
Abstract
Mutations in SLC26A4 cause a broad phenotypic spectrum, from typical Pendred syndrome to nonsyndromic hearing loss associated with enlarged vestibular aqueduct. Identification of these mutations is important for accurate diagnosis, proper medical management and appropriate genetic counseling and requires updated information regarding spectrum, clinical characteristics and genotype–phenotype correlations, based on a large cohort. In 100 patients with bilateral enlarged vestibular aqueduct among 1511 Japanese hearing loss probands registered in our gene bank, goiter data were available for 79, of whom 15 had Pendred syndrome and 64 had nonsyndromic hearing loss. We clarified the mutation spectrum for the SLC26A4 mutations and also summarized hearing levels, progression, fluctuation and existence of genotype–phenotype correlation. SLC26A4 mutations were identified in 82 of the 100 patients (82.0%). Of the Pendred syndrome patients, 93% (14/15) were carriers, as were 77% (49/64) of the nonsyndromic hearing loss patients. Clinical characteristics of patients with SLC26A4 mutations were congenital, fluctuating and progressive hearing loss usually associated with vertigo and/or goiter. We found no genotype–phenotype correlations, indicating that, unlike in the case of GJB2 mutations, the phenotype cannot be predicted from the genotype. Our mutation analysis confirmed the importance of mutations in the SLC26A4 gene among hearing loss patients with enlarged vestibular aqueduct and revealed the mutation spectrum, essential information when performing genetic testing.
Similar content being viewed by others


Introduction
Based on our genetic screening, SLC26A4 is the second most common responsible gene in Japanese deafness patients.1 Mutations in the SLC26A4 gene are known to be responsible for a broad phenotypic spectrum, from typical Pendred syndrome to nonsyndromic hearing loss with enlarged vestibular aqueduct (EVA). The prevalent association of SLC26A4 mutations in these patients (90% in Pendred syndrome and 78.1% in nonsyndromic hearing loss associated with EVA) indicates the importance of this gene in the pathophysiology of this category of hearing impairment.2 More than 160 mutations have been found in SLC26A4 (Pendred/BOR Homepage, http://www.healthcare.uiowa.edu/labs/pendredandbor/), and different mutational spectrums among different ethnic groups have been reported.2 The identification of SLC26A4 mutations enables more appropriate genetic counseling and proper medical management for these patients. For such clinical application, updated information regarding mutation spectrum, clinical characteristics and genotype–phenotype correlations based on a large cohort is needed. In addition to our previous reports,1, 2, 3, 4, 5, 6, 7 the present study was performed using a large cohort of patients to collect updated data and summarize these data to enable more precise decision making by ear, nose and throat clinicians.
Materials and methods
Subjects
Data on 1511 independent probands and 1545 family members were collected from 33 ear, nose and throat departments nationwide in Japan and registered in our gene bank. All subjects or next of kin, caretakers or guardians on behalf of the minors/children gave prior written informed consent for participation in the project, and the Shinshu University Ethical Committee as well as the respective Ethical Committees of the other participating institutions of the Deafness Gene Study Consortium (Hokkaido University, Hirosaki University, Iwate Medical University, Tohoku University, Yamagata University, Fukushima Medical University, Jichi Medical University, Gunma University, Nihon University, Nippon Medical School, Nippon Medical School Tama Nagayama Hospital, Jikei University, Toranomon Hospital, Kitasato University, Hamamatsu Medical University, Mie University, Shiga Medical Center for Children, Osaka Medical College, Hyogo College of Medicine, Kobe City Medical Center General Hospital, Wakayama Medical University, Okayama University, Yamaguchi University, Ehime University, Kyushu University, Fukuoka University, Nagasaki University, Kanda ENT Clinic, Miyazaki Medical College, Kagoshima University and Ryukyus University) approved the study.
Computerized tomography scan was used to diagnose EVA (according to the criteria of EVA: a diameter of >1.5 mm at the midpoint between the common crus and the external aperture), and they were clinically well characterized by repeated auditory examinations.
The 100 subjects (51 males and 49 females) from among the 1511 probands who met the criteria of bilateral EVA and who ranged in age from 0 to 59 years with a mean age of 13.9 years at the time of examination were enrolled in the current study. Fifteen subjects had Pendred syndrome and 64 had nonsyndromic hearing loss.
The controls were 192 unrelated Japanese healthy individuals with normal hearing evaluated by auditory testing.
Mutation analysis
To identify SLC26A4 mutations, a DNA fragment containing all the exons of SLC26A4, including flanking intronic sequences, was sequenced as described elsewhere.4 New variants were tested in 192 unrelated normal hearing controls.
Possible pathologic mutations were defined as (1) mutations found to be homozygotes or compound heterozygotes (and determined by segregation study); (2) variants that were not found, or were very few, in the 192 control subjects; and (3) amino acids that were well conserved among various species.
Clinical evaluations
Hearing levels were determined by pure-tone audiometry in adults. For the young patients, conditioned orientation response audiometry or auditory steady-state response was used. Clinical data, including hearing loss progression, fluctuation, episodes of tinnitus and vestibular dysfunction (vertigo, dizziness) and goiter, were collected by anamnestic evaluation. For genotype–phenotype correlation analysis, one-way analysis of variance (Tukey’s honest significant difference (HSD) test), Kruskal–Wallis test and multivariate statistics (multiple regression analysis and logistic regression analysis) were used.
Results
SLC26A4 mutation spectrum
There were a total of 39 SLC26A4 mutations found in the probands with bilateral EVA (Table 1). These mutations were either homozygous, compound heterozygous or heterozygous with no other mutations being detectable. There were two nonsense mutations (p.S610X, p.L727X), three deletion frameshift mutations (c.322delC, c.917delT, c.1219delCT) and three insertion frameshift mutations (c.139insC, c.1652insT, c.2111ins GCTGG). Seven splice site mutations were found (c.416-1G>A, c.600+1G>T, c.601-1G>A, c.919-2A>G, c.1001+1G>A, c.1002-9A>G and c.1707+5G>A).
There were 24 missense mutations (p.P76S, p.T94I, p.P123S, p.M147V, p.P297Q, p.K369E, p.A372V, p.N392Y, p.G396E, p.T410M, p.A434T, p.G439R, p.S448L, p.T527P, p.I529S, p.S532I, p.C565Y, p.R581S, p.S657N, p.V659L, p.S666F, p.T721M, p.H723R and p.H723Y). To evaluate the evolutionary conservation of the amino acids affected by these missense mutations, we made an alignment of the SLC26A4 amino acid sequence of four mammalian species: human, rat, cow and mouse. On the basis of this alignment, all missense mutations had changed evolutionary conserved amino acids. Of these mutations, nine variants had not been reported. We checked the 192 control subjects with normal hearing, but with the exception of p.H723R in 1 case, no mutations were detected.
Sequencing identified mutations in 82 of the 100 patients (82.0%). Mutations were detected in 93% of those with Pendred syndrome (14/15) and 77% (49/64) of those with nonsyndromic hearing loss. Of these, 15/100 (15.0%) were homozygous, 51/100 (51.0%) were compound heterozygous and 16/100 (16.0%) were heterozygous (Table 2).
The most frequent mutation was p.H723R that accounted for 36.0%, and the second was c.919-2A>G found in 7.0%, followed by c.1707+5G>A (4.0%). Frequency of the other 36 mutations was very low (0.5–2.0%).
Clinical findings
Table 2 shows the clinical details for the 100 subjects.
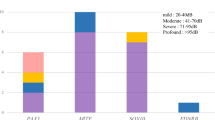
The subjects had an average hearing level of 80.9 dB (7.5–112.5 dB), with hearing loss that was mild in 5, moderate in 22, severe in 37, profound in 19 and unknown in 12. Regarding onset age of hearing loss, 45 patients were congenital, 18 were prelingual (1–3 years old), 20 were from 4 to 14 years and 17 were unknown. These results clearly indicated that early onset is dominant in patients with EVA. Also, 70 patients (70%) showed progressive hearing loss and 56 patients (56.0%) felt fluctuation of hearing. With regard to the 79 patients for whom data on vertigo were available, 41 patients complained of vertigo and 38 did not. Of the 79 patients for whom data on goiter were available, 15 had goiter and 64 did not, with an onset age from 12 to 33 years. As to family history, all families were recessive inheritance or sporadic cases.
Genotype–phenotype correlations for diagnostic age, fluctuation, vertigo, tinnitus and goiter are summarized in Figure 1.

Genotypes and phenotypes (diagnostic age, fluctuation, vertigo, tinnitus and goiter) in the current study. NT/(−), heterozygote of nontruncating mutation; NT/NT, nontruncating/nontruncating; NT/T, nontruncating/truncating; T/(−), heterozygote of truncating mutation; T/T, truncating/truncating; (−), wild type.
We defined nonsense or frameshift mutations as truncating (T) and missense mutations as nontruncating (NT) and classified the genotypes as truncating/truncating (T/T), truncating/nontruncating (T/NT) or nontruncating/nontruncating (NT/NT). Significant differences were not found between the groups in any of the clinical features (Tukey’s HSD test was used for diagnostic age and Kruskal–Wallis test was used for fluctuation, vertigo, tinnitus and goiter, all tests indicated P>0.05; Figure 1). Figure 2 shows the relationship between hearing loss severity and the mutation (T or NT) that also showed no significant differences (Tukey’s HSD test, P>0.05). We also performed multivariate statistics (multiple regression analysis and logistic regression analysis) and we found that only the age of the patients correlated with the hearing loss severity while the genotype of SLC26A4 mutations did not significantly affect the hearing loss severity (P>0.05).

The relationship between hearing level at the lower frequencies and genotype. Hearing level was the average of 125, 250 and 500 Hz. NT/(−), heterozygote of nontruncating mutation; NT/NT, nontruncating/nontruncating; NT/T, nontruncating/truncating; T/(−), heterozygote of truncating mutation; T/T, truncating/truncating; (−), wild type.
Discussion
The present large cohort study revealed a high prevalence (82%; 82/100) of SLC26A4 mutations in sensorineural hearing loss patients with EVA in Japanese. The frequency (8.7%) is the second most common next to GJB2 that is found in 16.2% of overall and 25.6% of congenital hearing loss patients.1
Our mutation analysis results confirmed the previous reports that indicated the importance of this gene among hearing loss patients with EVA. This study also added novel mutations and summarized updated data for the precise molecular diagnosis.
First, the high prevalence (82%) of SLC26A4 mutations in EVA patients is compatible with the high prevalence of SLC26A4 mutations reported in eastern Asians; that is, 97.9% in Chinese,8 and 92% in Koreans.9 These frequencies are higher than those reported in Caucasoid populations (20% in Americans,10 40.0% in French11 and 28.4% in Spanish12). It is still an open question whether other genes are involved in the EVA patients without SLC26A4 mutations. Mutations in FOXI1,13 a modulatory gene of SLC26A4, were not found in our series of patients (data not shown). As seen in previous mutation screening reports, we encountered a significant number of heterozygous cases without a second mutation even after direct sequencing of the coding region of the gene. It is highly likely that there is one more occult mutation somewhere because patients with heterozygous mutation are associated with EVA.
Second, it is evident that the mutation spectrum found in the Japanese population is quite different from that in Caucasoid populations, but similar to the mutation spectrum reported in the Asian populations, especially Koreans.8, 9, 10, 11, 12, 14 There are two frequent mutations in east Asians, namely p.H723R and c.919-2A>G. p.H723R is most prevalent in the Japanese and Korean populations,8 whereas c.919-2A>G is most common in the Chinese.7
The existence of a genotype–phenotype correlation is still controversial.6, 12, 15 Mutations in SLC26A4 can cause a broad phenotypic spectrum, from typical Pendred syndrome to nonsyndromic hearing loss associated with EVA. In the present study, various features of the phenotype were compared with the genotypes. We defined nonsense or frame shift mutations as truncating (T) and missense mutations as non-truncating (NT) and classified the genotypes as truncating/truncating (T/T), truncating/non-truncating (T/NT), or non-truncating/non-truncating (NT/NT). However, statistical differences were not found between the groups in any of the clinical features (χ2 tests, P>0.05; Figure 1).
Concerning the relationship between the severity of hearing loss and individual SLC26A4 mutations, several functional studies have demonstrated the property of transporter function.16, 17, 18 Furthermore, retention of improperly folded Pendrin mutants in the endoplasmic reticulum has been suggested as the major pathological mechanism for Pendred syndrome.19, 20 In this study, we compared not only the difference between the T and NT mutations, but also compared the individual mutations and severity of hearing. However, there were no correlations (data not shown). Indeed, there was great variation regarding hearing loss severity even with the same mutations. For example, in the patients homozygous for the most prevalent mutation, p.H723R, hearing level at low frequency varied from 61 to 99 dB (Table 2). In addition, many reports have described intrafamilial phenotypic variation.8, 9, 10, 11, 12 Therefore, phenotype may be determined not only by SLC26A4 mutations but also other factors (genetic as well as environmental), contributing to such variability (Figure 2).
Unlike in the case of GJB2, phenotype cannot be predicted from the genotype;6 however, the clarification of clinical features will enable more appropriate genetic counseling and proper medical management for these patients.
The present study confirmed clinical characteristics of 66 patients with EVA caused by biallelic SLC26A4 mutations. These included congenital (5/63, 7.9%), fluctuated (42/52, 80.8%) and progressive (49/56, 87.5%) hearing loss usually associated with vertigo (35/52, 67.3%) and/or goiter (12/53, 22.6%) during long-term follow-up, in accordance with our previous study.6 It is known that goiter sometimes becomes apparent between 10 and 20 years of age. The present cohort included young children, and therefore the frequency of goiter may be underestimated. As seen in Figure 3, in 66 patients with biallelic mutations for whom data were available, onset of hearing loss was likely to be early onset, and progressive with age.

The relationship between hearing level and age in subjects with biallelic SLC26A4 mutations. Hearing level was calculated as the average of 250, 500, 1000 and 2000 Hz in both sides.
Conclusions
Pendred syndrome and nonsyndromic hearing loss associated with EVA are a continuum of disease characterized as being associated with congenital, fluctuating and progressive hearing loss, and most patients have vertigo and/or goiter. However, in the present study, no genotype–phenotype correlation was found. The results obtained from the present study will facilitate accurate molecular diagnosis and better genetic counseling.
References
Usami, S, Nishio, S, Nagano, M, Abe, S & Yamaguchi, T, Deafness Gene Study Consortium Simultaneous screening of multiple mutations by invader assay improves molecular diagnosis of hereditary hearing loss: a multicenter study. PLoS One 7, e31276 (2012).
Tsukamoto, K, Suzuki, H, Harada, D, Namba, A, Abe, S & Usami, S Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. Eur. J. Hum. Genet. 11, 916–922 (2003).
Usami, S, Abe, S, Weston, MD, Shinkawa, H, Van Camp, G & Kimberling, WJ Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum. Genet. 104, 188–192 (1999).
Namba, A, Abe, S, Shinkawa, H, Kimberling, WJ & Usami, S Genetic features of hearing loss associated with ear anomalies: PDS and EYA1 mutation analysis. J. Hum. Genet. 46, 518–521 (2001).
Iwasaki, S, Tsukamoto, K, Usami, S, Misawa, K, Mizuta, K & Mineta, H Association of SLC26A4 mutations with clinical features and thyroid function in deaf infants with enlarged vestibular aqueduct. J. Hum. Genet. 51, 805–810 (2006).
Suzuki, H, Oshima, A, Tsukamoto, K, Abe, S, Kumakawa, K, Nagai, K et al. Clinical characteristics and genotype-phenotype correlation of hearing loss patients with SLC26A4 mutations. Acta Otolaryngol. 127, 1292–1297 (2007).
Tsukada, K, Nishio, S & Usami, S, Deafness Gene Study Consortium A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin. Genet. 78, 464–470 (2010).
Wang, QJ, Zhao, YL, Rao, SQ, Guo, YF, Yuan, H, Zong, L et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin. Genet. 72, 245–254 (2007).
Park, HJ, Lee, SJ, Jin, HS, Lee, JO, Go, SH, Jong, HS et al. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin. Genet. 67, 160–165 (2005).
Dai, P, Stewart, AK, Chebib, F, Hsu, A, Rozenfeld, J, Huang, D et al. Distinct and novel SLC26A4/Pendrin mutations in Chinese and U.S. patients with nonsyndromic hearing loss. Physiol. Genomics 38, 281–290 (2009).
Albert, S, Blons, H, Jonard, L, Feldmann, D, Chauvin, P, Loundon, N et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur. J. Hum. Genet. 14, 773–779 (2006).
Pera, A, Villamar, M, Viñuela, A, Gandía, M, Medà, C, Moreno, F et al. A mutational analysis of the SLC26A4 gene in Spanish hearing-impaired families provides new insights into the genetic causes of Pendred syndrome and DFNB4 hearing loss. Eur. J. Hum. Genet. 16, 888–896 (2008).
Yang, T, Vidarsson, H, Rodrigo-Blomqvist, S, Rosengren, SS, Enerback, S, Smith, RJ et al. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am. J. Hum. Genet. 80, 1055–1063 (2007).
Park, HJ, Shaukat, S, Liu, XZ, Hahn, SH, Naz, S, Ghosh, M et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J. Med. Genet. 40, 242–248 (2003).
Pryor, SP, Madeo, AC, Reynolds, JC, Sarlis, NJ, Arnos, KS, Nance, WE et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J. Med. Genet. 42, 159–165 (2005).
Pera, A, Dossena, S, Rodighiero, S, Gandía, M, Bottà, G, Meyer, G et al. Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA. Proc. Natl Acad. Sci. USA 105, 18608–18613 (2008).
Yoon, JS, Park, HJ, Yoo, SY, Namkung, W, Jo, MJ, Koo, SK et al. Heterogeneity in the processing defect of SLC26A4 mutants. J. Med. Genet. 45, 411–419 (2008).
Dossena, S, Rodighiero, S, Vezzoli, V, Nofziger, C, Salvioni, E, Boccazzi, M et al. Functional characterization of wild-type and mutated pendrin (SLC26A4), the anion transporter involved in Pendred syndrome. J. Mol. Endocrinol. 43, 93–103 (2009).
Taylor, JP, Metcalfe, RA, Watson, PF, Weetman, AP & Trembath, RC Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J. Clin. Endocrinol. Metab. 87, 1778–1784 (2002).
Rotman-Pikielny, P, Hirschberg, K, Maruvada, P, Suzuki, K, Royaux, IE, Green, ED et al. Retention of pendrin in the endoplasmic reticulum is a major mechanism for Pendred syndrome. Hum. Mol. Genet. 11, 2625–2633 (2002).
Coucke, PJ, Van Hauwe, P, Everett, LA, Demirhan, O, Kabakkaya, Y, Dietrich, NL et al. Identification of two different mutations in the PDS gene in an inbred family with Pendred syndrome. J. Med. Genet. 36, 475–477 (1999).
Coyle, B, Reardon, W, Herbrick, JA, Tsui, LC, Gausden, E, Lee, J et al. Molecular analysis of the PDS gene in Pendred syndrome. Hum. Mol. Genet. 7, 1105–1112 (1998).
Acknowledgements
We thank the participants of the Deafness Gene Study Consortium: Drs Norihito Takeichi and Satoshi Fukuda (Hokkaido University), Drs Atsushi Namba and Hideichi Shinkawa (Hirosaki University), Drs Yumiko Kobayashi and Hiroaki Sato (Iwate Medical University), Drs Tetsuaki Kawase and Toshimitsu Kobayashi (Tohoku University), Drs Tomoo Watanabe, Tsukasa Ito and Masaru Aoyagi (Yamagata University), Drs Hiroshi Ogawa and Koichi Omori (Fukushima Medical University), Drs Kotaro Ishikawa and Keiichi Ichimura (Jichi Medical University), Drs Kyoko Nagai and Nobuhiko Furuya (Gunma University), Drs Shuntaro Shigihara, Yasuyuki Nomura and Minoru Ikeda (Nihon University School), Drs Tetsuo Ikezono and Toshiaki Yagi (Nippon Medical School), Dr Shunichi Tomiyama (Nippon Medical School Tama Nagayama Hospital), Drs Hiromi Kojima, Yuika Sakurai and Hiroshi Moriyama (Jikei University), Dr Kozo Kumakawa (Toranomon Hospital), Drs Hajime Sano and Makito Okamoto (Kitasato University), Dr Satoshi Iwasaki (Hamamatsu Medical University), Dr Kazuhiko Takeuchi (Mie University), Dr Masako Nakai (Shiga Medical Center for Children), Drs Masahiko Higashikawa and Hiroshi Takenaka (Osaka Medical College), Drs Yuko Saito, Masafumi Sakagami (Hyogo College of Medicine), Dr Yasushi Naito (Kobe City Medical Center General Hospital), Drs Keiji Fujihara, Akihiro Sakai and Noboru Yamanaka (Wakayama Medical University), Drs Kunihiro Fukushima, and Kazunori Nishizaki (Okayama University), Drs Kazuma Sugahara and Hiroshi Yamashita (Yamaguchi University), Drs Naoto Hato and Kiyofumi Gyo (Ehime University), Drs Yasuhiro Kakazu and Shizuo Komune (Kyushu University), Drs Mayumi Sugamura and Takashi Nakagawa (Fukuoka University), Dr Haruo Takahashi (Nagasaki University), Dr Yukihiko Kanda (Kanda ENT Clinic), Drs Hirokazu Kawano and Tetsuya Tono (Miyazaki Medical College), Drs Ikuyo Miyanohara andYuichi Kurono (Kagoshima University), Drs Akira Ganaha and Mikio Suzuki (Ryukyus University), for providing samples of their patients. We also thank AC Apple-Mathews for help in preparing the manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Miyagawa, M., Nishio, Sy., Usami, Si. et al. Mutation spectrum and genotype–phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: a large cohort study. J Hum Genet 59, 262–268 (2014). https://doi.org/10.1038/jhg.2014.12
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.12
Keywords
This article is cited by
-
The correlation between deafness progression and SLC26A4 mutations in enlarged vestibular aqueduct patients
European Archives of Oto-Rhino-Laryngology (2024)
-
Comprehensive genetic testing improves the clinical diagnosis and medical management of pediatric patients with isolated hearing loss
BMC Medical Genomics (2022)
-
The genetic etiology of hearing loss in Japan revealed by the social health insurance-based genetic testing of 10K patients
Human Genetics (2022)
-
Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss
Human Genetics (2022)
-
Insights into phenotypic differences between humans and mice with p.T721M and other C-terminal variants of the SLC26A4 gene
Scientific Reports (2021)
