
Abstract
Stressful stimuli in healthy subjects trigger activation of a consistent and reproducible set of brain regions; yet, the notion that there is a single and constant stress neuromatrix is not sustainable. Indeed, after chronic stress exposure there is activation of many brain regions outside that network. This suggests that there is a distinction between the acute and the chronic stress neuromatrix. Herein, a new working model is proposed to understand the shift between these networks. The understanding of the factors that modulate these networks and their interplay will allow for a more comprehensive and holistic perspective of how the brain shifts ‘back and forth’ from a healthy to a stressed pattern and, ultimately, how the latter can be a trigger for several neurological and psychiatric conditions.
Similar content being viewed by others

What is stress?
As pointed out by Hans Selye1 ‘Stress is a scientific concept which has received the mixed blessing of being too well known and too little understood’. Indeed, there is still no perfect definition of stress that is commonly viewed as the brain’s response to a demand and/or challenge (from now on here referred to as stressors). Stressors can be real or perceived. More so, stressors are not only subject dependent (value attribution), but also have distinct temporal dynamics (recurring, short term or prolonged) and may vary in their intensity (or at least in the individual’s perception of it). That is, stressors can be mild and relatively harmless or result from major events and may have immediate and/or long-term effects on the subject’s well-being. Thus, it is basically unavoidable that most individuals have/will feel stressed, at least from time to time.2 However, it is also of critical importance to highlight that not all stress is ‘bad’ and/or detrimental. In fact, stress has been crucial to our very own survival as a species, being intrinsically linked to evolution: survival through adaptation. Indeed, all animals, and even other organisms, such as plants, have a stress response; however, as with other aspects in life, if the stress response is not moderate and controlled, it may cause harm. This prolonged, maladaptive, stress response is the focus of this review.
Some characteristics of stressors (or stress response) are determinant variables for the installation of the maladaptive response to stress: timing, individual variability, predictability and controllability (Figure 1). Timing, viewed not only as the temporal dynamics of the stress effects in the brain, but also how the brain will orchestrate the response to stressors in different states, will be in the center of this review. Individual variability is also critical as different individuals experience stress in different ways. Symptoms vary from anxious and/or depressed mood, anger and/or irritability to digestive or skin complains or even immunosuppression.3 Importantly, there are also significant variations in the way distinct subjects cope with stress and this is a critical element for the establishment of maladaptive stress and for its impact on mental and physical health.4 Predictability, given that challenges are common events, the stress response has evolved in order to be tightly controlled and regulated. In other words, if an individual faces repeatedly the same stressor, the organism typically develops an adaptive response.5, 6 This is of the utmost relevance, particularly in the (translational) approaches used to study stress. A substantial number of studies use protocols of prolonged stress composed by a single stressor; these typically will not only measure the response to that stressor, but also represent the capacity of the subject to develop an adaptation to that stressor. The fourth factor to have into consideration is controllability. As shown by work from different laboratories,7, 8, 9 to have, or not to have, control over the stressor is critical for the installation of its detrimental effects. In fact, there are elegant studies reporting that animals with control over the stressor do not display any major signs or symptoms of stress.7, 8, 9 Thus, the monitoring of these factors is critical for the correct interpretation of the effects of stress in the brain.

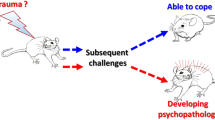
Proposed working model for a more dynamic, comprehensive and holistic perspective on how the brain shifts ‘back and forth’ from a healthy to a stressed pattern. In this working model, there are independent, although interacting, steps ((1) response; (2) susceptibility; (3) transition and (4) maintenance) that explain the dynamics from the acute stress condition to the chronic stress brain construct; here the critical questions related with the determination of the key nodes for the transition to chronicity and ‘points of no-return’. Whereas the response to stress depends on some factors (timing, individual variability, predictability, controllability), the transition to chronicity and the recovery are dependent on multifactorial determinants of individual susceptibility/resistance; in this case, the major research challenge is the ability to determine predictors of such individual response patterns.
Yet, these are not the only critical aspects to understand how stress can affect, and shift, the structure of the brain. In fact, the concepts of multistability, metastability and criticality will be key to understand how an individual brain network shifts (learns): depending on the individual’s initial repertoire (see below), adaptive changes are governed by a shift mechanism (or a bi- or tri-furcation mechanism),10, 11 where distinct routes occur. These alternative routes depend on each other and may be viewed as two successive phases of the learning/shifting process. A possible reason is that a minimum level of multistability (attained through the bifurcation mechanism) is needed to evolve gradually through the shift route; only later does the shift mechanism kick in giving rise to gradual behavioral change.10, 11, 12
Obviously, brain circuits can become unstable leading to the emergence of novel states. Multistable coordination dynamics confers a capacity on the brain to lock into one of several available patterns. It goes without saying that locking in and switching capabilities can be adaptive and useful, or maladaptive and harmful. In coordination dynamics, metastability is the simultaneous realization of two competing tendencies: the tendency of the individual components to couple together and the tendency for the components to express their independent behavior. Metastability emerges from multistability and, importantly, in the metastable brain, the activity of individual elements obeys neither the intrinsic dynamics of the elements nor the dynamics dictated by the assembly.10
Systems operating at the critical point of transition between ordered and random behavior are metastable with respect to a set of control parameters, and are capable of rapid qualitative change in response to fluctuations of external input.13 At or near the point of phase transition, the systems exhibit complex patterns of fluctuations on all scales of space and time, this being one of the indicators of an impending phase transition.14 Systems at criticality exhibit an optimal dynamical range for information processing, but shift as a result of plastic adaptation of synaptic strengths.15 These attributes of criticality are the reason for its rapid switching between different cooperative neuron collectives. A remarkable consequence of phase transitions is the generation of qualitative novelty in the form of networks with new properties.13
A new model for understanding stress disorders
Stressful stimuli in healthy subjects give rise to a consistent and reproducible activation of a set of brain regions16, 17, 18 (Figure 2). This can be considered as the stress ‘neurosensorial-matrix’. It is of importance to note that that this network is necessary, and probably also sufficient, for stressor perception and valuation. The nature of the stressor determines the sensorial pathway that is initially recruited. If the stressor is of physical nature (for example, a painful stimuli, hypovolemia, exposure to inflammatory cytokines, hypoglycemia), activation of the brain stem nuclei, or of circumventricular organs, takes place. These will, via ascending projections, ultimately activate corticotropin-releasing hormone and arginine vasopressin (AVP) releasing neurons in the paraventricular nucleus of the hypothalamus that control the release of adrenocorticotropin hormone in the anterior pituitary and, in turn, the release of corticosteroids in the adrenal cortex. If the stressor is a psychosocial stimulus, activation may occur in the amygdala, hippocampus and/or frontal cortex, among other limbic brain structures that modulate the activity of distinct nuclei in the bed nucleus of the stria terminalis or of the nucleus of the solitary tract, dorsomedial hypothalamic nucleus, arcuate nucleus or peri-paraventricular nucleus zone and, subsequently, the activity of the paraventricular nucleus (Figure 2). These constitute some of the initial nodes/networks of activation in response to stressors.16, 18 Understanding how these acute stress-related networks operate is probably helpful in uncovering pathways mediating pathological stress-related conditions. Therefore, it is not surprising that many tried to unravel functional properties and distinctions between the activated areas by studying which areas better correlate with stress intensity and which are better modulated by the exposure to previous stressful stimuli.

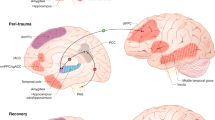
Schematic representation of some of the brain areas, and some of their connections, controlling one of the main aspects of the stress response: the activation of the paraventricular nucleus (PVN) of the hypothalamus. On the left panel, the normal pattern of connectivity that controls the response in an acute stress condition is illustrated. On the right side, a scheme of the altered pattern of connectivity in a chronic stress situation, where some of the major corticolimbic areas that modulate the stress response suffered a significant morphophysiological change that leads to a permanent shift in their pattern of connectivity (the stressed neuromatrix) that determines an altered behavior per se but also an inadequate control of the stress response. Please note that only some of the areas (critical nodes) that are instrumental for this shift (prefrontal cortex (PFC), central amygdaloid nucleus (CeA), amygdala (AMY: medial amygdaloid nucleus (MeA), ventral hippocampus (vHIP)) and their changes, are represented, given that many are still to be revealed. ARC, arcuate nucleus; BST, bed nucleus of the stria terminalis; DMH, dorsomedial hypothalamic nucleus: ventrolateral (vl) and dorsomedial (dm); E, epinephrine; NE, norepinephrine, NTS, nucleus of the solitary tract; peri-PVN, peri-paraventricular nucleus zone. Please see refs. 5, 16, 18 for further details.
However, there is growing evidence that with time, the way the brain deals with stressors, in terms of regions and patterns of activation, suffers a remarkable shift, suggesting that there is a distinction between the acute and the chronic neuromatrix. In fact, in the chronic maladaptive stress state, in contrast to acute or subacute states, there is an involvement of additional neurons with distinct projections, resulting in unique modifications of stress control nodes and networks (discussed in detail below). Moreover, this transition from acute to chronic stress involves a time-dependent neural structural and functional reorganization, initiating a series of events that potentiate one neuronal pathway at the cost of another. Thus, anchored in these premises, a new working model is here proposed (Figure 1); in this model there are independent, although interacting, steps that are modulated by factors that may explain the dynamics of the chronic stress brain construct: (1) susceptibility; (2) response and initial injury; (3) transition to chronicity; and (4) maintenance of a ‘stressed-brain’.
This model was inspired in research developed in the pain field by Melzack19 and Apkarian et al.20 More specifically, in this chronic pain there are specific brain areas and properties that are reliably linked with distinct chronic pain conditions; this pattern of activity is the result of a long-term and continued condition-specific reorganization of the brain across chronic pain that justifies the notion that chronic pain is a maladaptive neuropathological disease state.21, 22 Similarly, and also because of the commonality between the neuronal networks in chronic pain and chronic stress,22, 23 a model that explains how the temporal component emerges from the interplay between the network where the stressor was first perceived/processed and the networks that convey the experience-related reorganization is herein suggested; such interplay will reorganize the brain circuitry to distinct states, some of which are common to the ones observed in clinical conditions for which stress is a trigger factor. As a result, in the chronic stress stage, perception and salience of a stressor is a modified emotional and hedonic construct, where threat/value assessment and memory traces of stressful experiences are incorporated, eventually in an ‘altered mode’. Indeed, according to this model, the transition from acute to chronic stress also entails a transition in the salience of a stressor from a simple sign of external threat/challenge into a pathological construct; this can be of relevance to understand how stress triggers several psychiatric conditions, namely depression and post-traumatic stress disorder.
Susceptibility or predisposition to maladaptive stress
As indicated before, there is a remarkable variability in the individual response and predisposition to the effects of stress. This variability has a multifactorial origin, but certainly genetic and epigenetic mechanisms are implicated in it (Figure 3). It is well established that genetically transmitted patterns of reaction to stressors are highly preserved within species because they are critical for survival and evolution.24, 25 More recently, several lines of experimental evidence have also shown the relevance of epigenetic mechanisms in the programming of stress brain circuits.26, 27, 28, 29 Indeed, data obtained in various rodent models, such as models of prenatal and perinatal stress, maternal deprivation or separation and variation in maternal care, have revealed that exposure to stressful conditions in early life may lead to neuroendocrine perturbations later in life, some of them proposed to be transgenerationally transmitted.30, 31

Schematic representation, along with different stages of neurodevelopment, of emblematic (epi)genetic determinants of individual susceptibility/resistance to develop a stressed neuromatrix. 11βHSD2, 11β-Hydroxysteroid dehydrogenase type 2; CRF, corticotropin-releasing factor; GR, glucocorticoid receptor; H3K9Ac, H3 lysine 9 acetylation; HPC, hippocampus; PVN, paraventricular nucleus of the hypothalamus.
Prenatal stress, or excess exogenous glucocorticoid exposure, have been consistently linked to adverse health outcomes including low birth weight, neuroendocrine dysfunction and increased risk of infectious, cardiometabolic and psychiatric diseases in later life (for reviews, see refs. 25, 27, 28, 29, 30, 31, 32). These observations derive from both animal and human studies and are concomitant with alterations in the activity of the hypothalamic–pituitary–adrenal (HPA) axis.28, 29 Stress programming, however, extends further into early postnatal life. Data from models of impairment of mother–infant interaction reveal a disruption in neuroendocrine regulation(s) as a consequence of the downregulation of the hippocampal glucocorticoid receptor (GR) and the hypothalamic corticotropin-releasing hormone, resulting in a HPA overactivation.33, 34 Such early stress-induced neuroendocrine alterations are linked to behavioral problems and/or disease-state in adulthood, such as impaired memory, learning and anxiety- and depressive-like behaviors.35 Lasting effects associated with natural variations in postnatal maternal care in rodents (namely, high or low levels of licking and grooming) have also been noted. In adulthood, male rats reared by high-licking/grooming dams exhibit lower levels of stress response, better cognitive performance and increased exploratory activity as compared with the offspring of low- licking/grooming dams.36, 37, 38 Nonetheless, more recent studies have proposed that negative experiences early in life might also confer some preparation to face stressors later in life.39 Certainly, the interplay of early-life events with long-lasting programming of brain circuits is much more complex than what preliminary data seem to have indicated, involving crucial ‘check and balances’ of individual variability, predictability, controllability and timing.
Epigenetic mechanisms, which started to be revealed in this context in the past two decades, seem to operate and be decisive contributors in mounting the stress response.40 It has been shown that these early-life events trigger a developmental deregulation of epigenetic pathways that result in discrete or genome-wide changes in gene expression in various tissues, including the brain. These may influence the connectivity and functioning of neural circuitry and conferring a risk for both psychiatric and physical disorders in later life.41 The observation of an increased methylation of a CpG-rich region in the promoter and exon 1F of the GR gene (NR3C1) in the cord blood of newborns of mothers with depressed mood during the third trimester of gestation constitutes one of the first examples in humans of a functional consequence of epigenetic variation on stress reactivity.42 Importantly, this pattern on NR3C1 gene methylation, which occurred only in the offspring, was correlated with levels of response to stress throughout and beyond infancy.42 Similarly, increased methylation of NR3C1 promoter was also observed in childhood-abused suicide victims in comparison with nonabused individuals; this was also linked to decreased nerve growth factor-IA-inducible gene transcription.43 These findings suggest a common effect of parental care in both rodents and humans on the epigenetic regulation of hippocampal GR expression. Moreover, to illustrate other potential effects of stress on the epigenome, there is evidence that the exposure to maternal depressed mood during the second trimester of gestation leads to decreased levels of methylation in the promoter of the SLC6A4 gene, which encodes for the serotonin transporter, in maternal peripheral leukocytes and in offspring umbilical cord leukocytes.44 Similar epigenetic effects have also been observed in cord blood as a consequence of pregnancy-related anxiety.45 Finally, prenatal glucocorticoid (GC) exposure also leads to differential methylation of dopamine receptor D2, suggesting that different neurotransmitter systems may be programmed by early-life stressors.46
Shifts in glucocorticoid levels affect globally gene transcription and cell function; these are also likely associated with epigenetic changes in a number of tissues. The GR has been shown to induce stable demethylation in and around glucocorticoid receptor binding sites, leading to an increased transcriptional sensitivity of the target gene.47 A similar mechanism may also mediate the lasting effects of childhood abuse on DNA demethylation at intronic glucocorticoid response element of the FKBP5 gene, a co-chaperone regulating GR sensitivity, in both peripheral blood cells and in a hippocampal progenitor cell line.48 Several mechanisms have been proposed for this transcription factor-guided active demethylation and involve protein–protein interactions with methyl-DNA binding proteins and DNA repair mechanisms as well as an intermediate introduction of hydroxymethylation marks.49, 50, 51, 52
Epigenetic modifications in response to traumatic experience and stress are emerging as important factors in the long-term biological trajectories leading to stress-related psychiatric disorders, reflecting both environmental influences and individual genetic predisposition.49 In addition, epigenetic modifications are now recognized to be highly dynamic and also occurring in fully differentiated neurons. Such new form of plasticity has been implicated in the formation and function of synapses in the developing and adult brain53 and in synaptic remodeling in stress disorders.54 An emerging view also supports the existence of a complex epigenetic modulation of the adult neurogenesis process.55 Indeed, it has been postulated that epigenetically controlled reprogramming restricts developmental options in proliferation and terminal cellular differentiation during neurogenesis. In this way, environmental stressors can affect the epigenetic regulation of adult neurogenesis, mediating neurogenesis imbalances that participate in the development of depressive symptomatology.56, 57, 58 In parallel, there have been advances in our knowledge on the regulation of epigenetic modifications and, as an example, it is now established that dynamic epigenetic regulation of the glial cell-derived neurotrophic factor (Gdnf) promoter plays an important role in determining both the susceptibility and the adaptation responses to chronic stressful events.59 It is, therefore, not surprising that later postnatal stressful experiences may also have potential for long-term programming and to increase the risk of developing physical and mental problems in adulthood (reviewed in refs. 60, 61, 62, 63, 64), namely when facing subsequent stressors later in life.65
Obviously, genetic variability is influenced by other mechanisms. In fact, certain polymorphisms are reported to change HPA axis reactivity and stress response; for example there are single nucleotide polymorphisms in the mineralocorticoid receptor gene,66 glucocorticoid receptor66 and arginine vasopressin receptor 1b (AVPR1b) gene single-nucleotide polymorphisms.67 Of note, such changes in stress responses are known to have important implications for the risk of developing mental disorders.
In summary, these observations show that previous exposure to stressful conditions induce substantial biological changes that modify the maturation of systems involved in allostasis (active process of adaptation and maintaining homeostasis), particularly those of the stress neuromatrix. This supports the notion that each individual is endowed with a particular stress response pattern and, more relevant, that the subsequent activation of this network later in life may confer a particular predisposition for the development of maladaptive stress-related disorders. This opens new avenues of research in the stress field (Figure 1): can the predictors of stress maladaptive responses be determined? If yes, can these risk factors be modulated? These are certainly open (and exciting) research questions for the field to tackle in the next decade. But now, it is important to review the networks implicated in the initial stress response and the transition to a stressed neuromatrix.
The roadmap from the initial stress injury to chronicity
As discussed previously, there are initial nodes/networks of activation in response to stressors.16, 18 Moreover, the activation of the HPA axis is a known target of programming effects that may result in distinct susceptibility to stressors. Such may translate into distinct levels of corticosteroids produced both in basal conditions and in response to stressful stimuli. Ultimately, these variations in the levels of corticosteroids, and other stress hormones, either direct or through indirect alterations, will trigger other nodes in the central nervous system and drive a new set of changes in the stress neuromatrix.
The hippocampus as the starting point of mapping of stress effects in the brain
In this scenario, the best way to map the activational effects of stress in the brain is to take into account the pattern of distribution of stress-mediating receptors, namely corticosteroids,68 that are important (although not exclusive) mediators of maladaptive response.2, 5 This exercise clearly highlights the hippocampal formation, rich in corticosteroid receptors,68 as a target of the stress. And, indeed, much of the research focused on the effects of stress in the brain has been directed to the hippocampal formation. Almost 30 years ago, Sapolsky et al.69 ‘kick-started’ the field with the notion that chronic stress disrupts hippocampal structure and function, eliciting a vicious cycle that results in an unabated secretion of GCs. The central idea indicated for the loss of corticosteroid receptor-bearing hippocampal neurons after prolonged exposure to high levels of GC,70 resulting in a loss of the feedback inhibition that this brain region drives over the HPA axis. Subsequently, this view was strengthened by observations that GC can directly promote apoptosis71 and render hippocampal neurons more vulnerable to both excitotoxicity72, 73 and oxidative stress.74 Importantly, links between high GC levels and hippocampal degeneration have also become evident from magnetic resonance imaging studies in humans. In fact, for instance, GC hypersecretion, resulting from either Cushing’s syndrome75 or recurrent major depression,76 triggers hippocampal atrophy and declarative memory deficits.77 Notably, cognitive improvements are seen after remedy of the hypercortisolismic state in Cushingoid subjects.78 More so, there is evidence toward restoration of hippocampal volumes in depressed patients who received pharmacological treatment.79, 80 Of note, the impact of stress or excessive GC exposure in the hippocampal formation is not confined to loss of neurons, as several studies have also shown that stress exposure strongly reduces neurogenesis in the subgranular zone of the hippocampal dentate gyrus.81, 82, 83
Whether the variation in neuron numbers is of relevance to the stress effects in the brain has been disputed,84 and the emerging view is that the plasticity of brain network in response to stress involves other mechanisms. A much more common, faster and dynamic plastic event in neuronal adaptation is the synaptic remodeling of neurons, including changes in synaptic proteins, astroglia contacts at the tripartite synapse and alterations in the number of dendrites and in the type of spines. The idea that stress impairs brain function by compromising neuronal plasticity stems from reports that chronic stress or prolonged exposure to high GC levels impair cognitive performance and trigger dendritic atrophy and synaptic loss in dorsal hippocampal circuits;85, 86, 87, 88 interestingly, more recently it was shown that an opposite phenomenon takes place in the ventral hippocampus,89 a critical node to stress and emotional response control.90 Notably, in this process two distinct outcomes can occur: if the stress context is sustained, changes at the dendritic and synaptic level tend to evolve to more definite states in neuronal structure and function, progressing along transsynaptical pathways;5 in contrast, in conditions in which the context that triggered the stress response is altered (either by an intervening stress-free period and/or through therapeutic intervention), these changes are largely reversible and are associated with functional rescuing.87 Thus, the transient effects of stress/GC on the structure and function of the hippocampus, as well as in other brain regions, tend to support the view that dendritic atrophy and synaptic changes, rather than variations in neuronal numbers, are the critical mechanism(s) through which the transition from the initial insult of stress to the chronicity phase takes place; a good example of this transition and temporal dynamics is seen in the stress effects on depressive-like behavior.91, 92, 93
From the hippocampus to other brain regions
As a result of this view highlighting the relevance of the interconnectivity patterns in dynamic neuronal networks, a growing interest in generating systematic maps of stress-responsive brain circuits has developed.5 These efforts take into account the classic slow actions triggered by stress/corticosteroids through cytoplasmatic receptors located in the cell membrane94 as well as the novel fast actions of corticosteroid receptors.94 They also include other stress modulators operating in the hippocampus and in brain regions to which the hippocampus is interconnected, namely through changes in glutamate release95 that trigger distinct glutamate receptors96 and contribute to a better understanding of the events that occur from the perception of stress to the adaptive neuroendocrine and behavioral responses. Certainly, the direct effects mediated by receptors are of relevance for the shifts in network structure and function, but ultimately there are other indirect effects resulting from the changes in the pattern of the activity of nodes within networks that per se will lead to changes in cascade in other nodes/networks. These effects on neuronal (or better said, on neuroglial) networks are likely to explain why the disruptive effects of stress are generally not restricted to a single location or function. For example, stress initially interferes with hippocampus-dependent declarative memory but, eventually, also impairs frontocortical-dependent cognitive functions (for example, working memory, behavior flexibility and decision making)97, 98, 99 as well as behavioral domains that are regulated by multiple brain areas (for example, mood, anxiety, fear).91, 100 Importantly, these stress-induced behavioral deficits are underpinned by the morphological reorganization of dendrites, spines and synapses in the medial prefrontal cortex (mPFC),98, 101, 102, 103 orbitofrontal cortex,97 dorsal and ventral striatum,46, 97, 104 amygdala100, 105 and bed nucleus of the stria terminalis.106
Interestingly, at this level of interconnectivity it is possible to recognize the spatiotemporal and step-wise manner by which stress disrupts behaviors that require more than one brain area. A previous study demonstrated that whereas acute stress (3 days) in rats only impairs spatial reference memory (a hippocampal-dependent function), chronic stress (28 days) produces impairments in both spatial working memory and behavioral flexibility (functions in which the mPFC is implicated).98 These structural alterations are paralleled by eletrophysiological changes, namely by decreased theta coherence (crucial for performance in spatial working memory and long-term memory107, 108), in the hippocampal-to-PFC link.109 Altogether, these findings point to the high sensitivity and/or earlier response of the hippocampus to the deleterious actions of this stress protocol; thereafter, the impact of stress progresses to the mPFC. The progressive pattern of the stress disruption within one brain region or within different brain regions is further supported by other studies. One example derives from studies in the mPFC where changes in spine and dendritic morphology in the apical trees of pyramidal neurons, namely in the infralimbic compartment, may be seen after acute exposure to stress.110, 111 Importantly, these structural changes are also accompanied by resistance to and retrieval of fear extinction111, 112 and by eletrophysiological alterations that also display a temporal progression.113 Another example is the demonstration that the effects of stress on the corticostriatal networks evolve in the first weeks of exposure to stress; specifically, electrophysiological alterations are first observed in the mPFC, and then progress to the dorsomedial striatum. As a result, the activity of corticostriatal associative network decreases in favor of the activity of sensorimotor network, underlying the observation that stress promotes habit formation.97 Notably, similar observations were described in the amygdala-to-PFC projection,114 highlighting the broad, but specific, effects of stress on neuronal networks. But even more important is the notion that this transsynaptic evolution of stress effects with time also applies to its implication in pathological processes. For example, regarding the role of stress as a risk factor of Alzheimer’s disease, a recent study reveals that the stress-induced amyloid precursor protein misprocessing and abnormal Tau hyperphosphorylation, characteristic neuropathological biomarkers of Alzheimer’s disease, are first observed in the hippocampus and parahippocampal regions, from where they spread to frontocortical areas.115
The unexplored areas of the stress neuromatrix
One critical question is whether these stress-triggered effects are (1) reversible and/or (2) whether there is a ‘point of no return’ (Figure 1). Unfortunately, although a few laboratories have been exploring the issue of reversibility, none has specifically addressed the establishment of the critical ‘point of no-return’, where changes become irreversible. More precisely, it was shown that similarly to the hippocampus87 the stress-induced changes in the PFC morphology are reversible when (young) animals are allowed a period of recovery after stress.116, 117, 118 Of note, the same experimental protocols failed to induce recovery in older animals.118 This indicates that the critical point of reversibility is probably influenced by several factors, one of which is age(ing) in itself. Also relevant is the observation that the spontaneous recovery of plasticity can be augmented by antidepressant drugs,91 including fast-acting antidepressants,119, 120, 121 a finding that may be relevant in the context of pharmacological interventions for the management of stress-induced disorders in which impaired neuroplasticity is implicated.122 In humans, it has also been shown that the impact of stress in corticostrial networks (both at structural and functional levels) is reversible,123 but that some sequel of the detrimental effect of stress on resting-state networks were still present after a stress-free period.124, 125 Importantly, therapeutic interventions, namely the use of antidepressants, are also known to produce important reorganization in the neuronal circuits,126, 127 relevant in the context of the stressed neuromatrix. As for the point of no return, its clinical relevance and whether all individuals experience it (and a similar or distinct level) are open questions for which future research is needed. Such studies should not only include brain functionality analysis of phase transition, but also longitudinal imaging studies where the impact of stress (and stress recovery) in brain networks can be followed in association with functional (endocrine, electrophysiological and behavioral) outcomes. Such approaches will determine the different factors influencing the point of no return in distinct individuals to stress (see more in refs. 116, 128) and, certainly, affect the way we deal with stress-related disorders.
The design of better, and eventually customized, therapeutic interventions that may induce recovery or remediation from stress exposure is critically dependent on our knowledge of the mechanisms underlying stress detrimental effects.129, 130 A large number of studies have been done on the molecular and cellular mechanisms mediating its effects at the synapse, in the extracellular matrix, at the cascades triggered by distinct corticosteroids receptors, in the production of neurotrophins and pathological mechanisms (reviewed in refs. 2, 5, 131, 132, 133). Herein, a couple of recent findings will be highlighted. The first relates with the dual effects of stress changes at the synaptic levels (Figure 4). On one hand, acute stress, by activating GRs, activates serum-and-glucocorticoid-inducible kinase (SGK) and increases Rab4 levels, augmenting the trafficking and function of NMDA (N-methyl-D-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors and leading to potentiated synaptic transmission.134 On the other, chronic stress reduces AMPA and NMDA receptor expression and synaptic transmission through the enhancement of ubiquitin/proteasome-mediated degradation of some of its subunits (GluR1 and NR1), a process that is under the control of the E3 ubiquitin ligase Nedd4-1 and Fbx2, respectively, leading to synaptic dysfunction and eventually synaptic loss134 (Figure 4). The second set of exciting findings in the field relate with unbiased proteomic and lipidomic analysis. A proteomic study using a dynamic approach135 showed that prenatal stress exposure led to two distinct patterns of changes in protein expression: some were transiently changed, whereas others were consistently altered. These findings support the progressive nature of stress changes in the brain, namely in proteins implicated in calcium homeostasis, redox status, secretory protein synthesis and glucose or energy deprivation. Although this study represents a whole-brain analysis, other studies on specific brain regions have also largely confirmed the impact of stress in proteins implicated in these functions,136, 137 even at the synaptic level138 and with potential predictive value for vulnerability to stress-related disorders.139 In addition, more recently, and largely due to the technical advances in lipid analysis, the impact of stress in brain lipidomics has been revealed. Specifically, it was shown that the PFC and the hippocampus presented significant alterations in their lipidome, with an impairment in sphingolipid and phospholipid metabolism.140 The identification of impaired lipid signaling pathways opens a new avenue of potential targets of stress pathology. Remarkably, decreasing the levels or inhibiting the lipid modulating enzyme acidic sphingomyelinase prevented chronic stress-induced behavior and pathological hippocampal alterations.141

Schematic representation of the different levels that anchor the transition from a healthy to a stressed neuromatrix. (a) At the molecular and synaptic level, there is growing evidence that stress has a dual effect on the glutamatergic transmission in prefrontal cortex (PFC) pyramidal neurons: on one hand, acute stress, by activating glucocorticoid receptors (GRs), increases serum-and-glucocorticoid-inducible kinase (SGK) and Rab4 that augments the trafficking and function of N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and leads to potentiated synaptic transmission; on the other, chronic stress reduces AMPA and NMDA receptor expression and synaptic transmission through the enhancement of ubiquitin/proteasome-mediated degradation of some of its subunits (GluR1 and NR1), a process that is under the control of the E3 ubiquitin ligase Nedd4-1 and Fbx2, respectively. (b) At the neuroanatomical level, the progressive exposure to stress triggers a morphological reorganization of dendritic arbors and spines; as an example of the complexity of the structural reorganization, although in the dorsal hippocampus chronic stress induces an atrophy of apical dendrites in CA3 pyramids, the opposite occurs in the ventral hippocampus. (c) At the functional level, an example of a power spectral density map in ventral hippocampus of controls (healthy), acute stressed (acute) and chronic unpredictable stressed (chronic) rats for delta (1–4 Hz), theta (4–12 Hz) and low gamma (20–40 Hz) frequency bands showing that exposure to stress progressively increases power in this brain node. At the behavioral level, an example of progressive spatial working memory deficits from a healthy condition to a chronic stressed state. (a) Is an illustration of results presented in refs. 134, 170, (b) in refs. 87, 89 and (c) in refs. 98, 109.
In summary, the above-described findings ultimately lead, in a yet to be better characterized spatiotemporal pattern, to a new ‘stress connectome’ paradigm. The existence of this stress neuromatrix has two distinct implications: (1) it is at the center/origin of the pattern of response to stress, of relevance for the maintenance and hyperactivation of stress nodes and networks, and (2) it facilitates the appearance of signs and symptoms typically associated with neuronal and psychiatric disorders, because these stress signaling and neuronal pathways are also implicated in the etiopathogenesis of such conditions.
The stressed connectome
Brain function results from the pattern of activity generated in interconnected brain nodes. This view is clearly observed in functional brain imaging research that permits to examine localized brain activity and integrate such activity into networks.142, 143, 144, 145, 146, 147 These approaches indicate that the brain can be regarded as a network with fast signal processing, able to synchronize. The resting brain state and its properties are recent extensions on the topic and, perhaps, some of the most exciting new developments in our understanding of the functional human brain as a dynamic network. In the next paragraphs, using the resting-state networks as an example, attention will be given to the following questions: is the chronically stressed brain a healthy brain? Even if not, it is still able to go back to a healthy state?
Recent studies show that stress increases the activation of the default mode network (DMN) at rest in different nodes, including the ventral mPFC, posterior cingulate cortex, adjacent precuneus and inferior parietal cortex. Based on previous studies highlighting the role of the DMN at rest,148, 149, 150, 151 these results suggest an augment in self-reflective thoughts and also an increased dynamic interaction between emotional processing (that is, ventral regions) and cognitive functions (that is, dorsal regions) in stressed individuals, as a result of increased activity in the anterior components of the DMN. The increases in the posterior regions of the DMN, particularly the posterior cingulate cortex and the inferolateral parietal lobes, are likely associated with longer processing of emotionally salient stimuli and episodic memory retrieval.152, 153 Interestingly, after stress recovery, a global rescuing of the resting functional connectivity in the DMN has been noted, except for connectivity of the right anterior cingulate cortex. Notably, a volumetric contraction, with specific reductions in the left posterior cingulate cortex and left and right parietal inferior regions, paralleled the increase in functional connectivity in the DMN, after chronic stress. This is likely to reflect the stress-induced atrophic effects in cortical regions, as observed in previous reports,124, 154 that appear more resistant to recovery.125
The characterization of changes in functional connectivity between brain networks subserving distinct psychophysiological functions is of relevance to understand the symptoms triggered by stress. For example, a recent study in rodents revealed an altered (increased) pattern of resting-state network connectivity in chronic stress rats.155 Similarly, in humans the finding of an increased functional connectivity between dorsal mPFC (part of the ‘dorsal nexus’) and posterior cingulate cortex in stressed participants124 has also been reported in depressed subjects156 and to be rescued by antidepressants.157, 158, 159 In addition, the observation of increased connectivity in nodes of the dorsal attention network in stressed individuals suggests alterations in emotional regulation and in vigilance and awareness, typical of stressed-induced hyperemotionality. Interestingly, the dorsal attention network did not reveal a functional recovery after the cessation of the exposure to stress,125 maintaining a sustained pattern of increased functional connectivity that might affect emotional regulation in response to future stressors or pathological conditions.
Differences in deactivation of resting-state networks after stress have also been described, particularly in the ventral attention network and DMN, that are of relevance for task control function160, 161, 162 and ‘‘salience’’ processing.163 Importantly, the greater functional connectivity found in the ventral attention network during resting-state functional magnetic resonance imaging in stressed participants104 suggests a greater difficulty in moving from more oriented, self-related processes toward a task-focused behavior. Given that the ventral attention network has an important role in cognitive control related to switching between the DMN and task-related networks,164 this has a potential impact on cognitive performance; for example, a stronger DMN deactivation in a working memory task predicts better performance.165, 166
Importantly, the triggering effect of an acute stress episode to the development of clinical disorders, namely post-traumatic stress disorder, is a matter of relevance. Several studies have addressed this issue and it seems clear that exposure to a major episode of stress is a relevant antecedent for the development of future acute and chronic forms of post-traumatic stress disorder.167, 168 The key question seems to be why some individuals develop such disorders whereas others seem to be more resilient to the acute stress episode. Several predictors (gender, previous psychiatric problem, intensity and nature of exposure to the traumatic event and lack of social support) are implicated in such variability. Importantly, individuals who face trauma do develop changes in the dynamics of functional brain networks, independently of developing clinical symptoms; some brain areas, such as the amygdala and parahippocampal cortex, distinguished post-traumatic stress disorder patients from other individuals.169 Again, issue-related multi- and metastability seem to be of relevance to understand such transitions.
In summary, altogether, these observations strengthen the view that the pattern of brain activity in chronic stress is quite distinct from the one observed in healthy conditions, reinforcing the view that a shift toward a stressed neuromatrix also underlies a transition in the salience of a stressor from a simple sign of external threat/challenge into a pathological construct.
Final remarks
Taking into account that the brain is a complex and dynamic matrix, where detailed connectivity is constantly being modified by the instantaneous experience of the organism, it becomes obvious that quantifying chronic stress as an activation of sensorial node perception and HPA outflow is simplistic and inadequate. Preliminary studies looking at the brain connectome in distinct stress states reveal both the dynamic alterations and the particular properties/responses that take place in the stressed neuromatrix (Figure 1). By integrating preclinical and clinical evidence, an overall working model for the transition from acute to chronic stress is proposed. This model can be adapted and, hence, mechanistically reflect differences between subjects experiencing stress to varying extents, including in the context of different clinical conditions.
References
Selye H . The Stress of Life. McGraw-Hill: New York, 1956.
McEwen BS, Gianaros PJ . Stress- and allostasis-induced brain plasticity. Annu Rev Med 2011; 62: 431–445.
McEwen BS . Brain on stress: how the social environment gets under the skin. Proc Natl Acad Sci USA 2012; 109: 17180–17185.
Wu G, Feder A, Cohen H, Kim JJ, Calderon S, Charney DS et al. Understanding resilience. Front Behav Neurosci 2013; 7: 10.
Sousa N, Almeida OF . Disconnection and reconnection: the morphological basis of (mal)adaptation to stress. Trends Neurosci 2012; 35: 742–751.
de Kloet ER . From receptor balance to rational glucocorticoid therapy. Endocrinology 2014; 155: 2754–2769.
Maier SF . Behavioral control blunts reactions to contemporaneous and future adverse events: medial prefrontal cortex plasticity and a corticostriatal network. Neurobiol Stress 2015; 1: 12–22.
Lucas M, Ilin Y, Anunu R, Kehat O, Xu L, Desmedt A et al. Long-term effects of controllability or the lack of it on coping abilities and stress resilience in the rat. Stress 2014; 17: 423–430.
Hartley CA, Gorun A, Reddan MC, Ramirez F, Phelps EA . Stressor controllability modulates fear extinction in humans. Neurobiol Learn Mem 2014; 113: 149–156.
Kelso JAS . Multistability and metastability: understanding dynamic coordination in the brain. Philos Trans R Soc Lond B Biol Sci 2012; 367: 906–918.
Tognoli E, Kelso JAS . Brain coordination dynamics: true and false faces of phase synchrony and metastability. Prog Neurobiol 2009; 87:, 31–40.
Buzsáki G, Draguhn A . Neuronal oscillations in cortical networks. Science 2004; 304: 1926–1929.
Werner G . Fractals in the nervous system: conceptual implications for theoretical neuroscience. Front Physiol 2010; 1: 15.
Scheffer M, Bascompte J, Brock WA, Brovkin V, Carpenter SR, Dakos V et al. Early-warning signals for critical conditions. Nature 2009; 46:, 53–59.
Beggs JM . The criticality hypothesis: how local cortical networks might optimize information processing. Phil Trans R Soc A 2008; 366: 329–343.
Ulrich-Lai YM, Herman JP . Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 2009; 10: 397–409.
Dedovic K, Duchesne A, Andrews J, Engert V, Pruessner JC . The brain and the stress axis: the neural correlates of cortisol regulation in response to stress. Neuroimage 2009; 47: 864–871.
Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC et al. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol 2003; 24: 151–180.
Melzack R . From the gate to the neuromatrix. Pain 1999; 6: S121–S126.
Apkarian AV, Baliki MN, Farmer MA . Predicting transition to chronic pain. Curr Opin Neurol 2013; 26: 360–367.
Davis KD, Moayedi M . Central mechanisms of pain revealed through functional and structural MRI. J Neuroimmune Pharmacol 2013; 8: 518–534.
Baliki MN, Apkarian AV . Nociception, pain, negative moods, and behavior selection. Neuron 2015; 87: 474–491.
Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, Polepalli JS et al. Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science 2014; 345: 535–542.
Drake AJ, Seckl JR . Transmission of programming effects across generations. Pediatr Endocrinol Rev 2011; 9: 566–578.
Meaney MJ, Szyf M, Seckl JR . Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol Med 2007; 13: 269–277.
Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmuhl Y, Fischer D et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci 2009; 12: 1559–1566.
Babenko O, Kovalchuk I, Metz GA . Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci Biobehav Rev 2015; 48: 70–91.
Rodrigues AJ, Leao P, Carvalho M, Almeida OF, Sousa N . Potential programming of dopaminergic circuits by early life stress. Psychopharmacology (Berl) 2011; 214: 107–120.
Reynolds RM, Jacobsen GH, Drake AJ . What is the evidence in humans that DNA methylation changes link events in utero and later life disease? Clin Endocrinol (Oxf) 2013; 78: 814–822.
Cottrell EC, Seckl JR . Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci 2009; 3: 19.
Mastorci F, Vicentini M, Viltart O, Manghi M, Graiani G, Quaini F et al. Long-term effects of prenatal stress: changes in adult cardiovascular regulation and sensitivity to stress. Neurosci Biobehav Rev 2009; 33: 191–203.
Nielsen NM, Hansen AV, Simonsen J, Hviid A . Prenatal stress and risk of infectious diseases in offspring. Am J Epidemiol 2011; 173: 990–997.
Lippmann M, Bress A, Nemeroff CB, Plotsky PM, Monteggia LM . Long-term behavioural and molecular alterations associated with maternal separation in rats. Eur J Neurosci 2007; 25: 3091–3098.
Korosi A, Baram TZ . Plasticity of the stress response early in life: mechanisms and significance. Dev Psychobiol 2010; 52: 661–670.
Curley JP, Jensen CL, Mashoodh R, Champagne FA . Social influences on neurobiology and behavior: epigenetic effects during development. Psychoneuroendocrinology 2011; 36: 352–371.
Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A et al. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science 1997; 277: 1659–1662.
Liu D, Diorio J, Day JC, Francis DD, Meaney MJ . Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci 2000; 3: 799–806.
Caldji C, Diorio J, Meaney MJ . Variations in maternal care in infancy regulate the development of stress reactivity. Biol Psychiatry 2000; 48: 1164–1174.
Patchev AV, Rodrigues AJ, Sousa N, Spengler D, Almeida OF . The future is now: early life events preset adult behaviour. Acta physiologica 2014; 210: 46–57.
Meaney MJ . Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci 2001; 24: 1161–1192.
Monk C, Spicer J, Champagne FA . Linking prenatal maternal adversity to developmental outcomes in infants: the role of epigenetic pathways. Dev Psychopathol 2012; 24: 1361–1376.
Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM . Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 2008; 3: 97–106.
McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonte B, Szyf M et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009; 12: 342–348.
Devlin AM, Brain U, Austin J, Oberlander TF . Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PLoS One 2010; 5: e12201.
Hompes T, Izzi B, Gellens E, Morreels M, Fieuws S, Pexsters A et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J Psychiatr Res 2013; 47: 880–891.
Rodrigues AJ, Leao P, Pego JM, Cardona D, Carvalho MM, Oliveira M et al. Mechanisms of initiation and reversal of drug-seeking behavior induced by prenatal exposure to glucocorticoids. Mol Psychiatry 2012; 17: 1295–1305.
Thomassin H, Flavin M, Espinas ML, Grange T . Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J 2001; 20: 1974–1983.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM et al. Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions. Nat Neurosci 2013; 16: 33–41.
Provencal N, Binder EB . The neurobiological effects of stress as contributors to psychiatric disorders: focus on epigenetics. Curr Opin Neurobiol 2015; 30: 31–37.
Guo JU, Su Y, Zhong C, Ming GL, Song H . Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle 2011; 10: 2662–2668.
Sultan FA, Sweatt JD . The role of the Gadd45 family in the nervous system: a focus on neurodevelopment, neuronal injury, and cognitive neuroepigenetics. Adv Exp Med Biol 2013; 793: 81–119.
Wu H, Zhang Y . Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 2014; 156: 45–68.
Van den Bergh BR . Developmental programming of early brain and behaviour development and mental health: a conceptual framework. Dev Med Child Neurol 2011; 53: 19–23.
Golden SA, Christoffel DJ, Heshmati M, Hodes GE, Magida J, Davis K et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat Med 2013; 19: 337–344.
Mateus-Pinheiro A, Pinto L, Sousa N . Epigenetic (de)regulation of adult hippocampal neurogenesis: implications for depression. Clin Epigenetics 2011; 3: 5.
Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009; 459: 55–60.
Adachi M, Autry AE, Covington HE 3rd, Monteggia LM . MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. J Neurosci 2009; 29: 4218–4227.
Jawerka M, Colak D, Dimou L, Spiller C, Lagger S, Montgomery RL et al. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol 2010; 6: 93–107.
Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T et al. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 2011; 69: 359–372.
Danese A, McEwen BS . Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiol Behav 2012; 106: 29–39.
Varese F, Smeets F, Drukker M, Lieverse R, Lataster T, Viechtbauer W et al. Childhood adversities increase the risk of psychosis: a meta-analysis of patient-control, prospective- and cross-sectional cohort studies. Schizophr Bull 2012; 38: 661–671.
Brent DA, Silverstein M . Shedding light on the long shadow of childhood adversity. JAMA 2013; 309: 1777–1778.
Kelly-Irving M, Mabile L, Grosclaude P, Lang T, Delpierre C . The embodiment of adverse childhood experiences and cancer development: potential biological mechanisms and pathways across the life course. Int J Public Health 2013; 58: 3–11.
Fagundes CP, Glaser R, Kiecolt-Glaser JK . Stressful early life experiences and immune dysregulation across the lifespan. Brain Behav Immun 2013; 27: 8–12.
Sasaki A, de Vega WC, McGowan PO . Biological embedding in mental health: an epigenomic perspective. Biochem Cell Biol 2013; 91: 14–21.
Derijk RH, van Leeuwen N, Klok MD, Zitman FG . Corticosteroid receptor-gene variants: modulators of the stress-response and implications for mental health. Eur J Pharmacol 2008; 585: 492–501.
van West D, Del-Favero J, Deboutte D, Van Broeckhoven C, Claes S . Associations between common arginine vasopressin 1b receptor and glucocorticoid receptor gene variants and HPA axis responses to psychosocial stress in a child psychiatric population. Psychiatry Res 2010; 179: 64–68.
Rosenfeld P, van Eekelen JA, Levine S, de Kloet ER . Ontogeny of corticosteroid receptors in the brain. Cell Mol Neurobiol 1993; 13: 295–319.
Sapolsky RM, Krey LC, McEwen BS . The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev 1986; 7: 284–301.
Sapolsky RM . A mechanism for glucocorticoid toxicity in the hippocampus: increased neuronal vulnerability to metabolic insults. J Neurosci 1985; 5: 1228–1232.
Crochemore C, Lu J, Wu Y, Liposits Z, Sousa N, Holsboer F et al. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol Psychiatry 2005; 10: 790–798.
Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R . Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci 1994; 14: 5373–5380.
Lu J, Goula D, Sousa N, Almeida OF . Ionotropic and metabotropic glutamate receptor mediation of glucocorticoid-induced apoptosis in hippocampal cells and the neuroprotective role of synaptic N-methyl-D-aspartate receptors. Neuroscience 2003; 121: 123–131.
Behl C, Trapp T, Skutella T, Holsboer F . Protection against oxidative stress-induced neuronal cell death—a novel role for RU486. Eur J Neurosci 1997; 9: 912–920.
Starkman MN, Gebarski SS, Berent S, Schteingart DE . Hippocampal formation volume, memory dysfunction, and cortisol levels in patients with Cushing's syndrome. Biol Psychiatry 1992; 32: 756–765.
Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW . Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 1996; 93: 3908–3913.
Starkman MN, Giordani B, Gebarski SS, Berent S, Schork MA, Schteingart DE . Decrease in cortisol reverses human hippocampal atrophy following treatment of Cushing's disease. Biol Psychiatry 1999; 46: 1595–1602.
Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP et al. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci 1998; 1: 69–73.
Frodl T, Jager M, Smajstrlova I, Born C, Bottlender R, Palladino T et al. Effect of hippocampal and amygdala volumes on clinical outcomes in major depression: a 3-year prospective magnetic resonance imaging study. J Psychiatry Neurosci 2008; 33: 423–430.
Colla M, Kronenberg G, Deuschle M, Meichel K, Hagen T, Bohrer M et al. Hippocampal volume reduction and HPA-system activity in major depression. J Psychiatr Res 2007; 41: 553–560.
Gould E, Woolley CS, McEwen BS . Adrenal steroids regulate postnatal development of the rat dentate gyrus: I. Effects of glucocorticoids on cell death. J Comp Neurol 1991; 313: 479–485.
Gould E, Tanapat P, Rydel T, Hastings N . Regulation of hippocampal neurogenesis in adulthood. Biol Psychiatry 2000; 48: 715–720.
Duman RS, Nakagawa S, Malberg J . Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology 2001; 25: 836–844.
Sousa N, Almeida OF, Holsboer F, Paula-Barbosa MM, Madeira MD . Maintenance of hippocampal cell numbers in young and aged rats submitted to chronic unpredictable stress. Comparison with the effects of corticosterone treatment. Stress 1998; 2: 237–249.
Woolley CS, Gould E, McEwen BS . Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res 1990; 531: 225–231.
Magarinos AM, Verdugo JM, McEwen BS . Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci USA 1997; 94: 14002–14008.
Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM . Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience 2000; 97: 253–266.
Cerqueira JJ, Taipa R, Uylings HB, Almeida OF, Sousa N . Specific configuration of dendritic degeneration in pyramidal neurons of the medial prefrontal cortex induced by differing corticosteroid regimens. Cereb Cortex 2007; 17: 1998–2006.
Pinto V, Costa JC, Morgado P, Mota C, Miranda A, Bravo FV et al. Differential impact of chronic stress along the hippocampal dorsal-ventral axis. Brain Struct Funct 2014; 220: 1205–1212.
Strange BA, Witter MP, Lein ES, Moser EI . Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 2014; 15: 655–669.
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA et al. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 2009; 14: 739.
Bessa JM, Mesquita AR, Oliveira M, Pego JM, Cerqueira JJ, Palha JA et al. A trans-dimensional approach to the behavioral aspects of depression. Front Behav Neurosci 2009; 3: 1.
Mateus-Pinheiro A, Pinto L, Bessa JM, Morais M, Alves ND, Monteiro S et al. Sustained remission from depressive-like behavior depends on hippocampal neurogenesis. Transl Psychiatry 2013; 3: e210.
Groeneweg FL, Karst H, de Kloet ER, Joels M . Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol 2012; 350: 299–309.
Treccani G, Musazzi L, Perego C, Milanese M, Nava N, Bonifacino T et al. Stress and corticosterone increase the readily releasable pool of glutamate vesicles in synaptic terminals of prefrontal and frontal cortex. Mol Psychiatry 2014; 19: 433–443.
Musazzi L, Milanese M, Farisello P, Zappettini S, Tardito D, Barbiero VS et al. Acute stress increases depolarization-evoked glutamate release in the rat prefrontal/frontal cortex: the dampening action of antidepressants. PLoS One 2010; 5: e8566.
Dias-Ferreira E, Sousa JC, Melo I, Morgado P, Mesquita AR, Cerqueira JJ et al. Chronic stress causes frontostriatal reorganization and affects decision-making. Science 2009; 325: 621–625.
Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N . The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci 2007; 27: 2781–2787.
Morgado P, Silva M, Sousa N, Cerqueira JJ . Stress transiently affects pavlovian-to-instrumental transfer. Front Neurosci 2012; 6: 93.
Pego JM, Morgado P, Pinto LG, Cerqueira JJ, Almeida OF, Sousa N . Dissociation of the morphological correlates of stress-induced anxiety and fear. Eur J Neurosci 2008; 27: 1503–1516.
Wellman CL . Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol 2001; 49: 245–253.
Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR et al. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience 2004; 125: 1–6.
Shansky RM, Hamo C, Hof PR, McEwen BS, Morrison JH . Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb Cortex 2009; 19: 2479–2484.
Bessa JM, Morais M, Marques F, Pinto L, Palha JA, Almeida OF et al. Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl Psychiatry 2013; 3: e266.
Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S . Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 2002; 22: 6810–6818.
Ventura-Silva AP, Pego JM, Sousa JC, Marques AR, Rodrigues AJ, Marques F et al. Stress shifts the response of the bed nucleus of the stria terminalis to an anxiogenic mode. Eur J Neurosci 2012; 36: 3396–3406.
Benchenane K, Peyrache A, Khamassi M, Tierney PL, Gioanni Y, Battaglia FP et al. Coherent theta oscillations and reorganization of spike timing in the hippocampal- prefrontal network upon learning. Neuron 2010; 66: 921–936.
Jones MW, Wilson MA . Theta rhythms coordinate hippocampal-prefrontal interactions in a spatial memory task. PLoS Biol 2005; 3: e402.
Oliveira JF, Dias NS, Correia M, Gama-Pereira F, Sardinha VM, Lima A et al. Chronic stress disrupts neural coherence between cortico-limbic structures. Front Neural Circuits 2013; 7: 10.
Brown SM, Henning S, Wellman CL . Mild, short-term stress alters dendritic morphology in rat medial prefrontal cortex. Cereb Cortex 2005; 15: 1714–1722.
Izquierdo A, Wellman CL, Holmes A . Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci 2006; 26: 5733–5738.
Wilber AA, Walker AG, Southwood CJ, Farrell MR, Lin GL, Rebec GV et al. Chronic stress alters neural activity in medial prefrontal cortex during retrieval of extinction. Neuroscience 2011; 174: 115–131.
Lee YA, Poirier P, Otani S, Goto Y . Dorsal-ventral distinction of chronic stress-induced electrophysiological alterations in the rat medial prefrontal cortex. Neuroscience 2011; 183: 108–120.
Jacinto LR, Reis JS, Dias NS, Cerqueira JJ, Correia JH, Sousa N . Stress affects theta activity in limbic networks and impairs novelty-induced exploration and familiarization. Front Behav Neurosci 2013; 7: 127.
Sotiropoulos I, Catania C, Pinto LG, Silva R, Pollerberg GE, Takashima A et al. Stress acts cumulatively to precipitate Alzheimer's disease-like tau pathology and cognitive deficits. J Neurosci 2011; 31: 7840–7847.
Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS et al. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience 2009; 164: 798–808.
Radley JJ, Rocher AB, Janssen WG, Hof PR, McEwen BS, Morrison JH . Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp Neurol 2005; 196: 199–203.
Bloss EB, Janssen WG, McEwen BS, Morrison JH . Interactive effects of stress and aging on structural plasticity in the prefrontal cortex. J Neurosci 2010; 30: 6726–6731.
Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry 2013; 74: 742–749.
Opal MD, Klenotich SC, Morais M, Bessa J, Winkle J, Doukas D et al. Serotonin 2C receptor antagonists induce fast-onset antidepressant effects. Mol Psychiatry 2014; 19: 1106–1114.
Machado-Vieira R, Soeiro-De-Souza MG, Richards EM, Teixeira AL, Zarate CA Jr . Multiple levels of impaired neural plasticity and cellular resilience in bipolar disorder: developing treatments using an integrated translational approach. World J Biol Psychiatry 2014; 15: 84–95.
Castren E . Is mood chemistry? Nat Rev Neurosci 2005; 6: 241–246.
Soares JM, Sampaio A, Ferreira LM, Santos NC, Marques F, Palha JA et al. Stress-induced changes in human decision-making are reversible. Transl Psychiatry 2012; 2: e131.
Soares JM, Sampaio A, Ferreira LM, Santos NC, Marques P, Marques F et al. Stress impact on resting state brain networks. PLoS One 2013; 8: e66500.
Soares JM, Sampaio A, Marques P, Ferreira LM, Santos NC, Marques F et al. Plasticity of resting state brain networks in recovery from stress. Front Hum Neurosci 2013; 7: 919.
Wang L, Xia M, Li K, Zeng Y, Su Y, Dai W et al. The effects of antidepressant treatment on resting-state functional brain networks in patients with major depressive disorder. Hum Brain Mapp 2015; 36: 768–778.
Dichter GS, Gibbs D, Smoski MJ . A systematic review of relations between resting-state functional-MRI and treatment response in major depressive disorder. J Affect Disord 2014; 172C: 8–17.
McEwen BS . The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron 2013; 79: 16–29.
Ponniah K, Hollon SD . Empirically supported psychological treatments for adult acute stress disorder and posttraumatic stress disorder: a review. Depress Anxiety 2009; 26: 1086–1109.
Bisson JI, Roberts NP, Andrew M, Cooper R, Lewis C . Psychological therapies for chronic post-traumatic stress disorder (PTSD) in adults. Cochrane Database Syst Rev 2013; 12: CD003388.
Popoli M, Yan Z, McEwen BS, Sanacora G . The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 2012; 13: 22–37.
Lucassen PJ, Pruessner J, Sousa N, Almeida OF, Van Dam AM, Rajkowska G et al. Neuropathology of stress. Acta Neuropathol 2014; 127: 109–135.
Sandi C . Glucocorticoids act on glutamatergic pathways to affect memory processes. Trends Neurosci 2011; 34: 165–176.
Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z . Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 2012; 73: 962–977.
Lui CC, Wang JY, Tain YL, Chen YC, Chang KA, Lai MC et al. Prenatal stress in rat causes long-term spatial memory deficit and hippocampus MRI abnormality: differential effects of postweaning enriched environment. Neurochem Int 2011; 58: 434–441.
Yang Y, Yang D, Tang G, Zhou C, Cheng K, Zhou J et al. Proteomics reveals energy and glutathione metabolic dysregulation in the prefrontal cortex of a rat model of depression. Neuroscience 2013; 247: 191–200.
Pan J, Liu H, Zhou J, Liu Z, Yang Y, Peng Y et al. Ipsilateral hippocampal proteomics reveals mitochondrial antioxidative stress impairment in cortical-lesioned chronic mild stressed rats. Curr Mol Med 2014; 14: 1186–1196.
Mallei A, Failler M, Corna S, Racagni G, Mathe AA, Popoli M . Synaptoproteomic analysis of a rat gene-environment model of depression reveals involvement of energy metabolism and cellular remodeling pathways. Int J Neuropsychopharmacol 2015; 18: pyu067.
Henningsen K, Palmfeldt J, Christiansen S, Baiges I, Bak S, Jensen ON et al. Candidate hippocampal biomarkers of susceptibility and resilience to stress in a rat model of depression. Mol Cell Proteomics 2012; 11: M111 016428.
Oliveira TG, Chan RB, Bravo FV, Miranda A, Silva RR, Zhou B et al. The impact of chronic stress on the rat brain lipidome. Mol Psychiatry 2016; 21: 80–88.
Gulbins E, Palmada M, Reichel M, Luth A, Bohmer C, Amato D et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat Med 2013; 19: 934–938.
Biswal B, Yetkin FZ, Haughton VM, Hyde JS . Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn Reson Med 1995; 34: 537–541.
Biswal BB, Mennes M, Zuo XN, Gohel S, Kelly C, Smith SM et al. Toward discovery science of human brain function. Proc Natl Acad Sci USA 2010; 107: 4734–4739.
Di X, Biswal BB . Dynamic brain functional connectivity modulated by resting-state networks. Brain Struct Funct 2015; 220: 37–46.
Rubinov M, Sporns O . Weight-conserving characterization of complex functional brain networks. Neuroimage 2011; 56: 2068–2079.
Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, Raichle ME . The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci USA 2005; 102: 9673–9678.
Marques P, Soares JM, Magalhães R, Santos NC, Sousa N . The bounds of education in the human brain connectome. Sci Rep 2015; 5: 12812.
Greicius MD, Krasnow B, Reiss AL, Menon V . Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci USA 2003; 100: 253–258.
Mason MF, Norton MI, Van Horn JD, Wegner DM, Grafton ST, Macrae CN . Wandering minds: the default network and stimulus-independent thought. Science 2007; 315: 393–395.
Raichle ME, Snyder AZ . A default mode of brain function: a brief history of an evolving idea. Neuroimage 2007; 37: 1083–1090.
Andrews-Hanna JR, Reidler JS, Sepulcre J, Poulin R, Buckner RL . Functional-anatomic fractionation of the brain's default network. Neuron 2010; 65: 550–562.
Maddock RJ, Garrett AS, Buonocore MH . Posterior cingulate cortex activation by emotional words: fMRI evidence from a valence decision task. Hum Brain Mapp 2003; 18: 30–41.
Wagner AD, Shannon BJ, Kahn I, Buckner RL . Parietal lobe contributions to episodic memory retrieval. Trends Cogn Sci 2005; 9: 445–453.
Liston C, McEwen BS, Casey BJ . Psychosocial stress reversibly disrupts prefrontal processing and attentional control. Proc Natl Acad Sci USA 2009; 106: 912–917.
Henckens MJ, van der Marel K, van der Toorn A, Pillai AG, Fernandez G, Dijkhuizen RM et al. Stress-induced alterations in large-scale functional networks of the rodent brain. Neuroimage 2015; 105: 312–322.
Sheline YI, Price JL, Yan Z, Mintun MA . Resting-state functional MRI in depression unmasks increased connectivity between networks via the dorsal nexus. Proc Natl Acad Sci USA 2010; 107: 11020–11025.
McCabe C, Mishor Z . Antidepressant medications reduce subcortical-cortical resting-state functional connectivity in healthy volunteers. Neuroimage 2011; 57: 1317–1323.
McCabe C, Mishor Z, Filippini N, Cowen PJ, Taylor MJ, Harmer CJ . SSRI administration reduces resting state functional connectivity in dorso-medial prefrontal cortex. Mol Psychiatry 2011; 16: 592–594.
Scheidegger M, Walter M, Lehmann M, Metzger C, Grimm S, Boeker H et al. Ketamine decreases resting state functional network connectivity in healthy subjects: implications for antidepressant drug action. PLoS One 2012; 7: e44799.
Dosenbach NU, Fair DA, Miezin FM, Cohen AL, Wenger KK, Dosenbach RA et al. Distinct brain networks for adaptive and stable task control in humans. Proc Natl Acad Sci USA 2007; 104: 11073–11078.
Dosenbach NU, Visscher KM, Palmer ED, Miezin FM, Wenger KK, Kang HC et al. A core system for the implementation of task sets. Neuron 2006; 50: 799–812.
Mantini D, Corbetta M, Perrucci MG, Romani GL, Del Gratta C . Large-scale brain networks account for sustained and transient activity during target detection. Neuroimage 2009; 44: 265–274.
Seeley WW, Menon V, Schatzberg AF, Keller J, Glover GH, Kenna H et al. Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci 2007; 27: 2349–2356.
Sridharan D, Levitin DJ, Menon V . A critical role for the right fronto-insular cortex in switching between central-executive and default-mode networks. Proc Natl Acad Sci USA 2008; 105: 12569–12574.
Uddin LQ, Kelly AM, Biswal BB, Castellanos FX, Milham MP . Functional connectivity of default mode network components: correlation, anticorrelation, and causality. Hum Brain Mapp 2009; 30: 625–637.
Mayer JS, Roebroeck A, Maurer K, Linden DE . Specialization in the default mode: task-induced brain deactivations dissociate between visual working memory and attention. Hum Brain Mapp 2010; 31: 126–139.
Bryant RA, Harvey AG . The relationship between acute stress disorder and posttraumatic stress disorder following mild traumatic brain injury. Am J Psychiatry 1998; 155: 625–629.
Brewin CR, Andrews B, Rose S, Kirk M . Acute stress disorder and posttraumatic stress disorder in victims of violent crime. Am J Psychiatry 1999; 156: 360–366.
Stark EA, Parsons CE, Van Hartevelt TJ, Charquero-Ballester M, McManners H, Ehlers A et al. Post-traumatic stress influences the brain even in the absence of symptoms: a systematic, quantitative meta-analysis of neuroimaging studies. Neurosci Biobehav Rev 2015; 56: 207–221.
Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS et al. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry 2011; 16: 156–170.
Acknowledgements
I thank my collaborators for support throughout the years; they have turned it into a wonderful journey. The work of the author’s lab was supported by FCT and COMPETE through the projects EXPL/NEU-OSD/2196/2013 and FCT-ANR/NEU-OSD/0258/2012 founded by FCT/MEC, by Fundação Gulbenkian through the project ‘PhenoTEMPO’ and by Fundo Europeu de Desenvolvimento Regional (FEDER) through ‘Programa Operacional Regional do Norte (ON.2–O Novo Norte), ao abrigo do Quadro de Referência Estratégico Nacional (QREN)’ and by the European Commission (FP7): ‘SwitchBox’ (Contract HEALTH-F2-2010-259772).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Sousa, N. The dynamics of the stress neuromatrix. Mol Psychiatry 21, 302–312 (2016). https://doi.org/10.1038/mp.2015.196
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2015.196
This article is cited by
-
The cortisol switch between vulnerability and resilience
Molecular Psychiatry (2023)
-
Manuelle Therapie und die zerebralen Netzwerke der Schmerzkomponenten
Manuelle Medizin (2023)
-
Adult hippocampal neurogenesis shapes adaptation and improves stress response: a mechanistic and integrative perspective
Molecular Psychiatry (2022)
-
Chronic stress causes striatal disinhibition mediated by SOM-interneurons in male mice
Nature Communications (2022)
-
Perceived stress modulates the activity between the amygdala and the cortex
Molecular Psychiatry (2022)
