
Abstract
Parvalbumin inhibitory interneurons (PVIs) are crucial for maintaining proper excitatory/inhibitory balance and high-frequency neuronal synchronization. Their activity supports critical developmental trajectories, sensory and cognitive processing, and social behavior. Despite heterogeneity in the etiology across schizophrenia and autism spectrum disorder, PVI circuits are altered in these psychiatric disorders. Identifying mechanism(s) underlying PVI deficits is essential to establish treatments targeting in particular cognition. On the basis of published and new data, we propose oxidative stress as a common pathological mechanism leading to PVI impairment in schizophrenia and some forms of autism. A series of animal models carrying genetic and/or environmental risks relevant to diverse etiological aspects of these disorders show PVI deficits to be all accompanied by oxidative stress in the anterior cingulate cortex. Specifically, oxidative stress is negatively correlated with the integrity of PVIs and the extracellular perineuronal net enwrapping these interneurons. Oxidative stress may result from dysregulation of systems typically affected in schizophrenia, including glutamatergic, dopaminergic, immune and antioxidant signaling. As convergent end point, redox dysregulation has successfully been targeted to protect PVIs with antioxidants/redox regulators across several animal models. This opens up new perspectives for the use of antioxidant treatments to be applied to at-risk individuals, in close temporal proximity to environmental impacts known to induce oxidative stress.
Similar content being viewed by others


Main
Proper cortical excitatory-inhibitory balance through GABAergic circuits is critical for supporting cognitive, affective and social functions.1, 2 Some of these inhibitory circuits are impacted in psychiatric disorders (for example, autism,3 schizophrenia and4 bipolar disorder5). In particular, alterations in parvalbumin-expressing interneurons (PVIs) are highly replicated findings in postmortem brains of schizophrenia patients,6, 7 and are reported in bipolar (BP)8 and autism spectrum disorders (ASD).9 Anomalies in PVIs therefore constitute a pathological hallmark common to several different psychiatric conditions. Elucidating mechanisms underlying the causes and consequences of these alterations may help to identify biomarkers and novel targets for the improved detection, prevention and treatment. Here, we review published data and present novel results which provide compelling evidence for the convergence of various genetic and environmental risk factors onto oxidative stress leading to PVI impairment. We propose this represents a 'common pathological pathway', which contributes to schizophrenia, and at least some forms of ASD.
Aberrant PVI circuits in psychiatric disorders
PVIs, which express parvalbumin (a high-affinity calcium-binding protein), are fast-spiking, non-adapting interneurons. Because their inhibitory terminals synapse onto the cell body and axon initial segment of their target neurons, PVIs control the output of postsynaptic neurons via feedforward and feedback inhibition. They coordinate the activity of neuronal assemblies, particularly in high-frequency synchrony.1 PVI-associated circuits shape the processing and flow of information required for sensory perception, working memory, attention, learning, memory and social behavior. They also drive the early refinement of cortical networks. Maturation of PVIs and their specialized extracellular matrix (the perineuronal net, PNN) enveloping most PVIs10 define critical periods of network plasticity during postnatal development.11
There is accumulating evidence for anomalies of PVI-associated circuits in schizophrenia, but also in BP and ASD. In schizophrenia, these alterations include reduced expression of parvalbumin, GAD67 (GABA synthesizing enzyme), Kv3.1-containing K+--channels, changes in their pre- and postsynaptic terminals,7, 12 and abnormal PNN13 known to promote interneuron maturation, synaptic/network stability14, 15 and protection against oxidative stress.16 Together with an immature developmental gene expression profile of PVIs in ASD and schizophrenia,17 this suggests a defect in the proper maturation of these interneurons. Dysfunction of PVI-associated networks may lead to impaired high-frequency neuronal synchrony,18 and contribute to altered sensory perception, social and cognitive deficits found in patients. Understanding the mechanism(s) driving PVI anomalies is therefore central to the development of novel therapeutic strategies, targeting in particular negative symptoms and cognition.
PVIs are affected by many genetic and environmental risk factors of neurodevelopmental disorders
Schizophrenia and ASD are disorders with heterogeneous etiologies, involving a large array of genetic and environmental vulnerabilities. In preclinical models, environmental risk factors (for example, prenatal maternal stress, immune challenge, hypoxia, early-life iron deficiency, maternal separation and social isolation)19, 20, 21, 22, 23, 24 and manipulations of genes associated with these diseases (for example, DISC1, DTNB1, ERBB4, NRG1, SHANK3 and CNTAP2)25, 26, 27, 28 affect PVIs. Because of their positioning within cortical networks, their activity-dependent maturation and the plasticity of their associated networks, PVIs might be highly sensitive to abnormal cortical organization. Changes in expression of parvalbumin, GAD67, or GABA receptors may thus reflect homeostatic adaptations to alterations of neuronal wiring caused by diverse independent mechanisms of genetic and/or environmental origins. Alternatively, PVI alterations induced by a large variety of genetic and environmental factors may be attributed to a single or a subset of pathological mechanism(s) common to all these conditions. For example, PVI dysfunction in prefrontal cortex could be secondary to reduced glutamatergic transmission.7, 29
Another potential mechanism could be oxidative stress, for which there is considerable evidence in schizophrenia, BP and ASD.30, 31, 32, 33, 34, 35 Novel in vivo NAD+/NADH 31P-MRS technique has confirmed redox dysregulation in schizophrenia patients.36 Indeed, PVIs are vulnerable to redox dysregulation/oxidative stress.37, 38, 39, 40 To examine the hypothesis that oxidative stress is part of a common pathway leading to PVI abnormalities, we performed a comparative immunohistological analysis of oxidative stress and PVIs in the anterior cingulate cortex (ACC) of a panel of genetic and/or environmental models relevant to schizophrenia and ASD. Oxidative stress was detected with an antibody against 8-oxo-2'-deoxyguanosine (8-oxo-dG), a marker for DNA oxidation. PVI integrity was assessed with an antibody against parvalbumin and with the lectin, Wisteria floribunda agglutinin (WFA), to label the PNN39 (for brief methods, see legend of Figure 1). Models included genetically engineered mice-bearing human copy number variations linked to increased risk for schizophrenia and/or ASD (deletion in the 22q11, 15q13 or 1q21 loci) or knockout (KO) for genes associated with these disorders (FMR1, serine racemase (SRR) and GRIN2A), rodents with deficient glutathione (GSH) synthesis (GCLM KO mice, mice with conditional GCLC KO in PVIs, rats with transient GSH depletion during development), mouse models of gene-environment interactions (early postnatal oxidative challenge in GCLM and GRIN2A KO mice), an environmental 'two-hit' mouse model of schizophrenia (maternal immune challenge+peripubertal stress), and neurodevelopmental rat models relevant to schizophrenia (neonatal ventral hippocampal lesion (NVHL) and methylazoxymethanol acetate (MAM)). All analyses were performed in late adolescent/young adult animals (2–3 months old), and the results are summarized below and in Figure 1.

Relationship between oxidative stress and PVI integrity in the ACC of 2–3 month-old animal models relevant to schizophrenia, autism and/or redox dysregulation. (a) Oxidative stress (assessed by the immunoreactivity intensity against 8-oxo-2'-deoxyguanosine (8-oxo-dG), a marker of mitochondrial DNA oxidation), number of PV-IR cells (PV cells) and number of PV cells enwrapped with a WFA-labeled PNN. # indicates models for which the presented data are already published elsewhere.39, 41 The references indexed below provide detailed descriptions of each investigated model and its control. 22q11: mice with a 22q11.2 deletion (LgDel/+)42 (n=7 animals per group); 15q13.3: mice with a 15q13.3 deletion (Df[h15q13]/+)43 (n=5, 7); 1q21: mice with a 1q21 deletion (Df[h1q21]/+) (from M Didriksen) (n=4 per group), SRR: serine racemace KO mice44 (n=5, 7); FMR1: FMR1 KO mice45 (n=7, 8); PV-GCLC: mice with conditional KO of GCLC (catalytic subunit of the key synthesizing enzyme of GSH) in PVIs40 (n=5 per group); GRIN2A: GRIN2A KO mice46 (n=7 per group); GCLM: GCLM KO mice47 (n=5 per group); GCLM GBR: GCLM KO mice treated with dopamine uptake inhibitor GBR12909 during postnatal development (P10-20)39 (n=5 per group); GRIN2A GBR: GRIN2A KO mice treated with GBR12909 during postnatal development (P10-20) (n=7 per group); ODS BSO: ODS rats treated with the specific inhibitor of GSH synthesis (BSO) during postnatal development (P5-16)38 (n=4 per group); NVHL: rats with a neonatal ventral hippocampal lesion41 (n=6, 7); MAM: rats treated on gestation day GD17 with MAM48 (n=4, 6); Poly(I:C): mice with a sub-threshold prenatal immune challenge (on GD9) with poly(I:C); +Str: poly(I:C)-treated mice stressed at preadolescence (P30-40)49 (n=5 per group). Data are depicted by the mean±s.d. (in red: animal models; in blue: their respective controls). *** P<0.001; ** P<0.01, * P<05. (b) Quantile density contours with linear regression (red) and smoothing spline (green) plots illustrating the relationships between changes in oxidative stress (8-oxo-dG-IR), in number of PV cells, and in number of PV cells with WFA-labeled PNN (PV cells+PNN) for all animal models relative to their respective controls (JMP11, SAS Institute, Cary, NC, USA). (c) As in (b) but illustrating the relationship between changes in number of PV cells+PNN and in number of PV cells for all animal models relative to their respective controls. Brief method description: perfused fixed brains from all animal models were sent to Lausanne where immunohistological preparation, image acquisition and analyses were performed blindly using the methods described previously.39 Three to four sections per animal were used for the analyses. Analyses of 8-oxo-dG-IR intensity, numbers of PV-IR cells and PV-IR cells surrounded with a WFA-labeled PNN were done in a region of interest comprising all layers of the ACC. Oxidative stress was assessed in all cells of ROI. Each animal model was compared with its own control animals. Only males were analyzed, except for the GRIN2A model where individuals from both sexes were used. On the basis of previously analyzed data,39 sample size was choosen to detect ~25% change in number of PV-IR cells and ~75% change in 8-oxo-dG intensity with a power of 80% at a significant α-value set to P=0.05. Statistical significance was tested by comparing means of the different models with their respective controls using the Dunnett’s test. When variances were not equal, we used the Welch’s test to give confidence and confirm the Dunnett’s test outcome. ACC, anterior cingulate cortex; BSO, buthionine sulphoximine; KO, knockout; MAM, methylazoxymethanol acetate; NVHL, neonatal ventral hippocampal lesion; ODS, osteogenic disorder Shionogi; PNN, perineuronal net; PV, parvalbumin; PV-IR, parvalbumin-immunoreactive; PVI, parvalbumin inhibitory interneuron; WFA, Wisteria floribunda agglutinin.
Oxidative stress and PVI integrity in preclinical models of genetic and environmental risks
Mice with the human 22q11.2 deletion (LgDel/+)
The 22q11.2 deletion represents the most common microdeletion, and causes craniofacial, cardiovascular abnormalities, immunodeficiency and cognitive dysfunctions.50 Microdeletion of this region accounts for ~0.3% of persons diagnosed with schizophrenia,51 and leads to structure and connectivity abnormalities in various brain regions including the cortex, hippocampus, and cerebellum. Mice modeling the human 22q11.2 deletion show cognitive deficits, impaired sensorimotor gating and fear conditioning.52 In addition, they have disrupted cortical neurogenesis, abnormal interneuron migration/placement, reduced density of parvalbumin-immunoreactive (PV-IR) synapses and abnormal formation of excitatory synapses.52 Our analysis shows that, relative to their control wild-type (WT) mice, adult LgDel/+ mice displayed higher oxidative stress (+125% 8-oxo-dG immunoreactivity intensity), decreased number of PV-IR cells (−22%), and PV-IR cells surrounded by WFA-labeled PNN (-38%).
Mice with the human 15q13.3 deletion (Df[h15q13]/+)
Deletion or duplication of the chromosome region, 15q11-13, cause severe neurological phenotypes. The 15q13.3 deletion confers high risk for schizophrenia,53 but also epileptic seizures,54 attention deficit, aggression, developmental delay and ASD.55 Mice with such microdeletion (Df[h15q13]/+) display several features resembling the clinical syndromes, including changes in schizophrenia relevant domains like reduced evoked gamma oscillations and seizure susceptibility.43 Relative to their control mice, adult Df[h15q13]/+ mice showed increased oxidative stress (+262% 8-oxo-dG immunolabeling intensity), lower number of PV-IR cells (-35%) and PV-IR cells enwrapped with PNN (-45%).
Mice with the human 1q21 deletion (Df[h1q21]/+)
The symptoms caused by a 1q21.1 microdeletion overlap with those seen in individuals with a 22q11.2 microdeletion. These include congenital heart defect, facial abnormalities, developmental delay, microcephaly and neuropsychiatric disorders,56 including schizophrenia.51 Compared to their control mice, adult Df[h1q21]/+ mice, whose full phenotyping is ongoing, did not show any significant oxidative stress, deficit in PV-IR cells and in WFA-labeled PNN.
FMR1 KO mice
The Fragile X syndrome results from the expansion of an unstable trinucleotide repeat, hypermethylation and transcriptional shutdown of the fragile X mental retardation 1 (FMR1) gene causing the lack of the FMR1 protein. Fragile X syndrome patients present a complex neuropsychiatric phenotype. Half of the patients meet the diagnostic criteria for ASD; many present anxiety, attention deficit, cognitive impairment and preservative behaviors.57 Rare mutations in the FRM1 gene are also associated with schizophrenia.58 FMR1 KO mice have phenotypes that resemble the human disorder45, 59 and display abnormalities in GABAergic circuits including reduced PV-IR cell density.60 In adult FMR1 KO mice, we observed a strong increase in oxidative stress (+307% 8-oxo-dG immunolabeling intensity relative to their control WT mice) together with a decreased number of PV-IR cells (-48%), and PV-IR cells surrounded by WFA-labeled PNN (−50%).
SRR KO mice
Genetic and biochemical findings suggest that SRR, the enzyme that converts L-serine to D-serine, and D-serine itself are reduced in schizophrenia.61 Low levels of D-serine, the dominant endogenous co-agonist of NMDA receptors (NMDAR) in forebrain, may thus cause NMDAR hypofunction as observed in SRR KO mice.44 Although a previous study reported no alteration of parvalbumin immunoreactivity in hippocampus, pre- and infralimbic cortices of SRR KO mice,62 our analysis revealed reduced number of PV-IR cells (−26% relative to their control WT mice) and PV-IR cells surrounded by PNN (−35%) in the ACC of adult SRR KO mice. This was accompanied by an increase in intensity of 8-oxo-dG immunoreactivity (+410%).
GRIN2A KO mice
A large genome-wide association study identified independent loci associated with schizophrenia63 that contain numerous brain-enriched genes involved in glutamatergic transmission, including GRIN2A coding for the NMDAR subunit NR2A. Mutations in GRIN2A gene have been identified in intellectual disabilities, epilepsy, ASD and schizophrenia.64 GRIN2A KO mice show learning deficits65 and altered emotional-like behaviors.66 Relative to their control WT mice, GRIN2A KO mice display neither significant oxidative stress nor PV-IR deficit. The number of PV-IR cells with PNN was however slightly reduced in KO mice (−11% relative to their control WT). However, when an additional oxidative challenge was applied during early postnatal development (from days 10 to 20), young adult GRIN2A KO mice showed significant oxidative stress (+159% relative to WT), reduced number of PV-IR cells (−17%) and PV-IR cells surrounded by WFA-labeled PNN (−36%). The oxidative insult consisted of 10-days administration of GBR12909 (GBR),39 a dopamine uptake inhibitor, leading to elevated extracellular dopamine levels and subsequent generation of reactive oxygen species (ROS) and reactive compounds.67 This treatment mimics to some extent dopamine release induced by environmental stress.68 Such GBR treatment did not have any significant effects however in WT mice.39 These findings indicate that an exogenous stress during development induces long-lasting oxidative stress along with PVI/PNN deficits in GRIN2A KO mice.
GCLM KO mice and other models of GSH deficiency
Altered levels of enzymatic and non-enzymatic antioxidants, including decrease in GSH, are reported in blood or brain of some patients suffering from schizophrenia, BP or ASD. Polymorphisms and copy number variations in genes related to the GSH synthesis and metabolism are associated with schizophrenia.50, 69, 70, 71, 72, 73, 74, 75 Moreover, subjects with high-risk genotype of the key GSH synthesizing gene GCLC presents low prefrontal GSH levels.76 Mice with a functional deletion of the modulatory subunit (GCLM) of the key GSH synthesizing enzyme constitute a model of redox dysregulation with 60–70% decreased brain GSH levels.47 These mice display neurochemical, and behavioral anomalies relevant to schizophrenia and BP.77 Relative to their control WT, young adult GCLM KO mice showed oxidative stress (+53% 8-oxo-dG immunolabeling intensity), but no PVI/PNN deficit. However, an early-life GBR-induced oxidative insult (from days 10 to 20) led to a further increase of 8-oxo-dG immunolabeling (+127% relative to WT mice), along with a decreased number of PV-IR cells (−44%) and PV-IR cells surrounded by PNN (−52%) in young adult GCLM KO mice, compared with WT controls. In osteogenic disorder Shionogi rats lacking the capacity for ascorbic acid synthesis, a transient GSH depletion induced pharmacologically with the specific inhibitor of GSH synthesis (BSO) during the postnatal period of PVI maturation (P5-P16) also sufficed to decrease the number of PV-IR cells (−13% relative to control rats) and to increase 8-oxo-dG immunolabeling (+77%) in the ACC of adult animals. Finally, a genetically induced blockade of GSH synthesis restricted to PVIs (PV:GCLC KO mice40) also led to reduced number of PV-IR cells (−26% relative to their control mice) and PV-IR cells surrounded by WFA-labeled PNN (−36%) in the ACC. Together, this demonstrates that a GSH deficiency confers a vulnerability to oxidative stress-induced PVI impairment.
Environmental 'two-hit' mouse model
Prenatal infection and exposure to traumatizing experiences during peripuberty have been associated with increased risk for neuropsychiatric disorders. Prenatal infection and psychological trauma in peripubertal life can also synergistically increase the risk for schizophrenia.78 Combining a sub-threshold prenatal immune challenge (at embryonic day E9) using the viral mimetic, poly(I:C) with sub-chronic unpredictable stress during pubescence (postnatal days 30–40) causes synergistic effects on adult mouse behaviors, neurochemistry and number of hippocampal PV-IR cells.49, 79 Here, it was found that the two environmental hits synergistically affected the oxidative stress (+190%), the number of PV-IR cells (−38%) and PV-IR cells enwrapped with WFA-labeled PNN (−63%) in the ACC of adult poly(I:C)/stressed relative to their naive control mice.
NVHL rat model
Developmental manipulations and insults inducing adult deficits in rodents provide an opportunity to explore mechanisms involved in a delayed emergence of anomalies driven by developmental alterations. A widely used tool to assess developmental trajectories of adult-onset prefrontal cortical deficit is the NVHL. This procedure yields adult animals with PFC-dependent neurochemical, electrophysiological and behavioral anomalies related to schizophrenia phenotypes, all of which emerge during adolescence.80 In young adult NVHL rats, there was a strong increase of 8-oxo-dG immunolabeling intensity (+450% relative to control sham rats) accompanied by a decrease in number of PV-IR cells (−43%) and PV-IR cells surrounded by PNN (−55%) in the ACC.41
MAM developmental rat model
Dopamine dysfunction has been regarded as the primary factor underlying psychosis. Administration in rats of the DNA-alkylating agent MAM at gestational day 17 affects hippocampal-midbrain-striatal circuits, causing an elevated subcortical dopamine function through changes in descending outputs from the medial temporal lobe.81 The MAM rat model incorporates the disruption of brain development, believed to be fundamental to psychotic disorders. Prenatal administration of MAM recapitulates a neurodevelopmental disruption leading to histological, neurophysiological, and behavioral deficits analogous to schizophrenia.81 This includes reduction of PV-IR cells within the medial prefrontal cortex and ventral hippocampus.48 Our analysis of the ACC revealed an increase of 8-oxo-dG immunolabeling intensity (+164%) in MAM relative to their control rats. In parallel, the number of PV-IR cells and PV-IR cells surrounded by WFA-labeled PNN were decreased (−44 and −49%, respectively).
Is oxidative stress causal to PVI impairments?
Overall, our analysis remarkably demonstrates that PVI deficit in the ACC was in all cases accompanied by oxidative stress. By contrast, significant oxidative stress and PVI deficit were absent in the 1q21 deletion model and GRIN2A KO mice. Young adult GCLM KO mice showed no PVI/PNN deficit despite small signs of oxidative stress. However, both GRIN2A and GCLM KO mice were susceptible to early postnatal oxidative challenge induced by GBR (from P10 to P20) as evidenced by a long-lasting increase in oxidative stress and PVI/PNN deficits. Considering all investigated animal models, increased oxidative stress correlated negatively with the relative decreased number of PV-IR cells or PV-IR cells with PNN (Figure 1b), suggesting a direct link between oxidative stress and PVI anomalies. Furthermore, there was a strong relationship between the decreases in number of PV-IR cells and number of PV-IR cells with PNN (Figure 1c), indicating that both reduction of PV immunoreactivity and weakening of PNN are coupled, thus reinforcing their relation to oxidative stress. The association between oxidative stress and PVI deficit in the medial prefrontal cortex has been also observed in other models not reported here: rodents treated with NMDAR antagonists,37, 82 isolated mice bearing a deletion of the NMDAR subunit NR1 in forebrain interneurons,83 isolated rats,23 socially defeated rats,84 mutant DISC1 mice85, 86 (Figure 2). Although we have focused on prefrontal cortex, several studies have shown that PVIs of other brain regions can be also susceptible to redox dysregulation/oxidative stress.37, 47, 84, 87 The nature of the genetic vulnerabilities and the timing of environmental insults during development might affect however differentially PVI networks across brain regions involved in cognitive, affective and social dimensions leading to heterogeneous clinical phenotypes.

A common mechanism of oxidative stress-induced PVI/PNN deficit in animals modeling genetic and/or environmental risks, and experimental disruptions of brain development relevant to schizophrenia and/or autism. Bold boxes and text represent animal models where PVI deficit has been linked to redox dysregulation/oxidative stress in the present study or in published works: 22q11: mice with a 22q11.2 deletion (LgDel/+); 15q13.3: mice with a 15q13.3 deletion (Df[h15q13]/+); 1q21: mice with a 1q21 deletion (Df[h1q21]/+), SRR: serine racemace KO mice; FMR1: FMR1 KO mice; PV-GCLC: mice with conditional KO of GCLC in PVIs; GRIN2A: GRIN2A KO mice; GCLM: GCLM KO mice; GCLM+hyperdopaminergia: GCLM KO mice treated with dopamine uptake inhibitor GBR12909 during postnatal development (P10-20); GRIN2A+hyperdopaminergia: GRIN2A KO mice treated with GBR12909 during postnatal development (P10-20); ODS+transient GSH deficit: osteogenic disorder Shionogi rats treated with the specific inhibitor of GSH synthesis (buthionine sulphoximine) during postnatal development (P5-16); NVHL: rats with a neonatal ventral hippocampal lesion; MAM: rats treated prenatally (GD17) with methylazoxymethanol acetate; Poly(I:C)+stress: mice with a sub-threshold prenatal immune challenge (on GD9) with poly(I:C) followed by chronic stress at preadolescence (P30-40); GRIN1+social isolation: isolated mice with conditional GRIN1 KO in forebrain interneurons;83 rats socially isolated;23 social defeated rats;84 ketamine administration;37 DISC1 mutant mice; and85, 86 selenoprotein P (SEPP1) KO mice87). Boxes with normal text represent animal models for which PVI deficit is very likely linked to redox dysregulation/oxidative stress. Indeed, PVI deficit and oxidative stress are reported in separate studies for the following models: prenatal stress;24, 31 maternal separation in rats;19, 31 early-life iron deficiency;20, 88 hypoxia;21, 31 DTNB1 mutant mice; and25, 89 PCP.90, 91 Mice with COX10 KO in PVIs (PV-COX10)92 and PGC1-α KO mice83, 93 are models of mitochondria impairment which likely display redox dysregulation/oxidative stress. Dotted boxes, animal models with PVI deficit for which redox dysregulation/oxidative stress may be expected based on literature data: CHRNA7 KO mice;94, 95 mice with disrupted NRG1/ErbB4 signaling. KO, knockout; NVHL, neonatal ventral hippocampal lesion; PCP, postnatal treatment with phencyclidine; PNN, perineuronal net; PV, parvalbumin; PV-IR, parvalbumin-immunoreactive; PVI, parvalbumin inhibitory interneuron.26, 96, 97
A causal effect of oxidative stress on PVI integrity or phenotype is strongly supported by the fact that oxidative stress precedes PVI deficit in the hippocampus47 and by the protective effect of antioxidants on PVIs in several models (GBR-treated GCLM mice,39 ketamine-treated mice,37 isolated rat,23 isolated mice with deletion of NR1 in forebrain interneurons,83 social defeated rats and84 NVHL rats41). Many early-life stressors relevant to environmental risk factors for schizophrenia (maternal separation, hypoxia and early-life iron deficiency) also lead to PVI deficit in medial prefrontal cortex and/or hippocampus of rodents19, 20, 21, 24 (Figure 2). Although the link between PVI deficit and oxidative stress has not been investigated in the above studies, it is a reasonable hypothesis that oxidative stress contributes to prefrontal and hippocampal PVI deficit induced by these environmental insults known to generate oxidative stress31, 88 and/or neuroinflammation.19, 98, 99
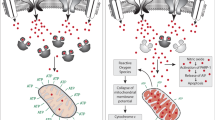
As fast-spiking neurons, PVIs contain many mitochondria to meet their high-energy demand.100 Therefore, they are vulnerable to weakened antioxidant defenses,39, 40, 87 excessive production of ROS37 and diminished mitochondrial support.92, 93 However, PVIs are also particularly susceptible to redox dysregulation/oxidative stress during their postnatal maturation when their fast-spiking properties are not yet fully developed101 and the formation of the protective PNN enwrapping them are not yet complete.16 Based on the fact that a peripubertal antioxidant treatment can abolish oxidative stress and rescue PVI/PNN deficit in the NVHL rat model,41 we propose a hypothesis in which redox dysregulation/oxidative stress halts normal PVI maturation in the prefrontal cortex without causing their death,82 possibly via epigenetic mechanisms.102, 103The deleterious effect of oxidative stress on PNN integrity16 may be also central to the failure of PV circuits to properly mature as PNN is crucial for PVI maturation and synapse formation. As consequence, oxidative stress might impair the PVI-associated network plasticity, that is, activity-dependent regulation of the number of excitatory and inhibitory synapses on PVIs.103, 104 This would be consistent with the observation of an immature-like transcriptome in PVIs of schizophrenia and autism patients.17
Convergence onto oxidative stress: interplay between glutamatergic, immune and redox systems
It is remarkable that oxidative stress is observed in so many models of genetic and environmental risks for psychiatric diseases. It is unlikely however that the mechanisms causing oxidative stress are similar in all models. Oxidative stress could be a consequence of perturbations within a number of systems known to be disrupted in these disorders, that is, glutamatergic, neuroimmune, dopaminergic, antioxidant and mitochondrial systems.105 Thus, NMDAR hypofunction causes a susceptibility to oxidative stress. This could be due to the neuronal production of IL-6 mediating NADPH oxidase activation106 and/or the weakening of the antioxidant capacities.37, 64, 83, 90 This might explain the effects observed in SRR and GRIN2A KO mice (Figure 2). Furthermore, phencyclidine administration results in hypermethylation of CpG sites in the promoter of PV gene which includes binding sites for Nrf2, the master regulator of antioxidant defense.102 This may also contribute to oxidative damage and PV deficits. Concerning the environmental 'two-hit' model, we propose that a sub-threshold immune challenge primes microglia to be activated at pre-pubertal stress49 resulting in ROS production and PVI/PNN deficits. Excess dopamine also generates ROS, which may not be efficiently neutralized when the antioxidant defenses are suboptimal as in GCLM and GRIN2A KO mice. Thus, a central role for oxidative stress is compatible with glutamatergic and dopaminergic dysregulations, as well with neuroinflammatory processes. The mechanisms by which oxidative stress and PVI deficits occur in several models that we analyzed (copy number variations mouse models, FMR1 KO mice, MAM and NVHL rat models) remain, however elusive, but may involve dysregulation within the systems mentioned above. Interestingly, the 22q11 locus contains the gene encoding COMT (a dopamine degrading enzyme) and six genes for proteins expressed in mitochondria,107 among which at least two (PRODH, and TRXR2) are implicated in the defense against oxidative stress.108 Likewise, MAM is known to decrease brain GSH levels.109
Our hypothesis that abnormal PVI-associated networks in schizophrenia and ASD has a neurodevelopmental origin and implicates redox dysregulation/oxidative stress should be however further tested in other models. For instance, mice with a deletion of α-7-nAChR, whose expression and function are affected in schizophrenia,110 display reduced expression of parvalbumin, GAD67, and NMDAR in PVIs.94 In this model, oxidative stress might also be the mechanism responsible for the PVI deficit, as activation of α-7-nAChR inhibits NF-kB-dependent pathways and mediates Nrf2-induced antioxidant responses, supporting an anti-inflammatory and neuroprotection role for this nicotinic receptor.95 Interestingly, the gene CHRNA7 coding for this receptor is located within the 15q13.3 loci and may contribute to the neuropsychiatric phenotype caused by a microdeletion of this chromosomal region.111
Likewise, impaired PVIs in mutant mice with reduced dysbindin-1 [ref. 25] could result from oxidative stress as dysbindin interacts with the antioxidant enzymes peroxiredoxins 1 and 2.89 The neuregulin-1/ErbB4-mediated pathway, whose involvement in the etiology of schizophrenia is strongly suspected, is also required for migration and appropriate wiring of PVIs in the postnatal cortex.26 Postnatal conditional KO of ERBB4 on PVIs leads to reduced glutamatergic drive and tends to diminish NMDAR function in these interneurons.96 Involvement of redox dysregulation/oxidative stress in the PVI anomalies mediated by the disruption of neuregulin-1/ErbB4 signaling remains speculative, although this cascade has been implicated in neuroprotection against oxidative stress112 and in stimulation of antioxidant defense.97
In contrast, aberrant migration of interneuron precursors and perturbed positioning of PVIs within the cortical circuits due to genetic and/or prenatal environmental factors may be mediated by mechanisms independent of oxidative stress. Manipulations of other genes associated with intellectual disability and ASD (for example, MECP2, NLGN3, Shank3; CNTNAP2) reveal diverse abnormalities within cortical GABAergic circuits.27, 28, 113, 114, 115 Both increase and decrease in number of PV-IR cells are reported. Moreover, Shank1 and Shank3B KO mice show a reduced number of PV-IR cells but no deficit in WFA-labeled PNN in the somatosensory cortex.27 In contrast to the schizophrenia models, this suggests no convergent mechanism responsible for PVI anomalies in ASD relevant models.
Conclusion
Redox dysregulation/oxidative stress is a common pathological mechanism affecting prefrontal PVIs during postnatal maturation in a strikingly diversified number of models relevant to schizophrenia and some forms of ASD. Although our analysis focused on oxidative stress-induced PVI deficit in the prefrontal cortex, this deficit may also hold for other brain regions like the hippocampus, as shown in some models of schizophrenia (for example, GCLM KO, ketamine, MAM, poly(I:C) and social defeat.22, 37, 47, 48, 84 This confluence suggests that oxidative stress could be the main agent responsible for PVI-associated network anomalies particularly in schizophrenia. The fact that peri-adolescent administration of antioxidants can restore or protect PVI integrity in some animal models provides a strong support that early antioxidant-based treatments might be beneficial to PVI-associated networks. Knowing the role of PVIs in cognition and social behavior, this could improve cognitive processing and ameliorate general functioning of patients. Such preventive intervention will require however the identification of reliable biomarkers for brain oxidative stress/redox dysregulation and PVI function in high-risk individuals.
References
Hu H, Gan J, Jonas P . Interneurons. Fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science 2014; 345: 1255263.
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011; 477: 171–178.
Zikopoulos B, Barbas H . Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci 2013; 7: 609.
Inan M, Petros TJ, Anderson SA . Losing your inhibition: linking cortical GABAergic interneurons to schizophrenia. Neurobiol Dis 2013; 53: 36–48.
Savitz JB, Price JL, Drevets WC . Neuropathological and neuromorphometric abnormalities in bipolar disorder: view from the medial prefrontal cortical network. Neurosci Biobehav Rev 2014; 42: 132–147.
Beasley CL, Reynolds GP . Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr Res 1997; 24: 349–355.
Lewis DA, Curley AA, Glausier JR, Volk DW . Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 2012; 35: 57–67.
Sakai T, Oshima A, Nozaki Y, Ida I, Haga C, Akiyama H et al. Changes in density of calcium-binding-protein-immunoreactive GABAergic neurons in prefrontal cortex in schizophrenia and bipolar disorder. Neuropathology 2008; 28: 143–150.
Hashemi E, Ariza J, Rogers H, Noctor SC, Martinez-Cerdeno V . The number of parvalbumin-expressing interneurons is decreased in the medial prefrontal cortex in autism. Cereb Cortex 2016. pii:bhw021.
Rossier J, Bernard A, Cabungcal JH, Perrenoud Q, Savoye A, Gallopin T et al. Cortical fast-spiking parvalbumin interneurons enwrapped in the perineuronal net express the metallopeptidases Adamts8, Adamts15 and Neprilysin. Mol Psychiatry 2015; 20: 154–161.
Do KQ, Cuenod M, Hensch TK . Targeting oxidative stress and aberrant critical period plasticity in the developmental trajectory to schizophrenia. Schizophr Bull 2015; 41: 835–846.
Yanagi M, Joho RH, Southcott SA, Shukla AA, Ghose S, Tamminga CA . Kv3.1-containing K channels are reduced in untreated schizophrenia and normalized with antipsychotic drugs. Mol Psychiatry 2014; 19: 573–579.
Mauney SA, Athanas KM, Pantazopoulos H, Shaskan N, Passeri E, Berretta S et al. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol Psychiatry 2013; 74: 427–435.
Bernard C, Prochiantz A . Otx2-PNN interaction to regulate cortical plasticity. Neural Plast 2016; 2016: 7931693.
Kwok JC, Dick G, Wang D, Fawcett JW . Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol 2011; 71: 1073–1089.
Cabungcal JH, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK et al. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci USA 2013; 110: 9130–9135.
Gandal MJ, Nesbitt AM, McCurdy RM, Alter MD . Measuring the maturity of the fast-spiking interneuron transcriptional program in autism, schizophrenia, and bipolar disorder. PloS One 2012; 7: e41215.
Uhlhaas PJ, Singer W . Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 2010; 11: 100–113.
Brenhouse HC, Andersen SL . Nonsteroidal anti-inflammatory treatment prevents delayed effects of early life stress in rats. Biol Psychiatry 2011; 70: 434–440.
Callahan LS, Thibert KA, Wobken JD, Georgieff MK . Early-life iron deficiency anemia alters the development and long-term expression of parvalbumin and perineuronal nets in the rat hippocampus. Dev Neurosci 2013; 35: 427–436.
Komitova M, Xenos D, Salmaso N, Tran KM, Brand T, Schwartz ML et al. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J Neurosci 2013; 33: 13375–13387.
Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J . Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun 2008; 22: 469–486.
Schiavone S, Sorce S, Dubois-Dauphin M, Jaquet V, Colaianna M, Zotti M et al. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry 2009; 66: 384–392.
Stevens HE, Su T, Yanagawa Y, Vaccarino FM . Prenatal stress delays inhibitory neuron progenitor migration in the developing neocortex. Psychoneuroendocrinology 2013; 38: 509–521.
Carlson GC, Talbot K, Halene TB, Gandal MJ, Kazi HA, Schlosser L et al. Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc Natl Acad Sci USA 2011; 108: E962–E970.
Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K et al. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 2010; 464: 1376–1380.
Filice F, Vorckel KJ, Sungur AO, Wohr M, Schwaller B . Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol Brain 2016; 9: 10.
Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011; 147: 235–246.
Le Magueresse C, Monyer H . GABAergic interneurons shape the functional maturation of the cortex. Neuron 2013; 77: 388–405.
Brown NC, Andreazza AC, Young LT . An updated meta-analysis of oxidative stress markers in bipolar disorder. Psychiatry Res 2014; 218: 61–68.
Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M . Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol 2009; 19: 220–230.
Flatow J, Buckley P, Miller BJ . Meta-analysis of oxidative stress in schizophrenia. Biol Psychiatry 2013; 74: 400–409.
Munkholm K, Vinberg M, Berk M, Kessing LV . State-related alterations of gene expression in bipolar disorder: a systematic review. Bipolar Disord 2012; 14: 684–696.
Rossignol DA, Frye RE . Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front Physiol 2014; 5: 150.
Yao JK, Keshavan MS . Antioxidants, redox signaling, and pathophysiology in schizophrenia: an integrative view. Antioxid Redox Signal 2011; 15: 2011–2035.
Kim SY, Cohen BM, Chen X, Lukas SE, Shinn AK, Yuksel AC et al. Redox dysregulation in schizophrenia revealed by in vivo NAD+/NADH measurement. Schizophr Bull 2017; 43: 197–204.
Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 2007; 318: 1645–1647.
Cabungcal JH, Nicolas D, Kraftsik R, Cuenod M, Do KQ, Hornung JP . Glutathione deficit during development induces anomalies in the rat anterior cingulate GABAergic neurons: relevance to schizophrenia. Neurobiol Dis 2006; 22: 624–637.
Cabungcal JH, Steullet P, Kraftsik R, Cuenod M, Do KQ . Early-life insults impair parvalbumin interneurons via oxidative stress: reversal by N-acetylcysteine. Biol Psychiatry 2013; 73: 574–582.
Morishita H, Cabungcal JH, Chen Y, Do KQ, Hensch TK . Prolonged period of cortical plasticity upon redox dysregulation in fast-spiking interneurons. Biol Psychiatry 2015; 78: 396–402.
Cabungcal JH, Counotte DS, Lewis EM, Tejeda HA, Piantadosi P, Pollock C et al. Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron 2014; 83: 1073–1084.
Long JM, LaPorte P, Merscher S, Funke B, Saint-Jore B, Puech A et al. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics 2006; 7: 247–257.
Fejgin K, Nielsen J, Birknow MR, Bastlund JF, Nielsen V, Lauridsen JB et al. A mouse model that recapitulates cardinal features of the 15q13.3 microdeletion syndrome including schizophrenia- and epilepsy-related alterations. Biol Psychiatry 2014; 76: 128–137.
Balu DT, Li Y, Puhl MD, Benneyworth MA, Basu AC, Takagi S et al. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc Natl Acad Sci USA 2013; 110: E2400–E2409.
The Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 1994; 78: 23–33.
Longordo F, Kopp C, Mishina M, Lujan R, Luthi A . NR2A at CA1 synapses is obligatory for the susceptibility of hippocampal plasticity to sleep loss. J Neurosci 2009; 29: 9026–9041.
Steullet P, Cabungcal JH, Kulak A, Kraftsik R, Chen Y, Dalton TP et al. Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J Neurosci 2010; 30: 2547–2558.
Lodge DJ, Behrens MM, Grace AA . A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci 2009; 29: 2344–2354.
Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 2013; 339: 1095–1099.
Kobrynski LJ, Sullivan KE . Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet 2007; 370: 1443–1452.
Rees E, Walters JT, Chambert KD, O'Dushlaine C, Szatkiewicz J, Richards AL et al. CNV analysis in a large schizophrenia sample implicates deletions at 16p12.1 and SLC1A1 and duplications at 1p36.33 and CGNL1. Hum Mol Gen 2014; 23: 1669–1676.
Meechan DW, Maynard TM, Tucker ES, Fernandez A, Karpinski BA, Rothblat LA et al. Modeling a model: mouse genetics, 22q11.2 deletion syndrome, and disorders of cortical circuit development. Prog Neurobiol 2015; 130: 1–28.
Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008; 455: 232–236.
Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet 2008; 40: 322–328.
Ben-Shachar S, Lanpher B, German JR, Qasaymeh M, Potocki L, Nagamani SC et al. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet 2009; 46: 382–388.
Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet 2008; 40: 1466–1471.
Penagarikano O, Mulle JG, Warren ST . The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet 2007; 8: 109–129.
Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014; 506: 185–190.
Santos AR, Kanellopoulos AK, Bagni C . Learning and behavioral deficits associated with the absence of the fragile X mental retardation protein: what a fly and mouse model can teach us. Learn Mem 2014; 21: 543–555.
Cea-Del Rio CA, Huntsman MM . The contribution of inhibitory interneurons to circuit dysfunction in Fragile X Syndrome. Front Cell Neurosci 2014; 8: 245.
Labrie V, Wong AH, Roder JC . Contributions of the D-serine pathway to schizophrenia. Neuropharmacology 2012; 62: 1484–1503.
Benneyworth MA, Roseman AS, Basu AC, Coyle JT . Failure of NMDA receptor hypofunction to induce a pathological reduction in PV-positive GABAergic cell markers. Neurosci Lett 2011; 488: 267–271.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
Hardingham GE, Do KQ . Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat Rev Neurosci 2016; 17: 125–134.
Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 1995; 373: 151–155.
Boyce-Rustay JM, Holmes A . Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology 2006; 31: 2405–2414.
Hirrlinger J, Schulz JB, Dringen R . Effects of dopamine on the glutathione metabolism of cultured astroglial cells: implications for Parkinson's disease. J Neurochem 2002; 82: 458–467.
Vaessen T, Hernaus D, Myin-Germeys I, van Amelsvoort T . The dopaminergic response to acute stress in health and psychopathology: a systematic review. Neurosci Biobehav Rev 2015; 56: 241–251.
Akyol O, Yanik M, Elyas H, Namli M, Canatan H, Akin H et al. Association between Ala-9Val polymorphism of Mn-SOD gene and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29: 123–131.
Gravina P, Spoletini I, Masini S, Valentini A, Vanni D, Paladini E et al. Genetic polymorphisms of glutathione S-transferases GSTM1, GSTT1, GSTP1 and GSTA1 as risk factors for schizophrenia. Psychiatry Res 2011; 187: 454–456.
Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P et al. Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci USA 2007; 104: 16621–16626.
Mehta D, Iwamoto K, Ueda J, Bundo M, Adati N, Kojima T et al. Comprehensive survey of CNVs influencing gene expression in the human brain and its implications for pathophysiology. Neurosci Res 2014; 79: 22–33.
Reif A, Herterich S, Strobel A, Ehlis AC, Saur D, Jacob CP et al. A neuronal nitric oxide synthase (NOS-I) haplotype associated with schizophrenia modifies prefrontal cortex function. Mol Psychiatry 2006; 11: 286–300.
Rodriguez-Santiago B, Brunet A, Sobrino B, Serra-Juhe C, Flores R, Armengol L et al. Association of common copy number variants at the glutathione S-transferase genes and rare novel genomic changes with schizophrenia. Mol Psychiatry 2010; 15: 1023–1033.
Tosic M, Ott J, Barral S, Bovet P, Deppen P, Gheorghita F et al. Schizophrenia and oxidative stress: glutamate cysteine ligase modifier as a susceptibility gene. Am J Hum Genet 2006; 79: 586–592.
Xin L, Mekle R, Fournier M, Baumann PS, Ferrari C, Alameda L et al. Genetic polymorphism associated prefrontal glutathione and its coupling with brain glutamate and peripheral redox status in early psychosis. Schizophr Bull 2016; 42: 1185–1196.
Kulak A, Steullet P, Cabungcal JH, Werge T, Ingason A, Cuenod M et al. Redox dysregulation in the pathophysiology of schizophrenia and bipolar disorder: insights from animal models. Antioxid Redox Signal 2013; 18: 1428–1443.
Debost JP, Larsen JT, Munk-Olsen T, Mortensen PB, Meyer U, Petersen L . Joint effects of exposure to prenatal infection and peripubertal psychological trauma in schizophrenia. Schizophr Bull 2017; 43: 171–179.
Giovanoli S, Weber L, Meyer U . Single and combined effects of prenatal immune activation and peripubertal stress on parvalbumin and reelin expression in the hippocampal formation. Brain Behav Immun 2014; 40: 48–54.
O'Donnell P . Adolescent onset of cortical disinhibition in schizophrenia: insights from animal models. Schizophr Bull 2011; 37: 484–492.
Modinos G, Allen P, Grace AA, McGuire P . Translating the MAM model of psychosis to humans. Trends Neurosci 2015; 38: 129–138.
Powell SB, Sejnowski TJ, Behrens MM . Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology 2012; 62: 1322–1331.
Jiang Z, Rompala GR, Zhang S, Cowell RM, Nakazawa K . Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol Psychiatry 2013; 73: 1024–1034.
Bouvier E, Brouillard F, Molet J, Claverie D, Cabungcal JH, Cresto N et al. Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Mol Psychiatry 2016 advance online publication, 20 September 2016; doi:10.1038/mp.2016.144.
Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci USA 2007; 104: 14501–14506.
Johnson AW, Jaaro-Peled H, Shahani N, Sedlak TW, Zoubovsky S, Burruss D et al. Cognitive and motivational deficits together with prefrontal oxidative stress in a mouse model for neuropsychiatric illness. Proc Natl Acad Sci USA 2013; 110: 12462–12467.
Pitts MW, Raman AV, Hashimoto AC, Todorovic C, Nichols RA, Berry MJ . Deletion of selenoprotein P results in impaired function of parvalbumin interneurons and alterations in fear learning and sensorimotor gating. Neuroscience 2012; 208: 58–68.
Walter PB, Knutson MD, Paler-Martinez A, Lee S, Xu Y, Viteri FE et al. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc Natl Acad Sci USA 2002; 99: 2264–2269.
Gokhale A, Larimore J, Werner E, So L, Moreno-De-Luca A, Lese-Martin C et al. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J Neurosci 2012; 32: 3697–3711.
Stojkovic T, Radonjic NV, Velimirovic M, Jevtic G, Popovic V, Doknic M et al. Risperidone reverses phencyclidine induced decrease in glutathione levels and alterations of antioxidant defense in rat brain. Prog Neuropsychopharmacol Biol Psychiatry 2012; 39: 192–199.
Wang CZ, Yang SF, Xia Y, Johnson KM . Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology 2008; 33: 2442–2455.
Inan M, Zhao M, Manuszak M, Karakaya C, Rajadhyaksha AM, Pickel VM et al. Energy deficit in parvalbumin neurons leads to circuit dysfunction, impaired sensory gating and social disability. Neurobiol Dis 2016; 93: 35–46.
Lucas EK, Markwardt SJ, Gupta S, Meador-Woodruff JH, Lin JD, Overstreet-Wadiche L et al. Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1alpha. J Neurosci 2010; 30: 7227–7235.
Lin H, Hsu FC, Baumann BH, Coulter DA, Anderson SA, Lynch DR . Cortical parvalbumin GABAergic deficits with alpha7 nicotinic acetylcholine receptor deletion: implications for schizophrenia. Mol Cell Neurosci 2014; 61: 163–175.
Egea J, Buendia I, Parada E, Navarro E, Leon R, Lopez MG . Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol 2015; 97: 463–472.
Yang JM, Zhang J, Chen XJ, Geng HY, Ye M, Spitzer NC et al. Development of GABA circuitry of fast-spiking basket interneurons in the medial prefrontal cortex of erbb4-mutant mice. J Neurosci 2013; 33: 19724–19733.
Zhang Y, Hodgson N, Trivedi M, Deth R . Neuregulin 1 promotes glutathione-dependent neuronal cobalamin metabolism by stimulating cysteine uptake. Oxid Med Cell Longev 2016; 2016: 3849087.
Garate I, Garcia-Bueno B, Madrigal JL, Caso JR, Alou L, Gomez-Lus ML et al. Stress-induced neuroinflammation: role of the Toll-like receptor-4 pathway. Biol Psychiatry 2013; 73: 32–43.
Kaur C, Rathnasamy G, Ling EA . Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J Neuroimmune Pharmacol 2013; 8: 66–78.
Kann O, Papageorgiou IE, Draguhn A . Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J Cereb Blood Flow Metab 2014; 34: 1270–1282.
Yang JM, Zhang J, Yu YQ, Duan S, Li XM . Postnatal development of 2 microcircuits involving fast-spiking interneurons in the mouse prefrontal cortex. Cereb Cortex 2014; 24: 98–109.
Fachim HA, Srisawat U, Dalton CF, Harte MK, Marsh S, Neill JC et al. Subchronic administration of phencyclidine produces hypermethylation in the parvalbumin gene promoter in rat brain. Epigenomics 2016; 8: 1179–1183.
Morishita H, Kundakovic M, Bicks L, Mitchell A, Akbarian S . Interneuron epigenomes during the critical period of cortical plasticity: Implications for schizophrenia. Neurobiol Learn Mem 2015; 124: 104–110.
Donato F, Rompani SB, Caroni P . Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 2013; 504: 272–276.
Steullet P, Cabungcal JH, Monin A, Dwir D, O'Donnell P, Cuenod M et al. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: a "central hub" in schizophrenia pathophysiology? Schizophr Res 2016; 176: 41–51.
Behrens MM, Ali SS, Dugan LL . Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci 2008; 28: 13957–13966.
Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC et al. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci 2008; 39: 439–451.
Natarajan SK, Zhu W, Liang X, Zhang L, Demers AJ, Zimmerman MC et al. Proline dehydrogenase is essential for proline protection against hydrogen peroxide-induced cell death. Free Radic Biol Med 2012; 53: 1181–1191.
Fonnum F, Lock EA . The contributions of excitotoxicity, glutathione depletion and DNA repair in chemically induced injury to neurones: exemplified with toxic effects on cerebellar granule cells. J Neurochem 2004; 88: 513–531.
Young JW, Geyer MA . Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatment of schizophrenia. Biochem Pharmacol 2013; 86: 1122–1132.
Gass N, Weber-Fahr W, Sartorius A, Becker R, Didriksen M, Stensbol TB et al. An acetylcholine alpha7 positive allosteric modulator rescues a schizophrenia-associated brain endophenotype in the 15q13.3 microdeletion, encompassingCHRNA7. Eur Neuropsychoph 2016; 26: 1150–1160.
Goldshmit Y, Erlich S, Pinkas-Kramarski R . Neuregulin rescues PC12-ErbB4 cells from cell death induced by H(2)O(2). Regulation of reactive oxygen species levels by phosphatidylinositol 3-kinase. J Biol Chem 2001; 276: 46379–46385.
Cellot G, Cherubini E . Reduced inhibitory gate in the barrel cortex of Neuroligin3R451C knock-in mice, an animal model of autism spectrum disorders. Physiol Rep 2014; 2: e12077.
Gogolla N, Takesian AE, Feng G, Fagiolini M, Hensch TK . Sensory integration in mouse insular cortex reflects GABA circuit maturation. Neuron 2014; 83: 894–905.
Tomassy GS, Morello N, Calcagno E, Giustetto M . Developmental abnormalities of cortical interneurons precede symptoms onset in a mouse model of Rett syndrome. J Neurochem 2014; 131: 115–127.
Acknowledgements
We thank Dr Sandra Giovanoli, Dr Aubin Michalon, Dr Kraftsik, Ms Adeline Cottier for excellent technical assistance and Dr Fulvio Magara and the technical staff of the Centre d’Etudes du Comportement, Center for Psychiatric Neuroscience, CHUV, for their support in animal facilities. We thank our financial supports: Swiss National Science Foundation (# 31-116689 to KQD and # 310030_135736/1 to KQD and PS), National Center of Competence in Research (NCCR) “SYNAPSY - The Synaptic Bases of Mental Diseases” from the Swiss National Science Foundation (no 51AU40_125759 to KQD), the NIMH Silvio Conte Center (P50MH094271) (to TKH), the Banque Lombard Odier & Cie SA and the Avina, Damm-Etienne, Pro Scientia et Arte and Alamaya Foundations. Roche covered materials, technical costs and confocal microscopy platform fees for the analysis of the FMR1 KO study. Experiments were approved by the local ethical and veterinary committees for each collaborator who provided us with their animal models.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MD is employed by Lundbeck A/S, LL is employed by F. Hoffmann-La Roche and PD is employed by Pfizer. J-HC received funding from Hoffmann-La Rochet to cover material, technical costs and confocal microscopy fees for the Fmr1 KO study. The remaining authors declare no conflict of interest.
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Steullet, P., Cabungcal, JH., Coyle, J. et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol Psychiatry 22, 936–943 (2017). https://doi.org/10.1038/mp.2017.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.47
This article is cited by
-
Real-time imaging of mitochondrial redox reveals increased mitochondrial oxidative stress associated with amyloid β aggregates in vivo in a mouse model of Alzheimer’s disease
Molecular Neurodegeneration (2024)
-
Variations of plasma oxidative stress levels in male patients with chronic schizophrenia. Correlations with psychopathology and matrix metalloproteinase-9: a case-control study
BMC Psychiatry (2024)
-
Use of ECT in Autism Spectrum Disorder and/or Intellectual Disability: A Single Site Retrospective Analysis
Journal of Autism and Developmental Disorders (2024)
-
Discrete hippocampal projections are differentially regulated by parvalbumin and somatostatin interneurons
Nature Communications (2023)
-
Cell type specific transcriptomic differences in depression show similar patterns between males and females but implicate distinct cell types and genes
Nature Communications (2023)
