the CD74 gene deletion.
Genome tiling arrays is another improving methodology that appears useful for future investigations into MHC epigenetics,130 SNPs,7 gene–gene interactions131 and gene expression activity132 both inside and outside the MHC genomic region by using high-density oligonucleotide arrays with probes chosen uniformly from both strands of the entire genome, including all genic and intergenic regions. Genome-wide protein profiling (proteomics) by using chips, arrays or high-throughput mass spectrometry is a rapidly emerging technology in disease and diversity studies to screen for protein activities such as protein–protein, protein–DNA, protein–drug and protein–peptide interactions; to identify enzyme substrates and to profile immune responses.133, 134 Some of these procedures have been applied specifically to MHC gene functions, particularly to detect and characterize antigen-specific T-cell populations in disease,135 HLA protein–peptide (antigen) interactions,136 targeting autoantibody/autoantigen targets137, 138 and to profile other immune responses.139 Bioinformatic and statistical algorithms are continually being developed to integrate the genomics of DNA variation, transcription and phenotypic data, to provide a system genetics view of disease and to enhance identification of the associations between DNA variation and diseases as well as to characterize those parts of the molecular networks that drive disease.140
MHC and disease associations
The main function of the MHC gene region is to protect itself and its organism against harmful infectious agents (to recognize and deal with foreign organisms and antigens) and to dispense with the damaged, dying or infected cells and tissues. The extremely high levels of polymorphism and heterozygosity within the MHC genomic region provide the immune system with a selective advantage against the diversity and variability of pathogens. However, the high level of polymorphisms and mutations in the MHC has the added risk of generating autoimmune diseases and other genetic disorders. Several hundred autoimmune and infectious diseases have been associated with the MHC since the first report in 1967 that HLA-B antigens were increased in frequency in patients with Hodgkin's lymphoma.141 At least another 40 different autoimmune diseases were linked to specific HLA types by the end of 1986.142, 143 In an update on the role of the MHC genes in disease, Shiina et al.31 presented an overview of 109 HLA-associated diseases. When PubMed online at NCBI was searched in September 2008 with the keywords ‘human MHC (or HLA) gene disease,’ 3151 journal publications were listed on the subject of HLA and disease. Using ‘HLA’ as a keyword to search the Genetic Association Database (GAD) (http://geneticassociationdb.nih.gov/cgi-bin/index.cgi), 500 journal publications were found on HLA gene association and disease between 1999 and 2007. The statistical, biological and medical significance of many of the MHC disease association studies, however, remain unclear or doubtful.
A number of recent reviews are available on HLA and infections,12, 144, 145, 146 as well as HLA and autoimmune diseases,11, 31, 32, 147, 148, 149, 150 and will not be considered in any detail here. OMIM is a database of human genes and genetic disorders that provides information and references on the discoverers, chromosomal location, molecular functions, mutations and associations between the genes and disease.151 There are at least 100 OMIM identifiers concerning the HLA region loci, mostly of expressed genes, that can be accessed through http://www.ncbi.nlm.nih.gov/ or through links from other sites, including Entrez Gene database at NCBI.36
The 31 HLA disease associations listed in Table 7 and sourced from the OMIM database152 are some examples of HLA-associated diseases that have a strong experimental or statistical association with reasonable reproducibility. At least 26 of these diseases have been associated with non-HLA genes encoded within the MHC, with the regulatory cytokines TNF and LTA contributing to a large number of disease associations by way of mutations or polymorphisms within the gene promoter or coding regions that might affect expression levels.153, 154, 155, 156 Ten of the diseases appear to be monogenic owing to mutations within one of the MHC genes. Adrenal hyperplasia is now well accepted to be the consequence of 21-hydroxylase deficiency and alterations in the CYP21A2 gene.157 Some of the CYP21A2 gene alterations may arise by transference of sequences to CYP21A2 from the neighboring non-coding CYP21A1P pseudogene by gene conversion.158 It is also generally well accepted that mutations within the NEU1 gene are responsible for neuraminidase deficiency and sialidosis, which is characterized by the progressive lysosomal storage of sialylated glycopeptides and oligosaccharides,159 and that C2 mutations cause C2 deficiency in the process of the complement cascade.160 Of the 21 multifactorial diseases listed in Table 7, 11 (type I diabetes (T1D), inflammatory bowel disease, multiple sclerosis (MS), AITD, PV, RA, celiac disease (CD), ankylosing spondylitis (AS), SLE, juvenile RA (JRA) and vitiligo (VIT)) were linked most significantly to the HLA region in a recent meta-analysis of 42 independent genome-wide linkage studies.161 In a recent genome-wide association study of seven common diseases using SNP markers, the MHC associations were strongest for RA, T1D, moderate for CD and weak or absent for bipolar disorder, coronary artery disease, hypertension and type II diabetes.162 In another recent review and pooled analysis of the MHC in autoimmunity, a number of overlapping HLA class II and TNF alleles and haplotypes were associated with the diseases MS, T1D, SLE, UC, CD and RA.11
Most of the 21 multifactorial diseases listed in Table 7 are polygenic with a few specified or unspecified MHC gene alleles possibly interacting in some unspecified way with other genes inside and/or outside the MHC region. The exact MHC genes involved with many of the diseases are still not clearly defined. For example, the association of an HLA genomic region with the onset or maintenance of psoriasis is definite, but which of a number of MHC candidate genes (or combination of genes) ranging between the MICA and CDSN loci is responsible remains uncertain.39, 58, 163, 164, 165, 166, 167, 168, 169
Only a few autoimmune diseases have been related just to the classical class I and II alleles, in spite of the continuing dogma that disease associations are caused by altered or faulty peptide presentation to T cells by polymorphic class I and II gene products. AS is primarily attributed to HLA-B27, with minor associations such as HLA-Cwl and -Cw2 or HLA-DR7 considered secondary because of LD or a hitchhiking effect. Similarly, HLA-B51 continues to be strongly associated with Behcet syndrome,170 although other chromosomal regions may be involved.171 In Caucasian populations of Northern European descent, the DR15 haplotype (DRB1*1501-DQA1*0102-DQB1*0602) is hypothesized to be the primary HLA genetic susceptibility factor for MS. Experiments with transgenic mice have confirmed the importance of the DRB5*0101 and DRB1*1501 allelic interactions in creating a mild form of MS-like disease,97 but more severe forms probably depend on other genes172 such as T-cell receptor beta, CTLA4, ICAM1 and SH2D2A. Schmidt et al.149 reviewed 72 publications on the HLA association with MS and found that most investigators reported a higher frequency of the DR15 haplotype and/or its component alleles for the MS cases than the controls, but the results may have been biased by poor study designs.
Owing to the difficulty in identifying a single MHC gene that is responsible for disease, some researchers prefer to examine the association between MHC haplotypes and disease susceptibility and resistance.46 Common Caucasian MHC haplotypes may be accounted for by a limited number of ancestral haplotypes using the alleles of five or more gene loci.173 The MHC ancestral haplotype (AH) 8.1, characterized by the alleles HLA-A*01, -B*08, -DRB1*03, -DQB1*02 and -DQA1*05 has been dubbed the ‘autoimmune haplotype’ because of its association with numerous autoimmune diseases, including T1D, CD, Graves’ disease, SLE and Myasthenia Gravis (MS).174 The complete MHC genomic sequences for eight haplotypes involved in autoimmune diseases, including the 8.1 AH, have been published.7 In this regard, Shiina et al.3 proposed, on the basis of comparative genomics between human haplotype sequences and the sequences of chimpanzee and rhesus macaque, that the rapid evolution of the MHC class I genes in primates is likely to have generated new disease alleles in humans through hitchhiking diversity.
The results of MHC disease association studies are complicated by race and population differences, influences of LD, the large polymorphism, copy number and InDel variations between different MHC haplotypes, disease severity and the need for large sample numbers to provide statistical significance. Fernando et al.11 noted in their review of six autoimmune diseases with genetically complex disease traits that nearly all association studies of the MHC in autoimmune and inflammatory disease have been limited to a subset of ∼20 genes and performed only in small cohorts of predominantly European origin. As highlighted in a recent review,5 the MHC association with complex disease phenotypes is dependent on the HLA and non-HLA genes, the genetic code (SNPs, CNV, InDels and inversions), the epigenetic code (DNA methylation and histone modification), biological effects (structural and biochemical changes in gene products and transcriptional regulation) and environmental factors (diet and antigen exposure). Modern HLA and whole genome association studies of SNPs, microsatellites, InDels and CNVs are now broadening toward elucidating gene interactions, epistasis, risk and penetrance of autoimmune diseases,162 although clear-cut results are often hampered by multiple testing errors and the statistical type I (false positives owing to multiple sample analysis) and statistical type II errors (false negatives owing to insufficient number of samples and other factors). Whole genome gene expression studies in combination with DNA variation and phenotypic data, as a single systematic study, have a greater potential for elucidating disease pathways and dissecting the role of individual genes and genomic loci, similar to the HLA super-locus, that interact in a molecular network. Such studies are still in their infancy, and much experimentation may be needed to overcome the potential data overload as we move rapidly toward a system genetics view of disease.140
HLA and cancer
The loss of HLA gene expression owing to viral infection, somatic mutations or other causes may have important effects on immune suppression and cancer development.175 To identify the molecular mechanisms involved in the maintenance of Epstein–Barr virus (EBV)-associated epithelial cancers, Sengupta et al.176 performed genome-wide expression profiling for all human genes and all latent EBV genes in a collection of 31 laser-captured, microdissected nasopharyngeal carcinoma (NPC) tissue samples and 10 normal nasopharyngeal tissues. They determined that all the HLA class I genes, TAP2 and HCG9 genes involved in regulating immune response through antigen presentation correlated negatively with increased EBV gene expression in NPC and concluded that antigen display is either directly inhibited by EBV, facilitating immune evasion by tumor cells and/or that tumor cells were selected for their EBV oncogene-mediated tumor-promoting actions. Global gene expression profiling of human papillomavirus (HPV)-positive and -negative head and neck cancers revealed a significant downregulation for two of the MHC genes, CDSN and LY6G6C, but not other MHC genes in HPV-16-positive head and neck squamous cell carcinomas.177
Non-viral tumors frequently lose expression of HLA molecules such as the reduction or total loss in colorectal carcinoma.178 Cells participating in immune response may fail to exert function without adequate MHC signaling in tumor cells, with the exception of NK cells, which may recognize MHC class I-negative tumor cells. Furthermore, soluble MHC class I-related (MIC) molecules play important roles in tumor immune surveillance through their interaction with the NKG2D receptor on NK, NKT and cytotoxic T cells.179, 180 Interestingly, genome-wide expression profiling has shown that non-steroidal anti-inflammatory drug (NSAID) treatment upregulated HLA class II genes in tumor tissue, but not in normal colon tissue, from the same patient.181 In total, 23 of the 100 most upregulated genes belonged to MHC class II; HLA-DM, -DO (peptide loading), HLA-DP, -DQ, -DR (antigen presentation), as did CD4+ T-helper cells, whereas HLA-A and -C expression were not increased by NSAID treatment.
In breast cancer, metastasis may be suppressed in part by the activity of the breast cancer metastasis suppressor 1 (BRMS1) gene, which can block development of metastasis without preventing tumor growth. In a comparison of gene expression patterns in BRMS1-expressing vs non-expressing human breast carcinoma cells, the BRMS1 expression in 435/BRMS1 cells was strongly correlated with an increased expression of MHC genes, HLA-DQB1, HLA-DRB1, HLA-DRB5, HLA-DMB, HLA-DQA1, HLA-DPA1, HLA-DRA, HLA-DRB4, HLA-DMA, C1S, HLA-B, HLA-C and HLA-F.182 Thus, the induction of MHC class I and II genes may be one mechanism by which 435/BRMS1 cells are kept at low populations, that is, by triggering an immune response that eliminates or reduces their metastasizing potential.
In an interesting paper by Rimsza et al.,183 gene expression profiling data were used to correlate the expression levels of MHCII genes with each other and their transcriptional regulator, CIITA (16p13), in 240 cases of diffuse large B-cell lymphoma (240 cases in the LLMPP data set). A correlation map was created for expression of the genes that are telomeric (HSPA1L, HSPA1A, BAT8, RDBP, CREBL1 and PBX2), within (MHCII genes, TAP1, TAP2, PSMB9 and BRD2) or centromeric (RXRB, RING1, RPS18, TAPBP, DAXX and BAK1) to the MHCII locus. Correlation coefficients among MHCII genes were high (0.73–0.92), whereas those between adjacent and intervening genes were low (0.12–0.49). The authors concluded that the loss of MHCII expression in non-immune-privileged site diffuse large B-cell lymphoma is highly coordinated and not due to chromosomal deletions or rearrangements. Furthermore, Dave et al.184 showed that gene expression profiling of MHC and non-MHC genes is an accurate, quantitative method for distinguishing Burkitt's lymphoma with the t(8;14) c-myc translocation from diffuse large-B-cell lymphoma. Burkitt's lymphoma was readily distinguished from diffuse large-B-cell lymphoma by the high-level expression of c-myc target genes and the low-level expression of all the MHC class I genes.
Conclusion
The human MHC genomic region is a super-locus composed of at least 250 coding and non-coding genes, the structural organization of which has evolved gradually, involving various mutation, duplication, deletion and genomic rearrangement events over a period of 450–520 Myr, at least from the time of the emergence of sharks (phylum Chordata, subphylum Vertebrata and class Chondrichthyes). A strong and progressive research interest remains toward haplotyping the entire human MHC genomic region by genomic resequencing for SNP, InDel and CNV analysis. The MHC genomic analysis was the prototype for many of the current procedures in genome-wide research, such as haplotyping, SNP and microsatellite analysis, and LD analysis for studies on human population diversity and disease association. The MHC genomic region is now part of the global systems analysis and network programs involved in the storage and dissemination of data on genome-wide gene expression at the level of the proteome, transcriptome, metabolome and phenotome, system and immune pathways, and disease associations using SNP, InDel and microsatellites as genomic markers or haplotype tags for statistical analysis. The degree and type of total MHC coordinated gene expression profiles have yet to be fully defined and understood in the processes of normal physiology, inflammatory and immune responses and autoimmune, chronic and infectious diseases. The field of MHC genomic research will clearly continue to expand into the future with the development of new procedures and studies to gain a better understanding of the intra- and extra-MHC gene interactions and their effects on human diversity and disease.
Website references
http://www.ncbi.nlm.nih.gov/sites/entrez Entrez Gene database
http://www.ncbi.nih.gov/entrez/query.fcgi?db=OMIM. OMIM: Online Mendelian Inheritance in Man
http://www.repeatmasker.org/ RepeatMasker program
http://www.ebi.ac.uk/imgt/hla/IMGT/HLA database: ImMunoGeneTics/HLA Sequence Database
http://espressosoftware.com/pages/sputnik.jsp Sputnik program
http://www.ncbi.nlm.nih.gov/gv/mhc/main.fcgi?cmd=initdbMHC database
http://www.ncbi.nlm.nih.gov/SNP/dbSNP database
http://projects.tcag.ca/variation/ Database of Genomic Variants
http://www.thebiogrid.org/index.php BioGRID: General Repository for Interaction Datasets
http://www.hprd.org/index_html HPRD: Human Protein Reference Database
http://www.ncbi.nlm.nih.gov/geo/ GEO: Gene Expression Omnibus
http://www.genego.com/pdf/PsoriasisCS.pdf MetaCore Applications
http://cgap.nci.nih.gov/Genes The Cancer Genome Anatomy Project
http://geneticassociationdb.nih.gov/cgi-bin/index.cgi GAD: Genetic Association Database
References
The MHC Sequencing Consortium. Complete structure and gene map of a human major histocompatibility complex (MHC). Nature 401, 921–923 (1999).
Smith, W. P., Vu, Q., Li, S. S., Hansen, J. A., Zhao, L. P. & Geraghty, D. E. Toward understanding MHC disease associations: partial resequencing of 46 distinct HLA haplotypes. Genomics 87, 561–571 (2006).
Shiina, T., Ota, M., Shimizu, S., Katsuyama, Y., Hashimoto, N., Takasu, M. et al. Rapid evolution of MHC class I genes in primates generates new disease alleles in man via hitchhiking diversity. Genetics 173, 1555–1570 (2006).
Muller-Hilke, B. & Mitchison, N. A. The role of HLA promoters in autoimmunity. Curr. Pharm. Des. 12, 3743–3752 (2006).
Traherne, J. A. Human MHC architecture and evolution: implications for disease association studies. Int. J. Immunogenet. 35, 179–192 (2008).
Solberg, O. D., Mack, S. J., Lancaster, A. K., Single, R. M., Tsai, Y., Sanchez-Mazas, A. et al. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: a meta-analytic review of 497 population studies. Hum. Immunol. 69, 443–464 (2008).
Horton, R., Gibson, R., Coggill, P., Miretti, M., Allcock, R. J., Almeida, J. et al. Variation analysis and gene annotation of eight MHC haplotypes: the MHC Haplotype Project. Immunogenetics 60, 1–18 (2008).
Fu, S., Zhao, H., Shi, J., Abzhanov, A., Crawford, K., Ohno-Machado, L. et al. Peripheral arterial occlusive disease: global gene expression analyses suggest a major role for immune and inflammatory responses. BMC Genomics 9, 369 (2008).
Claas, F. H. J. & Duquesnoy, R. J. The polymorphic alloimmune response in clinical transplantation. Editorial overview. Curr. Opin. Immunol. 20, 566–567 (2008).
Choo, S. Y. The HLA system: genetics, immunology, clinical testing, and clinical implications. Yonsei Med. J. 48, 11–23 (2007).
Fernando, M. M., Stevens, C. R., Walsh, E. C., De Jager, P. L., Goyette, P., Plenge, R. M. et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 4, e1000024 (2008).
Martin, M. P. & Carrington, M. Immunogenetics of viral infections. Curr. Opin. Immunol. 17, 510–516 (2005).
Fellay, J., Shianna, K. V., Ge, D., Colombo, S., Ledergerber, B., Weale, M. et al. A whole-genome association study of major determinants for host control of HIV-1. Science 317, 944–947 (2007).
Knapp, L. A. The ABCs of MHC. Evol. Anthropol. 14, 28–37 (2005).
Ziegler, A., Kentenich, H. & Uchanska-Ziegler, B. Female choice and the MHC. Trends Immunol. 26, 496–502 (2005).
Xiao, B. G. & Link, H. Immune regulation within the central nervous system. J. Neurol. Sci. 157, 1–12 (1998).
Huh, G. S., Boulanger, L. M., Du, H., Riquelme, P. A., Brotz, T. M. & Shatz, C. J. Functional requirement for class I MHC in CNS development and plasticity. Science 290, 2155–2159 (2000).
Boulanger, L. M. & Shatz, C. J. Immune signaling in neural development, synaptic plasticity and disease. Nat. Rev. Neurosci. 5, 521–531 (2004).
Cullheim, S. & Thams, S. The microglial networks of the brain and their role in neuronal network plasticity after lesion. Brain Res. Rev. 55, 89–96 (2007).
Ohtsuka, M., Inoko, H., Kulski, J. K. & Yoshimura, S. Major histocompatibility complex (Mhc) class Ib gene duplications, organization and expression patterns in mouse strain C57BL/6. BMC Genomics 9, 178 (2008).
Matsuo, R., Asada, A., Fujitani, K. & Inokuchi, K. LIRF, a gene induced during hippocampal long-term potentiation as an immediate-early gene, encodes a novel RING finger protein. Biochem. Biophys. Res. Commun. 289, 479–484 (2001).
Patiño-Lopez, G., Hevezi, P., Lee, J., Willhite, D., Verge, G. M., Lechner, S. M. et al. Human class-I restricted T cell associated molecule is highly expressed in the cerebellum and is a marker for activated NKT and CD8+ T lymphocytes. J. Neuroimmunol. 171, 145–155 (2006).
Goddard, C. A., Butts, D. A. & Shatz, C. A. Regulation of CNS synapses by neuronal MHC class I. Proc. Natl Acad. Sci. USA 104, 6828–6833 (2007).
Tonelli, L. H., Postolache, T. T. & Sternberg, E. M. Inflammatory genes and neural activity: involvement of immune genes in synaptic function and behavior. Front Biosci. 10, 675–680 (2005).
Lengen, C., Regard, M., Joller, H., Landis, T. & Lalive, P. Anomalous brain dominance and the immune system: do left-handers have specific immunological patterns? Brain Cogn. (2008) (e-pub ahead of print, 30 August 2008).
O’Keefe, G. M., Nguyen, V. T. & Benveniste, E. N. Regulation and function of class II major histocompatibility complex CD40, and B7 expression in macrophages and microglia: implications in neurological diseases. J. Neurovirol. 8, 496–512 (2002).
Raha-Chowdhury, R., Andrews, S. R. & Gruen, J. R. CAT 53: a protein phosphatase 1 nuclear targeting subunit encoded in the MHC class I region strongly expressed in regions of the brain involved in memory, learning, and Alzheimer’s disease. Brain Res. Mol. Brain Res. 138, 70–83 (2005).
Cohly, H. H. & Panja, A. Immunological findings in autism. Int. Rev. Neurobiol. 71, 317–341 (2005).
Bailey, S. L., Carpentier, P. A., McMahon, E. J., Begolka, W. S. & Miller, S. D. Innate and adaptive immune responses of the central nervous system. Crit. Rev. Immunol. 26, 149–188 (2006).
McElroy, J. P. & Oksenberg, J. R. Multiple sclerosis genetics. Curr. Top Microbiol. Immunol. 318, 45–72 (2008).
Shiina, T., Inoko, H. & Kulski, J. K. An update of the HLA genomic region, loci information and disease associations: 2004. Tissue Antigens 64, 631–649 (2004).
Horton, R., Wilming, L., Rand, V., Lovering, R. C., Bruford, E. A., Khodiyar, V. K. et al. Gene map of the extended human MHC. Nat. Rev. Genet. 5, 889–899 (2004).
Trowsdale, J. HLA genomics in the third millennium. Curr. Opin. Immunol. 17, 498–504 (2005).
Stewart, C. A., Horton, R., Allcock, R. J., Ashurst, J. L., Atrazhev, A. M., Coggill, P. et al. Complete MHC haplotype sequencing for common disease gene mapping. Genome Res. 14, 1176–1187 (2004).
Wheeler, D. L ., Barrett, T ., Benson, D. A ., Bryant, S. H ., Canese, K ., Chetvernin, V et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 35 (Database issue), D5–D12 (2007).
Maglott, D., Ostell, J., Pruitt, K. D. & Tatusova, T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 33, D54–D58 (2005).
Kulski, J. K. & Dawkins, R. L. The P5 multicopy gene family in the MHC is related in sequence to human endogenous retroviruses HERV-L and HERV-16. Immunogenetics 49, 404–412 (1999).
Colombo, S., Rauch, A., Rotger, M., Fellay, J., Martinez, R., Fux, C. et al. Swiss HIV Cohort Study. The HCP5 single-nucleotide polymorphism: a simple screening tool for prediction of hypersensitivity reaction to abacavir. J. Infect. Dis. 198, 864–867 (2008).
Liu, Y., Helms, C., Liao, W., Zaba, L. C., Duan, S., Gardner, J. et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 4, e1000041 (2008).
Chang, Y. T., Chou, C. T., Shiao, Y. M., Lin, M. W., Yu, C. W., Chen, C. C. et al. Psoriasis vulgaris in Chinese individuals is associated with PSORS1C3 and CDSN genes. Br. J. Dermatol. 155, 663–669 (2006).
Semple, J. I., Ribas, G., Hillyard, G., Brown, S. E., Sanderson, C. M. & Campbell, R. D. A novel gene encoding a coiled-coil mitochondrial protein located at the telomeric end of the human MHC class III region. Gene 314, 41–54 (2003).
Ostrov, D. A., Barnes, C. L., Smith, L. E., Binns, S., Brusko, T. M., Brown, A. C. et al. Characterization of HKE2: an ancient antigen encoded in the major histocompatibility complex. Tissue Antigens 69, 181–188 (2007).
de Vet, E. C., Aguado, B. & Campbell, R. D. G6b, a novel immunoglobulin superfamily member encoded in the human major histocompatibility complex, interacts with SHP-1 and SHP-2. J. Biol. Chem. 276, 42070–42074 (2001).
Adams, P. C. & Barton, J. C. Haemochromatosis. Lancet 370, 1855–1860 (2007).
Kulski, J. K. & Inoko, H. MHC genes in The Encyclopedia of the Human Genome 778–785 (Nature Publishing Group. Macmillan Publishers Ltd, Houndmills, Basingstoke, Hampshire, UK, 2003).
Dawkins, R., Leelayuwat, C., Gaudieri, S., Tay, G., Hui, J., Cattley, S. et al. Genomics of the major histocompatibility complex: haplotypes, duplication, retroviruses and disease. Immunol. Rev. 167, 275–304 (1999).
Bahram, S. MIC genes: from genetics to biology. Adv. Immunol. 76, 1–60 (2000).
Kulski, J. K., Shiina, T., Anzai, T., Kohara, S. & Inoko, H. Comparative genomic analysis of the MHC: the evolution of class I duplication blocks, diversity and complexity from shark to man. Immunol. Rev. 190, 95–122 (2002).
Shiina, T., Tamiya, G., Oka, A., Takishima, N., Yamagata, T., Kikkawa, E. et al. Molecular dynamics of MHC genesis unraveled by sequencing analysis of the 1,796,938 bp HLA class I region. Proc. Natl Acad. Sci. USA 96, 13282–13287 (1999).
García, A., Senis, Y. A., Antrobus, R., Hughes, C. E., Dwek, R. A., Watson, S. P. et al. A global proteomics approach identifies novel phosphorylated signaling proteins in GPVI-activated platelets: involvement of G6f, a novel platelet Grb2-binding membrane adapter. Proteomics 6, 5332–5343 (2006).
Jady, B. E. & Kiss, T. Characterisation of the U83 and U84 small nucleolar RNAs: two novel 2′-O-ribose methylation guide RNAs that lack complementarities to ribosomal RNAs. Nucleic Acids Res. 28, 1348–1354 (2000).
Lestrade, L. & Weber, M. J. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 34, D158–D162 (2006).
Kiss-Laszlo, Z., Henry, Y., Bachellerie, J. P., Caizergues-Ferrer, M. & Kiss, T. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell 85, 1077–1088 (1996).
Florence, B. & Faller, D. V. You bet-cha: a novel family of transcriptional regulators. Front. Biosci. 6, d1008–d1018 (2001).
Kohany, O., Gentles, A. J., Hankus, L. & Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7, 474 (2006).
Gourraud, P. A., Mano, S., Barnetche, T., Carrington, M., Inoko, H. & Cambon-Thomsen, A. Integration of microsatellite characteristics in the MHC region: a literature and sequence based analysis. Tissue Antigens 64, 543–555 (2004).
Matsuzaka, Y., Tounai, K., Denda, A., Tomizawa, M., Makino, S., Okamoto, K. et al. Identification of novel candidate genes in the diffuse panbronchiolitis critical region of the class I human MHC. Immunogenetics. 54, 301–309 (2002).
Oka, A., Tamiya, G., Tomizawa, M., Ota, M., Katsuyama, Y., Makino, S. et al. Association analysis using refined microsatellite markers localizes a susceptibility locus for psoriasis vulgaris within a 111 kb segment telomeric to the HLA-C gene. Hum. Mol. Genet. 8, 2165–2170 (1999).
Aly, T. A., Eller, E., Ide, A., Gowan, K., Babu, S. R., Erlich, H. A. et al. Multi-SNP analysis of MHC region: remarkable conservation of HLA-A1-B8-DR3 haplotype. Diabetes 55, 1265–1269 (2006).
de Bakker, P. I., McVean, G., Sabeti, P. C., Miretti, M. M., Green, T., Marchini, J. et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 38, 1166–1172 (2006).
Smith, W. P., Vu, Q., Li, S. S., Hansen, J. A., Zhao, L. P. & Geraghty, D. E. Toward understanding MHC disease associations: partial resequencing of 46 distinct HLA haplotypes. Genomics 87, 561–571 (2006).
Romero, V., Larsen, C. E., Duke-Cohan, J. S., Fox, E. A., Romero, T., Clavijo, O. P. et al. Genetic fixity in the human major histocompatibility complex and block size diversity in the class I region including HLA-E. BMC Genet. 8, 14 (2007).
Bjorkman, P. J. & Parham, P. Structure, function, and diversity of class I major histocompatibility complex molecules. Annu. Rev. Biochem. 59, 253–288 (1990).
Okamoto, K., Makino, S., Yoshikawa, Y., Takaki, A., Nagatsuka, Y., Ota, M. et al. Identification of I kappa BL as the second major histocompatibility complex-linked susceptibility locus for rheumatoid arthritis. Am. J. Hum. Genet. 72, 303–312 (2003).
Valentonyte, R., Hampe, J., Huse, K., Rosenstiel, P., Albrecht, M., Stenzel, A. et al. Sarcoidosis is associated with a truncating splice site mutation in BTNL2. Nat. Genet. 37, 357–364 (2005).
Vincek, V., Klein, D., Figueroa, F., Hauptfeld, V., Kasahara, M., O’hUigin, C. et al. The evolutionary origin of the HLA-DR3 haplotype. Immunogenetics 35, 263–271 (1992).
Marsh, S. G. WHO Nomenclature Committee for Factors of the HLA System. Nomenclature for factors of the HLA system, update July 2000. Tissue Antigens 56, 476–477 (2000).
Yang, Y., Chung, E. K., Wu, Y. L., Savelli, S. L., Nagaraja, H. N., Zhou, B. et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 80, 1037–1054 (2007).
Tuzun, E., Sharp, A. J., Bailey, J. A., Kaul, R., Morrison, V. A., Pertz, L. M. et al. Fine-scale structural variation of the human genome. Nat. Genet. 37, 727–732 (2005).
Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D. et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006).
Conrad, D. F., Andrews, T. D., Carter, N. P., Hurles, M. E. & Pritchard, J. K. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 38, 75–81 (2006).
McCarroll, S. A., Hadnott, T. N., Perry, G. H., Sabeti, P. C., Zody, M. C., Barrett, J. C. et al. International HapMap Consortium. Common deletion polymorphisms in the human genome. Nat. Genet. 38, 86–92 (2006).
Mills, R. E., Luttig, C. T., Larkins, C. E., Beauchamp, A., Tsui, C., Pittard, W. S. et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 16, 1182–1190 (2006).
Simon-Sanchez, J., Scholz, S., Fung, H. C., Matarin, M., Hernandez, D., Gibbs, J. R. et al. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum. Mol. Genet. 16, 1–14 (2007).
Wong, K. K., deLeeuw, R. J., Dosanjh, N. S., Kimm, L. R., Cheng, Z., Horsman, D. E. et al. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 80, 91–104 (2007).
Korbel, J. O., Urban, A. E., Affourtit, J. P., Godwin, B., Grubert, F., Simons, J. F. et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 318, 420–426 2007.
Levy, S., Sutton, G., Ng, P. C., Feuk, L., Halpern, A. L., Walenz, B. P. et al. The diploid genome sequence of an individual human. PLoS Biol. 5, e254 (2007).
Perry, G. H., Ben-Dor, A., Tsalenko, A., Sampas, N., Rodriguez-Revenga, L., Tran, C. W. et al. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 82, 685–695 (2008).
Kidd, J. M., Cooper, G. M., Donahue, W. F., Hayden, H. S., Sampas, N., Graves, T. et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64 (2008).
Komatsu-Wakui, M., Tokunaga, K., Ishikawa, Y., Kashiwase, K., Moriyama, S., Tsuchiya, N. et al. MIC-A polymorphism in Japanese and a MIC-A–MIC-B null haplotype. Immunogenetics 49, 620–628 (1999).
Kulski, J. K. & Dunn, D. S. Polymorphic Alu insertions within the major histocompatibility complex class I genomic region: a brief review. Cytogenet. Genome Res. 110, 193–202 (2005).
Takasu, M., Hayashi, R., Maruya, E., Ota, M., Imura, K., Kougo, K. et al. Deletion of entire HLA-A gene accompanied by an insertion of a retrotransposon. Tissue Antigens 70, 144–150 (2007).
Kulski, J. K., Shigenari, A., Shiina, T., Ota, M., Hosomichi, K., James, I. et al. Human endogenous retrovirus (HERVK9) structural polymorphism with haplotypic HLA-A allelic associations. Genetics 180, 445–457 (2008).
Cordell, H. J. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 11, 2463–2468 (2002).
Navarro, A. & Barton, N. H. Effects of multilocus balancing selection on neutral variability. Genetics 161, 849–863 (2002).
Miretti, M. M., Walsh, E. C., Ke, X., Delgado, M., Griffiths, M., Hunt, S. et al. A high-resolution linkage–disequilibrium map of the human major histocompatibility complex and first generation of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 76, 634–646 (2005).
Blomhoff, A., Olsson, M., Johansson, S., Akselsen, H. E., Pociot, F., Nerup, J. et al. Linkage disequilibrium and haplotype blocks in the MHC vary in an HLA haplotype specific manner assessed mainly by DRB1*03 and DRB1*04 haplotypes. Genes Immun. 7, 130–140 (2006).
Mathivanan, S., Periaswamy, B., Gandhi, T. K., Kandasamy, K., Suresh, S., Mohmood, R. et al. An evaluation of human protein–protein interaction data in the public domain. BMC Bioinformatics 7 (Suppl 5), S19 (2006).
Gomez, J. A., Majumder, P., Nagarajan, U. M. & Boss, J. M. X box-like sequences in the MHC class II region maintain regulatory function. J. Immunol. 175, 1030–1040 (2005).
Müller-Hilke, B. & Mitchison, N. A. The role of HLA promoters in autoimmunity. Curr. Pharm. Des. 12, 3743–3752 (2006).
Christova, R., Jones, T., Wu, P. J., Bolzer, A., Costa-Pereira, A. P., Watling, D. et al. P-STAT1 mediates higher-order chromatin remodelling of the human MHC in response to IFNgamma. J. Cell Sci. 120, 3262–3270 (2007).
Kumar, P. P., Bischof, O., Purbey, P. K., Notani, D., Urlaub, H., Dejean, A. et al. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat. Cell Biol. 9, 45–56 (2007).
Brown, J. H., Jardetzky, T. S., Gorga, J. C., Stern, L. J., Urban, R. G., Strominger, J. L. et al. Three dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364, 33–39 (1993).
Strominger, J. L. Human histocompatibility proteins. Immunol. Rev. 185, 69–77 (2002).
Momburg, F. & Tan, P. Tapasin––the keystone of the loading complex optimizing peptide binding by MHC class I molecules in the endoplasmic reticulum. Mol. Immunol. 39, 217–233 (2002).
Sadegh-Nasseri, S., Chen, M., Narayan, K. & Bouvier, M. The convergent roles of tapasin and HLA-DM in antigen presentation. Trends Immunol. 29, 141–147 (2008).
Gregersen, J. W., Kranc, K. R., Ke, X., Svendsen, P., Madsen, L. S., Thomsen, A. R. et al. Functional epistasis on a common MHC haplotype associated with multiple sclerosis. Nature 443, 574–577 (2006).
Martin, A. M., Kulski, J. K., Witt, C., Pontarotti, P. & Christiansen, F. T. Leukocyte Ig-like receptor complex (LRC) in mice and men. Trends Immunol. 23, 81–88 (2002).
Thananchai, H., Gillespie, G., Martin, M. P., Bashirova, A., Yawata, N., Yawata, M. et al. Cutting edge: allele-specific and peptide-dependent interactions between KIR3DL1 and HLA-A and HLA-B. J. Immunol. 178, 33–37 (2007).
Parham, P. Influence of KIR diversity on human immunity. Adv. Exp. Med. Biol. 560, 47–50 (2005).
Khakoo, S. I. & Carrington, M. KIR and disease: a model system or system of models? Immunol. Rev. 214, 186–201 (2006).
Swaroop, A., Branham, K. E., Chen, W. & Abecasis, G. Genetic susceptibility to age-related macular degeneration: a paradigm for dissecting complex disease traits. Hum. Mol. Genet. 16 (spec. no. 2), R174–R182 (2007).
Lester, S., McLure, C., Williamson, J., Bardy, P., Rischmueller, M. & Dawkins, R. L. Epistasis between the MHC and the RCA alpha block in primary Sjögren syndrome. Ann. Rheum. Dis. 67, 849–854 (2008).
Sánchez, F. O., Linga Reddy, M. V., Sakuraba, K., Ståhle, M. & Alarcón-Riquelme, M. E. IFN-regulatory factor 5 gene variants interact with the class I MHC locus in the Swedish psoriasis population. J. Invest. Dermatol. 128, 1704–1709 (2008).
Niewold, T. B., Kelly, J. A., Flesch, M. H., Espinoza, L. R., Harley, J. B. & Crow, M. K. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 58, 2481–2487 (2008).
Sugawara, T., Shimizu, H., Hoshi, N., Nakajima, A. & Fujimoto, S. Steroidogenic acute regulatory protein-binding protein cloned by a yeast two-hybrid system. J. Biol. Chem. 278, 42487–42494 (2003).
Tiala, I., Suomela, S., Huuhtanen, J., Wakkinen, J., Hölttä-Vuori, M., Kainu, K. et al. The CCHCR1 (HCR) gene is relevant for skin steroidogenesis and downregulated in cultured psoriatic keratinocytes. J. Mol. Med. 85, 589–601 (2007).
Kulski, J. K., Kenworthy, W., Bellgard, M., Taplin, R., Okamoto, K., Oka, A. et al. Gene expression profiling of Japanese psoriatic skin reveals an increased activity in molecular stress and immune response signals. J. Mol. Med. 83, 964–975 (2005).
Zhou, X., Krueger, J. G., Kao, M. C., Lee, E., Du, F., Menter, A. et al. Novel mechanisms of T-cell and dendritic cell activation revealed by profiling of psoriasis on the 63 100-element oligonucleotide array. Physiol. Genomics 13, 69–78 (2003).
Mee, J. B., Johnson, C. M., Morar, N., Burslem, F. & Groves, R. W. The psoriatic transcriptome closely resembles that induced by interleukin-1 in cultured keratinocytes: dominance of innate immune responses in psoriasis. Am. J. Pathol. 171, 32–42 (2007).
Nomura, I., Gao, B., Boguniewicz, M., Darst, M. A., Travers, J. B. & Leung, D. Y. Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J. Allergy Clin. Immunol. 112, 1195–1202 (2003).
Zhang, Z. H., Wang, Z. M., Crosby, M. E., Wang, H. F., Xiang, L. H., Luan, J. et al. Gene expression profiling of porokeratosis. J. Cutan. Pathol. 35, 1058–1062 (2008).
Koczan, D., Guthke, R., Thiesen, H. J., Ibrahim, S. M., Kundt, G., Krentz, H. et al. Gene expression profiling of peripheral blood mononuclear leukocytes from psoriasis patients identifies new immune regulatory molecules. Eur. J. Dermatol. 15, 251–257 (2005).
Chaussabel, D., Quinn, C., Shen, J., Patel, P., Glaser, C., Baldwin, N. et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity 29, 150–164 (2008).
Haider, A. S., Lowes, M. A., Suárez-Fariñas, M., Zaba, L. C., Cardinale, I., Khatcherian, A. et al. Identification of cellular pathways of ‘type 1,’ Th17T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J. Immunol. 180, 1913–1920 (2008).
Nzeusseu Toukap, A., Galant, C., Theate, I., Maudoux, A. L., Lories, R. J., Houssiau, F. A. et al. Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 56, 1579–1588 (2007).
Bovin, L. F., Rieneck, K., Workman, C., Nielsen, H., Sørensen, S. F., Skjødt, H. et al. Blood cell gene expression profiling in rheumatoid arthritis. Discriminative genes and effect of rheumatoid factor. Immunol. Lett. 93, 217–226 (2004).
van der Pouw Kraan, T. C., van Baarsen, L. G., Rustenburg, F., Baltus, B., Fero, M. & Verweij, C. L. Gene expression profiling in rheumatology. Methods Mol. Med. 136, 305–327 (2007).
Devauchelle, V., Marion, S., Cagnard, N., Mistou, S., Falgarone, G., Breban, M. et al. DNA microarray allows molecular profiling of rheumatoid arthritis and identification of pathophysiological targets. Genes Immun. 5, 597–608 (2004).
van der Pouw Kraan, T. C., van Gaalen, F. A., Huizinga, T. W., Pieterman, E., Breedveld, F. C. & Verweij, C. L. Discovery of distinctive gene expression profiles in rheumatoid synovium using cDNA microarray technology: evidence for the existence of multiple pathways of tissue destruction and repair. Genes Immun. 4, 187–196 (2003).
Batliwalla, F. M., Li, W., Ritchlin, C. T., Xiao, X., Brenner, M., Laragione, T. et al. Microarray analyses of peripheral blood cells identifies unique gene expression signature in psoriatic arthritis. Mol. Med. 11, 21–29 (2005).
Fu, S., Zhao, H., Shi, J., Abzhanov, A., Crawford, K., Ohno-Machado, L. et al. Peripheral arterial occlusive disease: global gene expression analyses suggest a major role for immune and inflammatory responses. BMC Genomics 9, 369 (2008).
Nisole, S., Stoye, J. P. & Saïb, A. TRIM family proteins: retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 3, 799–808 (2005).
Uchil, P. D., Quinlan, B. D., Chan, W. T., Luna, J. M. & Mothes, W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 4, e16 (2008).
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T. et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007).
Iafrate, A. J., Feuk, L., Rivera, M. N., Listewnik, M. L., Donahoe, P. K., Qi, Y. et al. Detection of large-scale variation in the human genome. Nat. Genet. 36, 949–951 (2004).
Fellermann, K., Stange, D. E., Schaeffeler, E., Schmalzl, H., Wehkamp, J., Bevins, C. L. et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am. J. Hum. Genet. 79, 439–448 (2006).
Hollox, E. J., Huffmeier, U., Zeeuwen, P. L., Palla, R., Lascorz, J., Rodijk-Olthuis, D. et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat. Genet. 40, 23–25 (2008).
Jiang, L., Yu, Z., Du, W., Tang, Z., Jiang, T., Zhang, C. et al. Development of a fluorescent and colorimetric detection methods-based protein microarray for serodiagnosis of TORCH infections. Biosens. Bioelectron 24, 376–382 (2008).
Tomazou, E. M., Rakyan, V. K., Lefebvre, G., Andrews, R., Ellis, P., Jackson, D. K. et al. Generation of a genomic tiling array of the human major histocompatibility complex (MHC) and its application for DNA methylation analysis. BMC Med. Genomics 1, 19 (2008).
Serrano, N. C., Millan, P. & Páez, M. C. Non-HLA associations with autoimmune diseases. Autoimmun. Rev. 5, 209–214 (2006).
Samanta, M. P., Tongprasit, W. & Stolc, V. In-depth query of large genomes using tiling arrays. Methods Mol. Biol. 377, 163–174 (2007).
Borrebaeck, C. A. & Wingren, C. High-throughput proteomics using antibody microarrays: an update. Expert Rev. Mol. Diagn. 7, 673–686 (2007).
Tao, S. C., Chen, C. S. & Zhu, H. Applications of protein microarray technology. Comb. Chem. High Throughput Screen. 10, 706–718 (2007).
Chen, D. S., Soen, Y., Stuge, T. B., Lee, P. P., Weber, J. S., Brown, P. O. et al. Marked differences in human melanoma antigen-specific T cell responsiveness after vaccination using a functional microarray. PLoS Med. 2, e265 (2005).
Fortier, M. H., Caron, E., Hardy, M. P., Voisin, G., Lemieux, S., Perreault, C. et al. The MHC class I peptide repertoire is molded by the transcriptome. J. Exp. Med. 205, 595–610 (2008).
Ho, P. P., Higgins, J. P., Kidd, B. A., Tomooka, B., Digennaro, C., Lee, L. Y. et al. Tolerizing DNA vaccines for autoimmune arthritis. Autoimmunity 39, 675–682 (2006).
Lueking, A., Huber, O., Wirths, C., Schulte, K., Stieler, K. M., Blume-Peytavi, U. et al. Profiling of alopecia areata autoantigens based on protein microarray technology. Mol. Cell Proteomics 4, 1382–1390 (2005).
EI Essawy, B., Otu, H. H., Choy, B., Zheng, X. X., Libermann, T. A. & Strom, T. B. Proteomic analysis of the allograft response. Transplantation 82, 267–274 (2006).
Sieberts, S. K. & Schadt, E. E. Moving toward a system genetics view of disease. Mamm. Genome 18, 389–401 (2007).
Amiel, J. L. Study of the leukocyte phenotypes in Hodgkin's disease in Histocompatibility Testing 79–81 (eds Curtoni, E.S., Mattiuz, P.L., Tosi, R.M.) (Munksgaard, Copenhagen, 1967).
Tiwari, J. L. & Terasaki, P. I. HLA and Disease Association (Springer-Verlag, New York, 1985).
Naito, S. The association of HLA with diseases in Japanese. J. Hum. Genet. 31, 323–329 (1986).
Geluk, A. & Ottenhoff, T. H. HLA and leprosy in the pre and postgenomic eras. Hum. Immunol. 67, 439–445 (2006).
Mehra, N. K. & Kaur, G. 14th International HLA and Immunogenetics Workshop: report on joint study on MHC and infection. Tissue Antigens 69, 226–227 (2007).
Goulder, P. J. & Watkins, D. I. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat. Rev. Immunol. 8, 619–630 (2008).
Lie, B. A. & Thorsby, E. Several genes in the extended human MHC contribute to predisposition to autoimmune diseases. Curr. Opin. Immunol. 17, 526–531 (2005).
Jones, E. Y., Fugger, L., Strominger, J. L. & Siebold, C. MHC class II proteins and disease: a structural perspective. Nat. Rev. Immunol. 6, 271–282 (2006).
Schmidt, H., Williamson, D. & Ashley-Koch, A. HLA-DR15 haplotype and multiple sclerosis: a HuGE review. Am. J. Epidemiol. 165, 1097–1109 (2007).
Jacobson, E. M., Huber, A. & Tomer, Y. The HLA gene complex in thyroid autoimmunity: from epidemiology to etiology. J. Autoimmun. 30, 58–62 (2008).
Hamosh, A., Scott, A. F., Amberger, J., Valle, D. & McKusick, V. A. Online Mendaian Inheritance in Man (OMIM). Hum. Mutat. 15, 57–61 (2000).
Hamosh, A., Scott, A. F., Amberger, J. S., Bocchini, C. A. & McKusick, V. A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517 (2005).
Naoum, J. J., Chai, H., Lin, P. H., Lumsden, A. B., Yao, Q. & Chen, C. Lymphotoxin-alpha and cardiovascular disease: clinical association and pathogenic mechanisms. Med. Sci. Monit. 12, RA121–RA124 (2006).
London, S. J. Gene-air pollution interactions in asthma. Proc. Am. Thorac. Soc. 4, 217–220 (2007).
Sharma, S., Ghosh, B. & Sharma, S. K. Association of TNF polymorphisms with sarcoidosis, its prognosis and tumour necrosis factor (TNF)-alpha levels in Asian Indians. Clin. Exp. Immunol. 151, 251–259 (2008).
Vallvé, J. C., Paredes, S., Girona, J., Uliaque, K., Ribalta, J., Hurt-Camejo, E. et al. Tumor necrosis factor-alpha −1031 T/C polymorphism is associated with smaller and more proatherogenic low density lipoprotein particles in patients with rheumatoid arthritis. J. Rheumatol. 35, 1697–1703 (2008).
Lee, H. H., Lee, Y. J., Wang, Y. M., Chao, H. T., Niu, D. M., Chao, M. C. et al. Low frequency of the CYP21A2 deletion in ethnic Chinese (Taiwanese) patients with 21 hydroxylase deficiency. Mol. Genet. Metab. 93, 450–457 (2008).
Lee, H. H., Tsai, F. J., Lee, Y. J. & Yang, Y. C. Diversity of the CYP21A2 gene:a 6.2-kb TaqI fragment and a 3.2-kb TaqI fragment mistaken as CYP21A1P. Mol. Genet. Metab. 88, 372–377 (2006).
Seyrantepe, V., Poupetova, H., Froissart, R., Zabot, M. T., Maire, I. & Pshezhetsky, A. V. Molecular pathology of NEU1 gene in sialidosis. Hum. Mutat. 22, 343–352 (2003).
Sjöholm, A. G., Jönsson, G., Braconier, J. H., Sturfelt, G. & Truedsson, L. Complement deficiency and disease: an update. Mol. Immunol. 43, 78–85 (2006).
Forabosco, P., Bouzigon, E., Ng, M. Y., Hermanowski, J., Fishe, R. S. A., Criswell, L. A. et al. Meta-analysis of genome-wide linkage studies across autoimmune diseases. Eur. J. Hum. Genet. 2008 (e-pub ahead of print, 10 September 2008).
Wellcome Trust Case Control Consortium. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 447, 661–678 2007.
Choi, H. B., Han, H., Youn, J. I., Kim, T. Y. & Kim, T. G. MICA 5.1 allele is a susceptibility marker for psoriasis in the Korean population. Tissue Antigens 56, 548–550 (2000).
Asumalahti, K., Veal, C., Laitinen, T., Suomela, S., Allen, M., Elomaa, O. et al. Psoriasis Consortium. Coding haplotype analysis supports HCR as the putative susceptibility gene for psoriasis at the MHC PSORS1 locus. Hum. Mol. Genet. 11, 589–597 (2002).
Chang, Y. T., Shiao, Y. M., Chin, P. J., Liu, Y. L., Chou, F. C., Wu, S. et al. Genetic polymorphisms of the HCR gene and a genomic segment in close proximity to HLA-C are associated with patients with psoriasis in Taiwan. Br. J. Dermatol. 150, 1104–1111 (2004).
Ameen, M., Allen, M. H., Fisher, S. A., Lewis, C. M., Cuthbert, A., Kondeatis, E. et al. Corneodesmosin (CDSN) gene association with psoriasis vulgaris in Caucasian but not in Japanese populations. Clin. Exp. Dermatol. 30, 414–418 (2005).
Helms, C., Saccone, N. L., Cao, L., Daw, J. A., Cao, K., Hsu, T. M. et al. Localization of PSORS1 to a haplotype block harboring HLA-C and distinct from corneodesmosin and HCR. Hum. Genet. 118, 466–476 (2005).
Martínez-Borra, J., Brautbar, C., González, S., Enk, C. D., López-Vázquez, A. & López-Larrea, C. The region of 150 kb telometic to HLA-C is associated with psoriasis in the Jewish population. J. Invest. Dermatol. 125, 928–932 (2005).
Nair, R. P., Stuart, P. E., Nistor, I., Hiremagalore, R., Chia, N. V., Jenisch, S. et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am. J. Hum. Genet. 78, 827–851 (2006).
Takemoto, T., Naruse, T., Namba, K., Kitaichi, N., Ota, M., Shindo, Y. et al. Re-evaluation of heterogeneity in HLA-B*510101 associated with Behçet's disease. Tissue Antigens 72, 347–353 (2008).
Karasneh, J., Gül, A., Ollier, W. E., Silman, A. J. & Worthington, J. Whole-genome screening for susceptibility genes in multicase families with Behçet's disease. Arthritis Rheum. 52, 1836–1842 (2005).
Oksenberg, J. R., Baranzini, S. E., Sawcer, S. & Hauser, S. L. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat. Rev. Genet. 9, 516–526 (2008).
Degli-Esposti, M. A., Leaver, A. L., Christiansen, F. T., Witt, C. S., Abraham, L. J. & Dawkins, R. L. Ancestral haplotypes: conserved population MHC haplotypes. Hum. Immunol. 34, 242–252 (1992).
Price, P., Witt, C., Allcock, R., Sayer, D., Garlepp, M., Kok, C. C. et al. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol. Rev. 167, 257–274 (1999).
Aptsiauri, N., Cabrera, T., Garcia-Lora, A., Lopez-Nevot, M. A., Ruiz-Cabello, F. & Garrido, F. MHC class I antigens and immune surveillance in transformed cells. Int. Rev. Cytol. 256, 139–189 (2007).
Sengupta, S., den Boon, J. A., Chen, I. H., Newton, M. A., Dahl, D. B., Chen, M. et al. Genome-wide expression profiling reveals EBV-associated inhibition of MHC class I expression in nasopharyngeal carcinoma. Cancer Res. 66, 7999–8006 (2006).
Schlecht, N. F., Burk, R. D., Adrien, L., Dunne, A., Kawachi, N., Sarta, C. et al. Gene expression profiles in HPV-infected head and neck cancer. J. Pathol. 213, 283–293 (2007).
Watson, N. F., Ramage, J. M., Madjd, Z., Spendlove, I., Ellis, I. O., Scholefield, J. H. et al. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int. J. Cancer 118, 6–10 (2006).
Wang, H., Yang, D., Xu, W., Wang, Y., Ruan, Z., Zhao, T. et al. Tumor-derived soluble MICs impair CD3(+)CD56(+) NKT-like cell cytotoxicity in cancer patients. Immunol. Lett. 120, 65–71 (2008).
Groh, V., Wu, J., Yee, C. & Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419, 734–738 (2002).
Lönnroth, C., Andersson, M., Arvidsson, A., Nordgren, S., Brevinge, H., Lagerstedt, K. et al. Preoperative treatment with a non-steroidal anti-inflammatory drug (NSAID) increases tumor tissue infiltration of seemingly activated immune cells in colorectal cancer. Cancer Immun. 8, 5 (2008).
Champine, P. J., Michaelson, J., Weimer, B. C., Welch, D. R. & DeWald, D. B. Microarray analysis reveals potential mechanisms of BRMS1-mediated metastasis suppression. Clin. Exp. Metastasis 24, 551–565 (2007).
Rimsza, L. M., Roberts, R. A., Campo, E., Grogan, T. M., Bea, S., Salaverria, I. et al. Loss of major histocompatibility class II expression in non–immune-privileged site diffuse large B-cell lymphoma is highly coordinated and not due to chromosomal deletions. Blood 107, 1101–1107 (2006).
Dave, S. S., Fu, K., Wright, G. W., Lam, L. T., Kluin, P., Boerma, E. J. et al. Lymphoma/Leukemia Molecular Profiling Project. Molecular diagnosis of Burkitt's lymphoma. N. Engl. J. Med. 354, 2431–2442 (2006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shiina, T., Hosomichi, K., Inoko, H. et al. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet 54, 15–39 (2009). https://doi.org/10.1038/jhg.2008.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2008.5
Keywords
This article is cited by
-
Contribution of HLA class I (A, B, C) and HLA class II (DRB1, DQA1, DQB1) alleles and haplotypes in exploring ethnic origin of central Tunisians
BMC Medical Genomics (2024)
-
Protection of cell therapeutics from antibody-mediated killing by CD64 overexpression
Nature Biotechnology (2023)
-
KiT-GENIE, the French genetic biobank of kidney transplantation
European Journal of Human Genetics (2023)
-
T-cell receptor diversity in minimal change disease in the NEPTUNE study
Pediatric Nephrology (2023)
-
HLA-SPREAD: a natural language processing based resource for curating HLA association from PubMed abstracts
BMC Genomics (2022)
