
Abstract
Spatial localization is a key determinant of cellular fate and behavior, but methods for spatially resolved, transcriptome-wide gene expression profiling across complex tissues are lacking. RNA staining methods assay only a small number of transcripts, whereas single-cell RNA-seq, which measures global gene expression, separates cells from their native spatial context. Here we present Seurat, a computational strategy to infer cellular localization by integrating single-cell RNA-seq data with in situ RNA patterns. We applied Seurat to spatially map 851 single cells from dissociated zebrafish (Danio rerio) embryos and generated a transcriptome-wide map of spatial patterning. We confirmed Seurat's accuracy using several experimental approaches, then used the strategy to identify a set of archetypal expression patterns and spatial markers. Seurat correctly localizes rare subpopulations, accurately mapping both spatially restricted and scattered groups. Seurat will be applicable to mapping cellular localization within complex patterned tissues in diverse systems.
Similar content being viewed by others



Main
A major focus of developmental biology is understanding the origin and features of different cell types in complex tissues, including the gene expression modules that underlie specific cell types and states, the regulatory circuits that set up these expression programs, and the cell's molecular signals and interactions. Genomics tools for dissecting such processes1,2,3 are not easily applied to three-dimensional tissues where heterogeneous cell populations are interleaved in close proximity. Spatial heterogeneity in developing organisms has typically been studied by RNA hybridization, immunohistochemistry, fluorescent reporters, or purification or induction of predefined subpopulations and subsequent genomic profiling (e.g., RNA-seq). Such approaches, however, currently rely on a small set of predefined markers, therefore introducing selection bias that limits discovery.
Emerging methods in single-cell genomics, especially single-cell RNA-seq4,5, provide new opportunities for developmental biology. Single-cell RNA-seq is quickly becoming an established experimental method, with ongoing cost and throughput improvements enabling applications from cell type discovery6,7 to regulatory network inference8,9 to the reconstruction of developmental processes10,11,12. However, high-throughput applications of single-cell RNA-seq to solid tissues rely on dissociating tissues into single cells7,10, separating cells from their native spatial context, such that further analyses lack crucial information on cells' environments and locations. Although new experimental approaches for sequencing cellular RNA in situ13,14 have been proposed, these require highly specialized experimental tools and do not yet offer the widespread applicability or molecular sensitivity of more established single-cell RNA-seq protocols.
Here we present an alternative approach to study spatial patterning of gene expression at the single-cell level, applying it in the context of the widely studied zebrafish embryo. In embryos at the late blastula stage, when cell fate is being decided15 on the basis of inputs from several morphogens16 whose gradients originate from different regions of the embryo, the spatial location of cells is paramount16,17. This stage has been extensively studied by in situ patterns for known drivers of embryonic patterning and gastrulation16,18,19. However, marker analysis can localize only a handful of genes simultaneously. We use single-cell RNA-seq to identify thousands of RNAs expressed in each cell and infer its spatial origin computationally. We implement our method as part of the Seurat R package for single-cell analysis, named for the artist Georges Seurat to invoke the analogy between the intricate spatial patterning of single cells and a pointillist painting. Seurat uses a statistical framework to combine cells' gene expression profiles, as measured by single-cell RNA-seq, with complementary in situ hybridization data for a smaller set of ‘landmark’ genes that guide spatial assignment. This addresses spatial localization more directly and generally than previous efforts, which used principal components to approximate spatial location20. Applying Seurat to a data set of 851 dissociated single cells from zebrafish embryos at a single developmental stage, we confirmed the method's accuracy with several experimental assays, used it to predict and validate patterns where in situ data were not available, identified and correctly localized rare cell populations—either spatially restricted or intermixed throughout the embryo—and defined additional markers of these populations.
Results
Combining RNA-seq and in situ hybridizations to infer spatial location
To identify the spatial position of dissociated cells, we developed a computational method (Fig. 1) implemented in Seurat (Supplementary Code) that takes as inputs: (i) the expression profiles of individual dissociated cells and (ii) a spatial reference map of gene expression for a small number of landmark genes. This requires the subdivision of the tissue of interest into discrete spatial domains (hereafter, ‘bins’) of user-defined geometry and size. For the map, landmark genes are defined as either ‘on’ or ‘off’ in each bin, for example, as determined from published in situ stainings. Seurat then uses the single-cell expression levels of the landmark genes to determine in which bins a cell likely originated.

As input, Seurat takes single-cell RNA-seq data (1) from dissociated cells (e.g., cells A–C), where information about the original spatial context was lost during dissociation, and (2) in situ hybridization patterns for a series of landmark genes. To generate a binary spatial reference map, the tissue of interest is divided into a discrete set of user-defined bins, and the in situ data are binarized to reflect the detection of gene expression within each bin, as is shown for genes X, Y and Z. (3) Seurat uses expression measurements across many correlated genes to ameliorate stochastic noise in individual measurements for landmark genes. As schematized, Seurat learns a model of gene expression for each of the landmark genes based on other variable genes in the data set, reducing the reliance on a single measurement, and mitigating the effect of technical errors. Seurat then builds statistical models of gene expression in each bin (4) by relating the bimodal expression patterns of the RNA-seq estimates to the binarized in situ data. Shown are probability distributions for genes X, Y and Z for three different embryonic bins. Finally, Seurat uses these models to infer the cell's original spatial location (5), assigning posterior probability of origin (depicted in shades of purple) to each bin. Seurat can map exclusively to one bin (e.g., cell C), or assign probability to multiple bins in some cases (e.g., cells A and B).
Seurat consists of the following steps. (i) It uses co-expression patterns across cells in the single-cell RNA-seq profiles to impute the expression of each landmark gene in each cell. This mitigates errors in detection of specific transcripts in individual cells due to technical limitations in single-cell RNA-seq21,22. (ii) It relates the continuous imputed RNA-seq expression levels of each landmark gene to the binary spatial expression values using a mixture model constrained by the proportion of cells expressing the gene in the reference map. (iii) For each bin, it constructs a multivariate normal model for the joint expression of the landmark genes based on these mixture models, the binary spatial reference map and an optional quantitative refinement step that estimates covariance parameters between all pairs of genes. (iv) Given these models, it infers the spatial origin of each profiled cell by calculating a posterior probability for each cell-bin pair, allowing determination of the cell's likely position(s) and confidence in the mapping. We describe each of these steps and associated computational challenges below, and then apply and validate Seurat by mapping cells in the zebrafish embryo.
Matching binary in situ hybridizations to continuous, noisy RNA-seq data
Seurat maps cells to their location by comparing the expression level of a gene measured by single-cell RNA-seq to its expression level in a three-dimensional tissue measured by in situ hybridization (Fig. 1). Although straightforward in principle, there are two primary challenges to address.
First, single-cell RNA-seq measurements are confounded by technical noise21,22, particularly false negatives and measurement errors for low-copy transcripts. As only a few landmark genes characterize each region of the spatial map, erroneous measurements for these genes in a given cell could interfere with its proper localization. To address this, Seurat leverages the fact that RNA-seq measures multiple genes that are co-regulated with the landmark genes and uses these genes to impute the values of the landmark genes. Specifically, Seurat uses the expression levels of all highly variable genes in the RNA-seq data set and an L1-constrained, LASSO (least absolute shrinkage and selection operator23) technique to construct separate models of gene expression for each of the landmark genes. In this way, expression measurements across many correlated genes ameliorate stochastic noise in individual measurements.
Second, for each landmark gene, Seurat must relate its continuous imputed RNA-seq expression levels to its binary state in the landmark map. As the in situ color deposition reaction is halted at an arbitrary point in standard protocols, and individual probes do not generate equivalent signals, each gene requires a separate conversion between gene expression level detected by RNA-seq and binary in situ interpretation. To this end, Seurat relates the typical bimodal distribution of its imputed expression measurements to the ‘on’ and ‘off’ modes of the spatial reference map. It models the imputed measurements as a mixture of two Gaussian distributions, initialized based on the percentage of cells where expression was detected in our binary in situ patterns. Seurat then fits the parameters describing the two modes using ‘expectation maximization’, followed by an additional heuristic step to better reflect the overall data (Online Methods; Supplementary Note).
Probabilistic inference of spatial origin
Seurat next constructs a model for the joint expression of the landmark genes in each bin based on the parameters of the mixture models and the binary spatial reference map. Intuitively, for each cell and landmark gene, Seurat calculates the likelihood that this cell's expression of the landmark gene reflects the ‘on’ state, and thus, a probability that this cell originated from bins marked as ‘on’ in the reference map. Seurat also implements a recommended refinement step that extends the quantitative nature of these models and also considers the covariance structure between all pairs of landmark genes. Finally, Seurat infers from these models a posterior probability that a cell originated from each of the bins in the map. When there is insufficient information to assign a cell exclusively to one bin, Seurat's probabilistic approach enables a cell to split its posterior probability across multiple bins.
Zebrafish blastula spatial reference map from RNA in situ hybridizations
We tested Seurat and demonstrated its utility using late blastula stage (50% epiboly) zebrafish embryos (Fig. 2a). We generated a reference spatial map by discretizing expression patterns for 47 genes obtained from published bright-field images of in situ hybridizations generated by standard colorimetric deposition, primarily from ZFIN's collection24 or high-throughput data sets25,26 (Supplementary Table 1). We divided the embryo into 128 bins (each ∼40–120 cells), equally sized along the dorsal-ventral axis, based on the most restricted expression domain in our in situ set, and unevenly sized along the animal-vegetal axis, broadening as they approached the animal pole where patterns were less complex and less sharply defined (Fig. 2b). Because the embryo still exhibits left-right symmetry at this stage, we collapsed the equivalent left and right bins in our analysis, treating the embryo as 64 bins. We ignored the depth axis (from surface to interior), because there are no major examples of gene expression differences along this axis at this stage of development, with the exception of the specialized enveloping layer and yolk syncytial layers17. We manually scored the in situ hybridizations in each bin once, before any data analysis (Fig. 2b). Binary discretization, although oversimplified, avoids overinterpretation, especially given that published images differ markedly in their resolution, lighting and extent of staining (Supplementary Fig. 1). As we show below, Seurat robustly mapped cells with high quality even based on an initial binary scoring.

(a) Zebrafish embryo at 50% epiboly, depicting cell layers (enveloping layer, EVL; deep cell layer, DEL; yolk syncytial layer, YSL), important structures (the margin), and the two major spatial axes (animal-vegetal and dorsal-ventral). (b) To create the spatial reference map, we used 47 colorogenic in situ hybridization patterns (i.e., landmark genes), which were previously published in the scientific literature. We subdivided the embryo into 64 bins and visually scored each landmark as ‘on’ or ‘off’ within each bin using in situ hybridizations oriented in both lateral and animal views. Shown here is an in situ for ta/no tail and its resultant binary representation. (c) Single-cell RNA-seq protocol modified to include Random Molecular Tags.
Single-cell RNA-seq of zebrafish embryos
To apply Seurat, we generated single-cell RNA-seq profiles of dissociated cells from developing zebrafish embryos. We used a modified, strand-specific, single-cell RNA-seq protocol based on the SMART template switching method4,5,8 (Fig. 2c), where the template-switch oligonucleotide included a stretch of five randomized nucleotides, thereby tagging each mRNA molecule with a random molecular tag (RMT) before PCR to mitigate amplification bias. Furthermore, we used a modified library preparation protocol that shares similarities with Soumillon et al.27 and is based on the Nextera Sample Preparation Kit. It selectively amplifies the 5′ transcript end, retains strand information and is compatible with standard Illumina sequencing primers. We pooled and sequenced libraries to an average depth of 530,000 reads per sample, where single-cell gene expression tends to saturate7,28,29. Following read alignment, we determined expression levels by counting the number of distinct RMTs associated with each gene and normalizing by the total number of RMTs in each cell. We prepared 1,152 libraries, but retained 945 single cells after excluding those where less than 2,000 expressed genes were detected. Finally, we observed a population of 94 cells expressing high levels of canonical markers of the enveloping layer (e.g., krt18, krt4, cldne), a single layer of differentiated squamous cells that cover the outside of the embryo30,31. These cells, identified by a principal component analysis (PCA), do not exhibit the same expression patterns of the landmark genes in our spatial reference map, and so were excluded from further consideration (Supplementary Fig. 2).
Overall, we analyzed 851 single-cell transcriptomes, obtained from cells that were isolated under three different experimental protocols. The vast majority (n = 682) were collected in an unbiased manner from 28 dissociated zebrafish embryos (Fig. 2c and Supplementary Movie 1). To reduce confounding transcriptional changes that occur as a result of dissociation, we collected and froze cells within 15 min of dissociation. In addition, for subsequent testing of Seurat's success, we included two internal controls: (i) 141 cells that were collected using a slightly modified dissection and dissociation protocol that enriches for cells nearer the embryonic margin (Supplementary Movie 2); and (ii) 28 ‘reference’ cells collected from intact embryos under a dissection microscope, such that their location is approximately known (Fig. 3c and Supplementary Movie 3). We used all 851 cells as input to Seurat, withholding from the method any information on how each individual cell was collected.

(a) Seurat maps cells throughout the embryo, consistent with the random dissociation of the tissue. Shown are cell centroids for randomly dissociated cells. (b) A smaller number of cells were prepared with a modified protocol that depletes for the animal cap (bin rows 6–8) (Supplementary Movie 2), and Seurat captures this depletion in its mapping of these cells. Shown are the fold-changes in localization percentages (y axis) between the randomly dissociated and animal-depleted cells along the margin to animal axis (x axis, as bins). (c) A small number of ‘reference’ cells were manually picked under a dissecting microscope so that their original spatial location could be estimated (Supplementary Movie 3). Because at 50% epiboly, dorsal-ventral (D-V) specification is not morphologically apparent, a cluster of previously transplanted fluorescent cells is used as a fiducial mark to track where cells were taken from and to deduce the cell's location once the dorsal-ventral axis becomes apparent at the shield stage. (d) Representative examples of Seurat's inferred location for reference cells (centroid: green; posterior bin probabilities: shades of purple, with the strongest color representing 100% posterior probability) versus experimentally annotated locations (pink). The embryonic margin is also depicted in khaki. The experimentally annotated sphere is drawn with a larger radius to reflect degree of confidence in the experimental measurement. (e) Histogram of distance errors between inferred and measured location for reference cells. The median error is two bins, divided equally between the animal-vegetal and dorsal-ventral axes. (f) For each landmark gene, we removed the gene from the input reference map, and then re-inferred its in situ pattern from Seurat's spatial patterns. Representative examples are compared to the binarized input pattern (top) and ROC scores (bottom). (g,h) ROC analysis of the accuracy of the inferred landmark in situ hybridizations versus the binarized input pattern (median ROC = 0.96), presented as curves for a selection of genes (g) and a summary of ROC performance for all landmarks (h).
Accurately inferred spatial assignments and in situ hybridizations
We determined that Seurat inferred cell location accurately by four complementary approaches. First, Seurat assigned the 682 randomly dissociated cells throughout the embryo (Fig. 3a and Supplementary Fig. 3), with roughly equal representation from the dorsal, ventral, marginal and animal regions, consistent with the randomized nature of embryonic dissociation. Second, as a low-resolution benchmark, we examined Seurat's localization of the cells that were produced using the modified dissociation protocol that strongly biases against the animal cap and enriches for the embryonic margin (Supplementary Movie 2). Indeed, Seurat's inferred locations overlapped considerably with the experimentally enriched area, exhibiting an approximately sevenfold depletion at the animal cap, and an accompanying enrichment at the margin, compared to the randomly dissociated cells (Fig. 3b). Third, we tested the inferred position of the 28 ‘reference’ cells that were manually isolated from intact embryos under a dissecting microscope, and hence we could visually approximate their original spatial location to an estimated precision of ± 1 bin margin in each axis (Fig. 3c and Supplementary Movie 3). Although throughput for this technique is too low for generating large numbers of cells, it enabled us to compare Seurat's inferred locations with an independent benchmark. Seurat's inferred location for these reference cells was, on average, within one bin of the registered location across both the dorsal-ventral and animal-vegetal axes, mirroring our own confidence in the collection of the cells (Fig. 3c–e).
Fourth, we generated an in silico catalog of inferred in situ patterns, by calculating for each gene its expected expression level in each of the 64 bins, as the weighted average of RNA-seq measurements for this gene based on Seurat's probabilistic assignment of cells to bins (Fig. 3f). In cross-validation, for each landmark gene, we removed that gene from the spatial map, re-inferred the cells' locations, and then created a simulated in situ pattern for the held-out gene. Our inferred patterns demonstrated remarkably high overlap with experimental data, defined as correctly classifying individual bins into the same ‘on’ or ‘off’ expression state as our binary interpretations of published in situ hybridizations (median receiver operating characteristic (ROC) = 0.96), with 12 of 47 genes exhibiting near-perfect classification (ROC > 0.98) (Fig. 3f–h). A rare subset of genes apparently performed poorly (e.g., chd, Fig. 3f), but further inspection of the literature revealed that these had a wide variety of published in situ patterns at this stage, some of which largely agreed with Seurat's predictions (Supplementary Fig. 1).
Clustering expression patterns reveal spatial archetypes
Extending this strategy, we next inferred in situ patterns for a larger set of 290 genes that were highly variable across single cells, clustered them and identified nine archetypal expression patterns (Fig. 4a and Supplementary Fig. 4). These archetypes are consistent with known patterning gradients at this embryonic stage16 and span the 47 genes in our reference spatial map. We selected for in situ validation 14 genes from the different archetypes whose expression patterns had not been previously characterized at 50% epiboly (Fig. 4b and Supplementary Fig. 5). Our experimentally determined in situ expression patterns exhibited overall high accordance with Seurat's predicted patterns (Fig. 4b). For example, genes predicted to be restricted to the very dorsal margin (tbr1b, slc25a33 and pkdcca) or to have dorsal enrichment (arl4ab) are indeed expressed with those patterns (Fig. 4b and Supplementary Fig. 5). All genes predicted to have marginal restriction (prickle1b and dusp4), marginal enrichment (irx7, ets2 and tcf3b) or ventral skew (nrarpa, id2a, insm1b and prdm12b) exhibited those predicted features in situ (Fig. 4b). Even unusual and complex predicted patterns were correctly predicted (Fig. 4b), such as irx7's lower expression in the animal cap and high expression close to the margin, especially on the lateral sides. In 2 of the 14 cases (ets2 and cpn1; Fig. 4b and Supplementary Fig. 5) the patterns were fundamentally correct, but were predicted to extend slightly farther than we observed in situ. Thus, Seurat can correctly transform single-cell RNA-seq data into spatial predictions for genes whose expression patterns are not known.

(a) We calculated imputed expression patterns based on Seurat's spatial mapping for 290 highly variable genes. Genes were then clustered by their imputed spatial localization (Supplementary Fig. 5) into nine ‘archetypes’ that broadly describe the patterns of multiple genes: RM, restricted to margin; VM, ventral margin; DEM, dorsally enriched margin; DRM, dorsally restricted margin; EM, extended margin; V, ventral; DA, dorsal animal; VA, ventral animal; A, animal. (b) RNA in situ hybridization of genes selected from various archetypes without published expression patterns at 50% epiboly (as of 4 September 2014). Top to bottom: Seurat's predicted expression pattern, a lateral view of the in situ (dorsal to the right), and an animal cap view of the in situ (dorsal to the right). Experimentally determined patterns exhibit high accord with Seurat's predictions, as described in the main text. Genes are connected to the archetype with which they clustered by black lines. Scale bar, 200 μm.
Spatially diverse landmark genes improve Seurat's mapping
To assess Seurat's sensitivity to the number and type of landmark genes in the spatial reference map, we downsampled the number of landmark genes used as input and performed a spatial power analysis (Supplementary Text and Supplementary Fig. 6). Seurat's spatial mappings began to stabilize after the inclusion of ∼30 landmark genes and were best when genes were sampled across all nine spatial archetypes, whereas maps drawn from a more spatially restricted set did not perform as well. Further analysis suggested that having two genes with overlapping spatial expression patterns is valuable, but additional redundancy has diminishing returns.
Seurat correctly localizes rare cell populations
Seurat's spatial inferences can be combined with unsupervised analysis of single-cell RNA-seq data to define and characterize both known and previously unidentified, rare subpopulations of cells within complex tissues. In this approach, putative subpopulations are first identified in an unsupervised manner and their identities are confirmed by examining the expression of known marker genes. Seurat is then used to determine the characteristic spatial patterning for each of these subpopulations.
To test this approach, we used Seurat to identify and localize three well-studied and rare subpopulations present near the embryonic margin: (i) prechordal plate progenitors (Fig. 5a, green), (ii) endodermal progenitors (Fig. 5a, blue), and (iii) primordial germ cells (PGC). We clearly identified the first two subpopulations in unsupervised analyses, with strong agreement between PCA and k-means clustering (Supplementary Fig. 7a–b). A distinct population of ten cells was distinguished by the second principal component and characterized by strong expression of the prechordal plate markers gsc and frzb (Fig. 5b and Supplementary Fig. 7c). The prechordal plate is located in the dorsal-most embryonic margin, and Seurat mapped all prechordal plate progenitors to this region (Fig. 5c, green). The PCA also uncovered another population of 19 putative endodermal progenitors, defined by high expression levels of sox32, cxcr4a and gata5 (Fig. 5b and Supplementary Fig. 7c). Seurat scattered the endodermal progenitors across the lowest tier of the embryonic margin (Fig. 5c, blue), consistent with their known localization and recapitulating their ‘salt-and-pepper’ pattern32. Finally, PGC cells only comprise ∼1 per 500 cells at this stage, and thus we could not uncover them through unsupervised analysis of our 851 cells. However, we identified one cell that expressed extremely high levels of the canonical PGC markers ddx4/vasa, nanos3, and dnd1 (ref. 33; Supplementary Fig. 7d). Seurat mapped this cell to a mid-margin location, consistent with the distribution of these cells at this stage (Supplementary Fig. 7e). Thus, Seurat successfully characterized the spatial distribution of known subpopulations with different characteristic localizations.

(a) Cartoon schematic of the prechordal plate progenitors (green) are co-localized at the dorsal margin, and endodermal progenitors (blue) are scattered along the embryonic margin. (b) Violin plots of the distribution of expression of classical endoderm markers (sox32, cxcr4a), classical prechordal plate marker (gsc) and proposed prechordal plate marker (ripply1), in the cell populations determined by PCA analysis: all marginal cells (margin), endodermal progenitors (endo) and prechordal plate progenitors (PCP). (c) Seurat localizes the endodermal progenitors (blue) and prechordal plate progenitors (green) to their characteristic locations. (d) Seurat's predicted expression pattern (left) and in situ validation (right) of the expression of ripply1. (e) Double in situ for gsc (orange) and ripply1 (blue) confirming that ripply1 is expressed in the prechordal plate progenitors. (f) Previously uncharacterized group of cells (magenta) distinguished by PC4 (by PCA), and expressing high levels of genes which are hallmarks of apoptosis. (g) Seurat's projected localization of these ‘apoptotic-like’ cells (magenta) are scattered around the embryo, but enriched toward the animal pole. (h) Violin plots of the distribution of expression of isg15 and mat2al, markers of the ‘apoptotic-like’ population in all the cells and the putative apoptotic-like cells. (i) In situ hybridization of four markers of the ‘apoptotic-like’ cells are expressed in similarly scattered patterns. Top: lateral view, bottom: animal pole view. (j) Double fluorescent in situ hybridization for aplnrb (magenta) and isg15 (green) reveals that these markers are co-expressed, as predicted by Seurat. Notably, cells appear to express high levels of either aplnrb or isg15 and lower levels of the other gene. Scale bars, 100 μm.
Seurat discovers markers of rare subpopulations
We next extended Seurat to discover markers of rare subpopulations, focusing on prechordal plate progenitors. A spatially naive approach comparing our ten prechordal plate progenitors to all other cells in the embryo was only partially successful. Although it identified known markers of the prechordal plate (e.g., gsc, nog1 (refs. 34,35)), it also identified many broader markers of the embryonic margin (e.g., osr1, mixl1). The spatially naive approach failed because the cells of interest belong to two restricted populations when considering the entire embryo—cells that are located along the embryonic margin and cells that are prechordal plate progenitors (which are a subset of those along the embryonic margin). To overcome this, we used Seurat's spatial inferences in a spatially aware marker selection strategy; we identified all marginally restricted cells, and then specifically searched for genes that were differentially expressed between the prechordal plate progenitors and the rest of the marginal cells. The spatially aware approach successfully rediscovered multiple well-characterized prechordal plate progenitor markers (e.g., gsc, nog1, klf17 and six3b34,35,36,37), avoided the broader, nonspecific markers above and also found candidate markers that were not previously annotated in the prechordal plate24, including ripply1 and ptf1a, whose expression patterns were not known at 50% epiboly. Although we were unable to detect ptf1a, which is expressed at a very low level, an in situ hybridization for ripply1 agreed with Seurat's prediction, and a ripply1/gsc double in situ hybridization showed that ripply1 is expressed only in a subset of gsc-expressing cells (Fig. 5d,e). Thus, we conclude that ripply1 is a bona fide marker of the prechordal plate progenitors at 50% epiboly, and the spatially aware approach discovers markers of rare subpopulations.
Seurat identifies dispersed, rare cell populations
Finally, we searched for potentially novel subpopulations present in our data set. Our PCA revealed a group of 12 cells (Fig. 5f–h, magenta, Supplementary Fig. 7f) which highly expressed genes that are hallmarks of apoptosis (foxo3b, tp53inp1, casp8 and ctsh), cellular stress (isg15, sesn3, mat2al and gadd45aa) and cell signaling (igf2a and aplnrb). Gene ontology analysis revealed a significant enrichment for targets of the p53 signaling pathway (FDR < 10−6). Seurat inferred that these ‘apoptotic-like’ cells were scattered throughout the developing embryo, although they originated more frequently toward the animal and ventral poles (Fig. 5g, magenta). Notably, these cells were not an artifact of the isolation process: they were identified in ten separate embryos, in each experimental batch, and previous in situ analysis for foxo3b, aplnrb and isg15 interdependently confirmed their individual scattered expression38,39.
We performed in situ analysis of casp8, gadd45aa, igf2a and tp53inp, and confirmed that these genes also exhibited similar scattered patterns in intact embryos (Fig. 5i). Namely, these genes are expressed in cells sprinkled throughout the embryo, observable at all depths (the EVL and throughout the DEL), generally more frequently toward the animal pole. The number of cells and their specific locations were different for each embryo, consistent with stochastic localization. We verified by double fluorescent in situ hybridization that at least two of these markers (aplnrb and isg15) are indeed co-expressed in the same cells (Fig. 5j). We conclude that these cells constitute a previously uncharacterized and stochastically localized population of cells whose gene expression profile suggests cell stress.
Discussion
To use single-cell RNA-seq within the context of complex, patterned and heterogeneous tissues, we developed Seurat, a computational method that uses a spatial reference map constructed from a small number of landmark in situ patterns to infer the spatial location of cells from their single-cell RNA-seq profiles. Seurat tackles several technical challenges, including the representation of in situ hybridizations for algorithmic input, handling stochastic noise in RNA-seq data for landmark genes and finding a correspondence between the two data types. We developed and tested Seurat on a data set of 851 cells in a developing zebrafish embryo and a reference map constructed from colorigenic in situ data for 47 genes. Our extensive validations indicate that Seurat performs well in this setting.
Seurat should be widely applicable, although different systems have distinct advantages and challenges associated with spatial mapping of single cells. For instance, Seurat relies on the spatial segregation of gene expression patterns in a tissue in order to construct a reference map; it may be challenging to apply it to tissues such as tumors where there is no guarantee of reproducible spatial patterning, or to tissues where cells with highly similar expression patterns are spatially scattered across a tissue (e.g., the adult retina, where each of the dozens of different cell types are distributed broadly and evenly along the dorsal-ventral and anterior-posterior axes).
In amenable systems, the use of binary interpretation of colorigenic in situ hybridizations without the need for an automated image processing pipeline creates a low barrier to entry for using Seurat. Extensive databases exist for many organisms (e.g., Caenorhabditis elegans, Drosophila melanogaster, D. rerio, Xenopus laevis and Mus musculus), and if they are insufficient, landmark in situ hybridizations can be easily generated. Additionally, most tissues should be amenable to binning because bins can be any shape, any size, noncontiguous and dissimilar; thus, any salient feature in the tissue of interest should be representable. Bins could even be reduced to the single-cell level in tissues where each cell in each position has a distinct and reproducible gene expression identity and position. Generation of such a fine-grained map would require the use of more finely resolved or more quantitative data. For instance, fluorescent in situ hybridization combined with confocal microscopy could create a more detailed input spatial map with more quantitative data that could be discretized into more than two states. Additional gene expression states could reduce the number of required in situ hybridizations, if they described new landmark patterns, rather than redundant spatial information. Such data are fully compatible with Seurat's computational approach by extending the mixture model fitting to parameterize models with more than two modes.
A complementary study (Achim et al.40) pursues a conceptually similar approach, but in the context of a reference map constructed from RNA fluorescent in situ hybridization (FISH) data in the brain of the annelid Platynereis dumerilii, where distinct cell types can be determined solely by the expression of a few highly expressed transcription factors (rather than the combinations of many often required in zebrafish). Thus, although the two approaches are conceptually similar, the computational hurdles presented by the individual data sets are distinct, and the two methods thus provide complementary approaches that will enable the spatial profiling of a wide range of tissues.
Finally, although our use of an existing spatial reference map requires previous knowledge, we envision two potential unbiased approaches for spatial mapping in less studied tissues. One is to use an iterative scheme starting with the generation of single-cell RNA-seq data. Our work indicates that standard techniques (e.g., unsupervised dimensionality reduction) could suggest the most relevant landmark genes to establish a preliminary input spatial map. Iterating this spatial mapping, combined with the generation of new in situ data, could quickly produce a high-quality spatial map of a tissue.
A second option is to generate an unbiased spatial reference map with emerging techniques that perform low-input RNA-seq on cryosections41,42. These techniques measure genome-wide expression across spatially resolved tissue slices, blending together the signal of many cells, but enabling unbiased discovery of spatial landmarks. As these are not single-cell experiments, they cannot resolve spatially intermixed populations, such as the endodermal progenitors or apoptotic-like cells we described here. However, the spatial reference maps generated by these techniques are highly complementary with Seurat's method, and combining these approaches represents a generalizable strategy for spatially reconstructing complex tissues at the single-cell level without prior knowledge of gene expression.
Finally, elements of Seurat's approach suggest a broader framework to integrate single-cell RNA-seq data with other complementary data sets based on a limited number of marker genes. Data imputation is a useful tool to be applied to these problems—specifically, leveraging correlated genes across the transcriptome improves the robustness of marker genes with complementary sources of information. Whereas Seurat focuses on inferring spatial origin, combining transcriptomics data with RNA-FISH, CyTOF mass spectrometer39 or flow cytometry41 data could help determine a cell's developmental state or disease phenotype and relate it to a rich body of published research. Seurat shares with these potential approaches the challenges and goal of learning the ‘metadata’ of each single cell, inferring its origins and history to better understand its behavior and future fate.
Methods
Animal models.
This study includes the use of live vertebrate embryos. Animals were handled according to National Institutes of Health (NIH) guidelines. All vertebrate animal work was performed at the facilities of Harvard University, Faculty of Arts & Sciences (HU/FAS) under protocol 25-08. The HU/FAS animal care and use program maintains full AAALAC accreditation, is assured with OLAW (A3593-01) and is currently registered with the US Department of Agriculture (USDA).
Collecting dissociated cells.
Cells from 28 embryos are in the final data set. Fertilized eggs from wild-type (Tupfel long fin/AB) in-crosses were incubated in 1 mg/ml pronase (protease from Streptomyces griseus, Sigma-Aldrich) in a glass dish for 4–5 min until the chorion began to blister. The embryos were submerged in ∼200 ml of embryo medium in a glass beaker without allowing them to contact air or plastic (both of which will cause the embryos to burst). The medium was poured off and new medium was vigorously added (again without allowing embryos to contact air) twice, in order to mechanically remove the chorions. The embryos were cultured in Petri dishes coated with 2% agarose (to avoid contact with plastic) either at 23 °C or 28 °C until they reached 50% epiboly (about 6 h post-fertilization at 28 °C). At 50% epiboly, single embryos were visually confirmed to be at the correct stage and were transferred to Petri dishes that had been coated with 2% agarose, filled with DMEM/F12 media (Gibco/Life Technologies), and allowed to soak for 3 h. Two pairs of watchmaker forceps were used to dissect the blastula cap of the embryo away from the yolk. First, one pair of forceps was used to hold and rotate the cap, while the other was used to cut and pinch away the yolk that extended below the blastula cap. Then, the blastula cap was cut slightly up the side, which exposed the yolk that was inside of the blastula cap, which could then be gently peeled away. The blastula cap was transferred by pipette into a microfuge tube that contained 60 μl of DMEM/F12 media. The cells were dissociated by vigorously flicking the tube ten times, and then pipetting the entire volume twice while visually confirming that dissociation had occurred. A timer was started at this time to track the amount of time the embryo had been dissociating. If cell clumps were still visible, the tube was flicked again. 180 μl of DMEM/F12 was added to dilute the cell mixture, then 120 μl of the diluted cell mixture was pipetted across the surface of a new agarose-coated dish filled with DMEM/F12 media. To collect cells, a P2 pipettor was used, while observing the cells under the dissecting scope, to collect 0.5 μl of media that contained a single cell. This was pipetted into 3 μl of TCL lysis buffer (Qiagen) in the lid of a PCR strip and mixed three times to ensure that lysis occurred. After eight cells were collected, the entire strip of lids was snapped into a 96-well plate and kept on dry ice.
For collections of margin-enriched populations, the same procedure was followed, except a scalpel was used to cut the embryo about halfway along the animal-vegetal axis and remove the animal cap before dissecting away the yolk and proceeding (Supplementary Movie 2).
Collecting single reference cells.
Fertilized eggs were collected from TL/AB in-crosses and dechorionated in 1 mg/ml pronase for 4–5 min. Cells from 21 embryos are in the final data set. A portion of the embryos was then injected at the one-cell stage with 1 picoliter of 0.3 mg/ml 3 kD dextran-Alexa 488 (Molecular Probes D34682). Both the dextran-injected and uninjected embryos were cultured at 28 °C in an agarose-coated Petri dish filled until they reached sphere stage. The plate was checked during culturing and damaged or abnormal embryos were removed. At sphere stage, embryos were transferred to an agarose-coated Petri dish with small wells (a transplantation dish) filled with 0.3× Danieau medium (final concentration: 17.4 mM NaCl, 0.21 mM KCl, 0.12 mM MgSO4, 0.18 mM Ca(NO3)2, 1.5 mM HEPES pH 7.6), and 10–20 cells were transplanted from the Alexa488-dextran–injected embryos to the uninjected15, placed in a cluster on one side of the embryo, generally about halfway between the margin and the animal pole. Damaged embryos were removed from the plate, and the transplanted embryos were divided into two populations; half were cultured at 28 °C, and the other half were cultured at room temperature so that the populations would develop asynchronously and provide a longer window when embryos would be at the proper stage for cell collection. When embryos reached 50% epiboly, a transplantation needle was used to remove a small cluster of cells from the embryo under a dissecting scope by mouth pipetting. The location from which the cluster was taken was immediately noted. It is important to note that at 50% epiboly stage, the dorsal-ventral axis is not yet morphologically apparent, so we used the fluorescent cells as a fiducial mark to later determine where cells came from on the dorsal-ventral axis. More specifically, the location of the cluster was judged in two ways (Fig. 3c): (i) tier was determined by visually counting the number of cells up from the embryonic margin (cells are readily visible under the dissecting scope if it has been set up with adequate contrast) and (ii) position in the DV axis was recorded as an angular measurement from the fluorescent cells that had been previously transplanted. The small cluster of cells that was sucked out of the embryo was injected into a clean, neighboring well. The cells were gently rinsed by ejecting a stream of buffer over them, and then a single cell was separated from the others preferably with a gentle stream of buffer, but occasionally by pipetting up and down. A single, isolated cell was transferred to 0.5 μl of 0.3× Danieau medium that had previously been placed in a PCR tube cap. The successful transfer of the cell was verified visually under the dissecting scope, and the cell was lysed by the addition of 3 μl of TCL lysis buffer (Qiagen). To preserve the embryos, generally a maximum of five cells were taken from any individual embryo. The embryos were returned to the incubator to develop to shield stage. At shield stage, gastrulation begins, which results in a thickening of the embryonic margin at the dorsal pole (the shield), allowing visual determination of the dorsal-ventral axis. Thus, at this stage, embryos were photographed from an animal cap view under a dissecting scope, where both the shield and fluorescent transplanted cells were clearly visible. To determine the location on the dorsal-ventral axis that the cells were picked from, an angle was drawn through the fluorescent cells, the center of the embryo, and the center of the shield providing an angular measure of where the cell originated along the dorsal-ventral axis.
Probe synthesis.
Fragments of the genes arl4ab, casp8, cpn1, dusp4, gadd45aa, id2a, igf2a, insm1b, irx7, isg15, pkdcca, prickle1b, ripply1, tbr1b, tcf3b and tp53inp1 were amplified using Hi-Fidelity Platinum Taq (Life Technologies; quarter-size reactions, but otherwise according to manufacturer's instructions) and the primers listed in Supplementary Table 2. These fragments were cloned into pSC-A plasmid using Strataclone PCR Cloning Kit (Agilent; half-size reactions, but otherwise according to manufacturer's instructions), used to transform the included cells, and plated on blue-white selection media. Colonies were selected, cultured, mini-prepped and sent for sequencing. Constructs cloned above as well as constructs for aplnrb43 and ets2 (ref. 44) were linearized with the appropriate restriction enzyme (Supplementary Table 2) and purified using PCR cleanup columns (Omega Cycle Pure Kit). The probe was synthesized according to manufacturer's instructions, using the appropriate polymerase (T3 or T7, Roche) and 10× RNA labeling mix (DIG or Fluorescein, Roche) (Supplementary Table 2). The transcription reactions were incubated for 3 h, purified using RNA cleanup columns (Omega E.Z.N.A. Total RNA Kit I), quantified using a Nanodrop, assessed on an agarose gel for successful transcription of a product of the expected size and normalized to 20 ng/μl in HM+ buffer (50% formamide, 5× saline-sodium citrate buffer (SSC), 0.1% Tween-20, citric acid to pH 6.0, 50 μg/ml heparin, 500 μg/ml tRNA), then stored at −20 °C.
Colorogenic in situ hybridization.
In situ hybridizations were done essentially as described45. Embryos were collected from TL/AB in-crosses, dechorionated, cultured to 50% epiboly in agarose-covered dishes at 28 °C. They were fixed in 4% formaldehyde (Sigma-Aldrich) at 4 °C overnight. They were rinsed twice for 10 min with PBST (1× PBS + 0.1% Tween-20 (OmniPur)), passed through a methanol dehydration series (10 min each, 67% PBST:33% methanol (Macron), 33% PBST: 67% methanol), then rinsed in methanol twice for 10 min, and permeabilized at −20° at least overnight. Embryos were rehydrated (10 min each, 75% methanol:25% PBST, 50% methanol:50% PBST, 25% methanol:75% PBST, four times for 10 min PBST). Embryos were then prehybridized in HM+ buffer (50% formamide, 5× SSC buffer, 0.1% Tween-20, citric acid to pH 6.0, 50 μg/ml heparin, 500 μg/ml tRNA) at 70 °C for at least 2 h. Probes were normalized to 1 ng/μl per probe (digoxigenin-incorporated for single in situ hybridizations or digoxigenin - and fluorescein-incorporated for double in situ hybridizations) in HM+ buffer and denatured at 70 °C for 10 min. The prehybridization HM+ buffer was replaced by the probe and embryos were incubated with the probe overnight.
The next morning, the probe was removed and returned to −20 °C for future re-use. Excess probe was removed with first a series of washes that had been prewarmed to 70 °C: 1× 10 min HM buffer (HM+ without heparin and tRNA), 1× 10 min 75% HM:25% 2× SSC, 1× 10 min 50% HM:50% 2× SSC, 1× 10 min 25% HM, 75% 2× SSC, 1× 10 min 0.2× SSC, 1× 30 min 0.2× SSC; then a series of room temperature washes: 1× 5 min 75% 0.2× SSC:25% PBST, 1× 5 min 50% 0.2× SSC:50% PBST, 1× 5 min 25% 0.2× SSC:75% PBST, 1× 5 min PBST. They were blocked for at least 3 h in blocking buffer: 2% Blocking Reagent (Roche, 11 096 176 001) in maleate buffer (150 mM maleic acid, 100 mM sodium chloride, pH 7.4). Finally, they were incubated overnight with anti-digoxigenin antibody coupled to alkaline phosphatase (Anti-Digoxigenin-AP Fab Fragments, Roche 11 093 274 910), diluted 1:5,000 in blocking buffer at 4 °C with gentle agitation.
The following morning, the antiserum was removed and discarded, and excess antibody was removed by rinsing embryos 6× 15 min in PBST. They were transferred into staining buffer (100 mM Tris-HCl pH 9.5, 50 mM magnesium chloride, 100 mM sodium chloride, 0.1% Tween-20) by rinsing 3× 5 min. Staining reagent was introduced (225 μg/ml Nitro Blue Tetrazolium and 175 μg/ml BCIP, Roche 11 383 213 001 and 11 383 221 001) and embryos were incubated in the dark, periodically checking their color development under a dissecting scope until the desired staining had been achieved (15 min–24 h). When the desired staining was achieved, the reaction was stopped by rinsing 3× 5 min.
For single in situ hybridizations, the embryos were dehydrated by passing through a methanol dehydration series, then stored overnight at −20 °C. They were cleared by replacing the methanol with BB/BA (2 parts benzyl benzoate: 1 part benzyl alcohol, Sigma-Aldrich) and imaged on a Zeiss Axioimager Z.1 with a 10× objective (100× total magnification).
For double in situ hybridizations, after the first staining reaction, the first antibody was removed by washing embryos for 2× 5 min with agitation in 0.1 M glycine-HCl pH 2.2, then rinsed 4× 5 min in PBST. The embryos were incubated overnight with anti-fluorescein-AP (Roche, 11 426 338 910) diluted 1:2,500 in blocking buffer at 4 °C overnight. The next morning, the antiserum was removed and discarded, and the embryos were washed 6× 15 min in PBST. They were then stained as above, except that the Nitro Blue Tetrazolium and BCIP were replaced with INT/BCIP Stock solution (Roche) diluted 1:133 in staining buffer. They were incubated in the dark and occasionally monitored for color development (1 h–8 h), and the reaction was stopped by washing 3× 5 min in PBST, then once in stop solution (50 mM phosphate buffer pH 5.8, 1 mM EDTA, 0.1% Tween-20). They were transferred to 80% glycerol and stored at 4 °C overnight to clear the embryos. They were imaged on the Zeiss Axioimager Z.1, as above.
Fluorescent in situ hybridization.
Fluorescent double in situ hybridizations were performed essentially as described46. Embryos were collected from TL/AB in-crosses, dechorionated, cultured to 50% epiboly in agarose-covered dishes at 28 °C. They were fixed in 4% formaldehyde at 4 °C overnight. They were rinsed 2× 10 min with PBST (1× PBS + 0.1% Tween-20), passed through a methanol dehydration series (10 min each, 67% PBST:33% methanol, 33% PBST: 67% methanol), rinsed in methanol 2× 10 min, and permeabilized at −20° at least overnight. Embryos were rehydrated (10 min each, 75% methanol:25% PBST, 50% methanol:50% PBST, 25% methanol:75% PBST, 4× 10 min PBST). Embryos were digested briefly in proteinase K (10 μg/ml, Bioline) for 30 s, then washed 3× 5 min in PBST and refixed for 20 min in 4% formaldehyde at room temperature. They were rinsed 5× 5 min in PBST. Embryos were prehybridized in HM+ buffer (50% formamide, 5× SSC buffer, 0.1% Tween-20, citric acid to pH 6.0, 50 μl/ml heparin, 500 μg/ml tRNA) at 70 °C for at least 2 h. Probes were normalized to 4 ng/μl per probe (digoxigenin - and fluorescein-incorporated) in HM+ buffer and denatured at 70 °C for 10 min. The prehybridization HM+ buffer was replaced by probe and embryos were incubated with probe overnight.
The next morning, probe mix was removed and returned to −20 °C for future re-use. Excess probe was removed with first a series of washes that had been prewarmed to 70 °C: 1× 10 min HM buffer (HM+ without heparin and tRNA), 1× 10 min 75% HM:25% 2× SSC, 1× 10 min 50% HM:50% 2× SSC, 1× 10 min 25% HM, 75% 2× SSC, 1× 10 min 0.2× SSC, 1× 30 min 0.2× SSC; then a series of room temperature washes: 1× 5 min 75% 0.2× SSC:25% PBST, 1× 5 min 50% 0.2× SSC:50% PBST, 1× 5 min 25% 0.2× SSC:75% PBST, 1× 5 min PBST. They were then blocked for at least 3 h in blocking buffer: 2% Blocking Reagent (Roche, 11 096 176 001) in maleate buffer (150 mM maleic acid, 100 mM sodium chloride, pH 7.4). Finally, they were incubated overnight with anti-fluorescein-HRP antibody (Anti-Fluorescein-POD Fab Fragments, Roche 11 426 346 910), diluted 1:400 in blocking buffer at 4 °C with gentle agitation.
The following morning, the antiserum was removed and discarded, and excess antibody was removed by rinsing embryos 3× 25 min in PBST. They were then stained by incubating in 100 μl of Cy5 tyramide reagent diluted 1:25 in amplification diluent (PerkinElmer TSA Plus Cyanine 5 System, NEL745001KT) for 45 min without agitation; this and all subsequent steps were done in the dark to protect fluorophores. Embryos were washed 3× 5 min in PBST. The remaining HRP antibody was inactivated by incubating in 1% hydrogen peroxide (VWR) in PBS for 20 min without agitation. Embryos were washed 3× 5 min in PBST. Antibody was removed by incubating in 0.1 M glycine pH 2.2 for 20 min without agitation. Embryos were washed 3× 5 min in PBST, then blocked in blocking buffer for at least 3 h at room temperature. They were incubated overnight at 4 °C with gentle agitation in 1:500 anti-digoxigenin-POD (Anti-Digoxigenin-POD Fab Fragments, Roche 11 207 733 910).
The following morning, the antiserum was removed and discarded, and excess antibody was removed by rinsing embryos 3× 25 min in PBST. They were stained by incubating in 100 μl of Cy3 tyramide reagent diluted 1:25 in amplification diluent (PerkinElmer TSA Plus Cyanine 3 System, NEL744001KT) for 45 min without agitation. Embryos were washed 8× 15 min in PBST and stored at 4 °C in PBST.
The in situ hybridizations were mounted in glass-bottom dishes in 1% low-melting agarose. They were imaged on a Zeiss LSM700 point-scanning confocal microscope. Imaging of the two channels was performed sequentially at 1,024 × 1,024 pixel resolution with a 20×/0.50 NA objective set at zoom 0.6 (1.5996 pixels per μm), and data were collected as 12-bit data with the photomultiplier tube set to gain 975, the pixel dwell-time at 1.272 and the pinhole set to 1 AU in the Cy3 channel. The sample was sequentially illuminated with 561 nm and 633 nm lasers. There was no signal carryover from the Cy5 to the Cy3 channel, as aplnrb-expressing cells (aplnrb probe in Cy5) at the margin that are not part of the apoptotic-like population do not exhibit any fluorescence in the Cy3 channel. Images were cropped to a 10 z-slice stack (20 μm) in ImageJ, z-projected and contrasted using a linear range with fewer than 5% of pixels saturated.
Binary interpretation of colorigenic in situ hybridizations.
Most of the in situ images scored were collected by searching the ZFIN Gene Expression Database (http://zfin.org/cgi-bin/webdriver?MIval=aa-xpatselect.apg) as well as other literature sources (Supplementary Table 1). Images were selected that were both from the animal pole orientation and lateral orientation in order to score expression accurately both around the embryonic margin and in the animal-vegetal axis. Scales were manually aligned to the in situ images that marked the defined bins—for lateral views, linear scales were used in the animal-vegetal and dorsal-ventral axes that had been corrected for spherical projection, and for animal views, a radial scale was used. In cases where fixation for in situ hybridization had caused the margin to be wavy, the animal-vegetal scale was realigned to the margin in each bin. In other tissues, bins can be defined in any manner that can be scored in the images. They need not be similar in size or shape, nor do they need to be contiguous. They can be cubic, spherical or even arbitrarily shaped, depending on what accurately describes the tissue of interest.
When multiple images were available, they were taken into account (for 14 genes, a single image was used, for the other 33, two to seven images were used, as detailed in Supplementary Table 1). Each image was scored separately, and the patterns were compared. In cases where there was a discrepancy, if there was a clear mode (i.e., out of four pictures, three agreed and one did not), the mode was followed. Otherwise, the broader expression pattern was chosen.
Single-cell RNA-seq: reverse transcription.
Single-cell lysates were transferred from the caps to the wells of the 96-well plate by first thawing the lysates at room temperature for 5 min and centrifuging in a tabletop centrifuge at 630g for 1 min. The lysate plate was transferred to an Agilent Bravo automated liquid handling platform, which automated the following steps. Lysates were mixed with 11 μl (2.2×) of Agencourt RNAClean XP SPRI beads (Beckman-Coulter) and incubated at room temperature for 10 min. The lysate plate was transferred to a magnet (DynaMag-96 Side Skirted Magnet, Life Technologies), the supernatant was removed, and the beads were washed twice in 75 μl of 80% ethanol, with care being taken to avoid loss of beads during the washes. The ethanol was removed, and the beads were left to dry at room temperature for 10 min. The beads were resuspended in 4 μl of Elution Mix (1 μl 10 μM RT primer (5′-AGACGTGTGCTCTTCCGATCT(T)30VN-3′, IDT), 1 μl 10 mM dNTP (Agilent), 0.1 μl SUPERaseIn RNase-Inhibitor (20 U/μl, Life Technologies) and 1.9 μl nuclease-free water). The plate was removed from the Bravo platform and the samples denatured at 72 °C for 3 min and placed immediately on ice afterwards. The plate was placed back on the Bravo platform and 7 μl Reverse Transcription Mix (2 μl 5× RT buffer (Thermo Scientific), 2 μl 5 M Betaine (Sigma-Aldrich), 0.9 μl 100 mM MgCl2 (Sigma-Aldrich), 1 μl 10 μM TSO (5′-AGACGTGTGCTCTTCCGATCTNNNNNrGrGrG-3′, IDT), 0.25 μl SUPERaseIn RNase-Inhibitor (20 U/μl, Life Technologies), 0.1 μl Maxima H Minus Reverse Transcriptase (200 U/μl, Thermo Scientific), and 0.75 μl nuclease-free water) were mixed with the resuspended beads. Reverse transcription was carried out by incubating the plate at 42 °C for 90 min, followed by ten cycles of (50 °C for 2 min, 42 °C for 2 min) and heat inactivation at 70° for 15 min.
Single-cell RNA-seq: PCR pre-amplification.
The plate was returned to the Bravo and 14 μl of PCR Mix (0.5 μl 10 μM PCR primer (5′AGACGTGTGCTCTTCCGATCT-3′, IDT), 12.5 μl 2x KAPA HiFi HotStart ReadyMix (KAPA Biosystems), 1 μl nuclease-free water) were added for a final PCR reaction volume of 25 μl. The reaction was carried out with an initial incubation at 98 °C for 3 min, followed by 18 cycles of (98 °C for 15 s, 67 °C for 20 s, and 72 °C for 6 min) and a final extension at 72° for 5 min. PCR products were purified using the Bravo by mixing with 20 μl (0.8×) Agencourt AMPureXP SPRI beads (Beckman-Coulter) and incubating for 5 min at room temperature. The plate was placed on a magnet for 5 min, the supernatant was removed, and the beads were washed twice with 75 μl of 70% ethanol, with care being taken to avoid loss of beads during the washes. The ethanol was removed, and the beads were left to dry at room temperature for 10 min. The beads were resuspended in 20 μl TE buffer (Teknova). The plate was placed on the magnet and supernatant containing the amplified cDNA was transferred to a new 96-well PCR plate. The concentration of amplified cDNA was measured on the Synergy H1 Hybrid Microplate Reader (BioTek) using High-Sensitivity Qubit reagent (Life Technologies), and the size distribution of select wells was checked on a High-Sensitivity Bioanalyzer Chip (Agilent). Expected quantification was around 0.5–2 ng/μl with size distribution sharply peaking around 2 kb.
Single-cell RNA-seq: library preparation.
Library preparation was carried out using the Nextera XT DNA Sample Kit (Illumina) with custom indexing adapters, allowing 384 libraries to be simultaneously generated in a 384-well PCR plate. For each library, the amplified cDNA was normalized to 0.15–0.20 ng/μl. The tagmentation reaction consisted of 0.625 μl of cDNA mixed with 1.25 μl Tagment DNA Buffer and 0.625 μl Tagment DNA enzyme mix. The 2.5 μl reaction was incubated at 55 °C for 10 min. The reaction was quenched with 0.625 μl Neutralize Tagment Buffer and incubated at room temperature for 5 min. The libraries were amplified by adding 1.875 μl Nextera PCR Master Mix, 0.625 μl 10 μM i5 adaptor (5′-AATGATACGGCGACCACCGAGATCTACAC(i5)TCGTCGGCAGCGTC-3′, IDT, where (i5) signifies the 8 bp i5 barcode sequence (see below for sequences), and 0.625 μl 10 μM i7 adaptor (5′-CAAGCAGAAGACGGCATACGAGAT(i7)GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGGG-3′, IDT, where (i7) signifies the reverse-compliment of the 8 bp i7 barcode sequence (see below for sequences). The PCR was carried out with an initial incubation at 72 °C for 3 min, 95 °C for 30 s, 12 cycles of (95 °C for 10 s, 55 °C for 30 s, 72 °C for 1 min) and a final extension at 72 °C for 5 min. Following PCR, 2 μl of each library were pooled in a 1.5 ml microcentrifuge tube. The pool was mixed with 690 μl (0.9×) Agencourt AMPureXP SPRI beads (Beckman-Coulter) and incubated at room temperature for 5 min. The pool was placed on a magnet (DynaMag-2, Life Technologies) and incubated for 5 min. The supernatant was removed, and the beads were washed twice in 1 ml of 70% ethanol. The ethanol was removed and the beads left to dry at room temperature for 10 min. The beads were resuspended in 50 μl of nuclease-free water. The tube was returned to the magnet, and the supernatant was transferred to a new 1.5 ml microcentrifuge tube. The concentration of the pooled libraries was measured using the High-Sensitivity DNA Qubit (Life Technologies), and the size distribution measured on a High-Sensitivity Bioanalyzer Chip (Agilent). Expected concentration of the pooled libraries was 10–30 ng/μl with size distribution of 300–700 bp.
i5 barcodes: AAGTAGAG, ACACGATC, TGTTCCGA, CATGATCG, CGTTACCA, TCCTTGGT, AACGCATT, ACAGGTAT, AGGTAAGG, AACAATGG, ACTGTATC, AGGTCGCA, GGTCCAGA, CATGCTTA, AGGATCTA, TCTGGCGA, AGGTTATC, GTCTGATG, CCAACATT, CAACTCTC, ATTCCTCT, CTAACTCG, CTGCGGAT, CTACCAGG.
i7 barcodes: CTACCAGG, CATGCTTA, GCACATCT, TGCTCGAC, AGCAATTC, AGTTGCTT, CCAGTTAG, TTGAGCCT, ACCAACTG, GGTCCAGA, GTATAACA, TTCGCTGA, AACTTGAC, CACATCCT, TCGGAATG, AAGGATGT.
Single-cell RNA-seq: read processing, alignment and gene quantification.
We sequenced our single-cell libraries on a HiSeq 2500 (Illumina) in rapid-run mode, obtaining an average of 530,000 paired-end reads per library. We sequenced 25 bp on the first read and 33 bp on the second read, as the first eight bases of the second read consist of (i) a 5 bp random molecular tag (RMT) and (ii) a GGG sequence introduced to the 5′ end of a transcript during template switching. We trimmed the first 8 bp from Read 2, leaving us with paired-end 25 bp reads, although we maintained a separate database linking each read-pair to its accompanying RMT.
To map reads, we modified the Zv9 reference transcriptome, as our reads were expected to originate from the 5′ end of each mRNA molecule, which could cause problems if the reference gene models had an incorrect annotation for the transcription start site (TSS). To address this, we extended the TSS for all Zv9 genes by 100 bases upstream to allow for minor fluctuations. We then aligned read-pairs directly to this modified reference transcriptome, using Bowtie with the following parameters: -q–phred33-quals -n 2 -l 25 -I 1 -X 2000 -a -m 200. As expected, following mapping, our reads overwhelmingly originated from near the TSS.
To quantify gene expression, we leveraged the 5 bp Random Molecular Tags (RMTs) that were associated with each sequencing read pair. For each annotated gene in Zv9, we identified all read-pairs that mapped to the correct strand and collected each of the 5 bp RMTs that were associated with these reads. We collapsed duplicate RMT sequences together to calculate the number of distinct RMTs associated with each gene. We quantified gene expression for 1,152 single-cell libraries and identified a subset of 207 failed/low-quality libraries with poor transcriptome complexity (<2,000 genes detected per cell). After excluding these (remaining data set of 945 cells), on average, we identified 47,000 unique molecules per sequencing library, corresponding to the detection of 3,400 genes per single cell.
To account for differences in the total number of molecules sequenced per library, we normalized Random Molecular Tags (RMTs) counts from each single cell by dividing by the total number of RMTs detected in that cell. Although these numbers are often multiplied by 1 × 106 (i.e., transcripts-per-million), we reasoned that a single cell was unlikely to contain one million transcripts. As we detected a maximum number of 135,000 UMIs across all the cells in our data set, we chose to multiply by 200,000 (i.e., transcripts-per-200,000 reads), but note that this scaling factor largely represents a consistent increase or decrease across all positive values in our data set. All downstream calculations were performed in log-space.
Identification of highly variable genes.
To increase the power of unsupervised dimensional reduction techniques, we first identified the set of genes that was most variable across our single-cell data set, after controlling for the relationship between mean expression and variability. We calculated the mean and a dispersion measure (variance/mean) for each gene across all single cells, and placed genes into 20 bins based on their average expression. Within each bin, we then z-normalized the dispersion measure of all genes within the bin to identify genes whose expression values were highly variable even when compared to genes with similar average expression. We used a z-score cutoff of 2 to identify 160 significantly variable genes, after excluding genes with very low average expression, or genes whose variability was explained primarily by differences between experimental batches. As expected, our highly variable genes consisted primarily of developmental and spatially regulated factors whose expression levels are expected to vary across the dissociated cells.
Principal components analysis.
We ran principal components analysis (PCA) as previously described8, using the prcomp function in R, after scaling and centering the data. We used only the previously identified ‘highly variable’ genes as input to the PCA to ensure robust identification of the primary structures in the data. However, as this encompassed only 160 genes, we extended the results of this analysis globally by projecting the PCA rotation matrix across the entire transcriptome. This additional projection does not enable us to discover new structures or patterns that are not present within the 160-gene PCA, but it does allow us to identify other genes with strong PCA loadings that may not have passed our stringent test for single-cell variation.
Identification of EVL cells.
When we ran PCA on the full data set of 945 cells, we observed that the second principal component was strongly defined by canonical markers for the embryonic enveloping layer (EVL), an epidermal shell that coats the embryo at this stage. We removed the cells through visual inspection and annotation of a PCA plot, resulting in a final data set of 851 single cells that was used as input to Seurat. After removal of the EVL cells, we recalculated the list of highly variable genes using the same procedure described above.
Constructing models of gene expression (data imputation).
Seurat leverages a spatial reference map consisting of a relatively small number of landmark genes in order to guide the inference of spatial origin. Single-cell measurements for a single gene, however, are inherently noisy, with extensive variability stemming from both biological and technical sources21,22. These sources of noise can introduce considerable error into the reconstruction process, where substantial value is placed upon the measurement of landmark genes.
To address this, we reasoned that instead of relying only on the measurements of the landmark genes, we could use our full RNA-seq profiles to improve our spatial inference. For example, the gene osr1 is a member of our spatial reference map and is known to be expressed in a tight band around the embryonic margin. Suppose we examine cell X, which is truly located at the margin, but where expression of osr1, either due to biological or technical noise, is detected at low level. However, other genes that are specific to the embryonic margin are highly expressed in cell X, consistent with its spatial origin. In this case, cell X's overall gene expression profile predicts a high level of osr1 expression, strongly suggesting that the measured value represents a technical error. Importantly, it is not necessary to know beforehand which other genes are expressed with osr1, nor any spatial information, as these genes can be directly identified based on their power to predict osr1 expression.
Thus, for each of the landmark genes, we constructed a linear model of single-cell expression for that gene, based on the measured values of all highly variable (z-score greater than two, see above) or ‘structured’ genes in the data set (genes with statistically significant loading scores, P < 10−5) for the first three principal components). We determined the set of genes with a significant PCA loading using a randomization approach (‘jack straw’) proposed by Chung and Storey47 and which we have previously applied to single-cell RNA-seq data29.
In principle, we could have many more predictive genes than cells, and so to avoid overfitting, we built L1-constrained models of gene expression using the LASSO technique, as implemented in the lars package in R. The LASSO algorithm requires a user-specified L1-constraint. While in principle this learning parameter could be set separately for each gene, we imposed a uniform parameter across all genes (n = 40 in the lasso.fit function, empirically determined). We then constructed a separate matrix of ‘imputed’ measurements for each of our landmark genes across all single cells. We note that although we use the LASSO approach here, Seurat's downstream analysis is widely compatible with any modeling or prediction technique, and we believe that implementing tailored machine-learning approaches to ameliorate technical noise in single-cell data sets represents a promising direction for future work.
Mixture model fitting to translate between RNA in situ hybridization and RNA-seq data.
Although our spatial reference map consisted of binary values for the landmark genes, our imputed measurements were on a continuous scale, and thus we needed a mapping to relate these two types of data. We reasoned that the bimodal distribution that characterized the imputed measurements represented an ‘on’ and ‘off’ mode of gene expression across single cells. Thus, we independently fit the distribution of imputed values for each landmark gene as a mixture of two Gaussian distributions, implemented using the normalmixEM function in the mixtools R package, with two modifications. First, in order to ensure coherence between the RNA-seq and the RNA in situ hybridization data, we constrained the mixing parameter to be equal to the percentage of bins represented in the ‘on’ state in the binarized in situ patterns. Second, our mixture models effectively divide cells into two clusters based on their imputed expression of a single landmark gene. To ensure that this subdivision was consistent with our overall data structure, we implemented one additional heuristic step, similar to a single step in a ‘greedy’ k-means approach. We calculated the cluster-means (vector representing the average expression of all highly variable or ‘significantly structured’ genes across n single cells in either the ‘on’ or the ‘off cluster’), calculated the L2 distance of each cell to the two cluster means, and reassigned cells to the closest population. Effectively, this step allowed a small number of cells to ‘flip’ between the on and off subpopulations in a manner that was consistent with the overall structure in the data, and we found that this improved the robustness of our mixture model fitting. Seurat then estimates the normal density parameters of the two modes by calculating the mean and variance of the imputed values.
Probabilistic inference of spatial origin.
Seurat leverages the spatial reference map and mixture models to build individual models of gene expression for each of the 64 bins. Specifically, Seurat models the imputed expression values across all landmark genes as a multivariate normal distribution, and therefore builds 64 distinct multivariate normal models. For our initial models, Seurat makes two simplifying assumptions to limit the resulting complexity. First, we assume that within a bin, imputed expression levels of the different landmark genes are independent of each other. This means that the off-diagonal elements of all covariance matrices are 0. Second, given the binary nature we have chosen for the input, we assume that for any given landmark gene, the mean and variance parameters can each take one of two possible values, taken directly from the mixture model, and depending on whether the gene is ‘on’ or ‘off’ in this bin in the spatial reference map. These assumptions strongly simplify the model, and result in extensive parameter sharing across all 64 bins.
Once these models have been estimated, Seurat examines the imputed landmark expression values for a cell and calculates the likelihood that these values originated from each of the 64 bins using the dmvnorm function (mixtools48 package in R). As the prior probability that a cell originated from any of the 64 bins is uniform, the likelihood is directly proportional to the posterior probability. Thus, Seurat calculates the posterior probability that a cell originated from each of 64 bins. These individual probabilities are retained, but also summarized to a single location by calculating the spatial centroid (specifically, the center of mass) of the spatial probability map. We therefore calculated spatial centroids for all 851 single cells in our data set.
Both of the assumptions made here do not perfectly reflect the biological nature of the zebrafish embryo at 50% epiboly. At this stage, developmental patterning genes often do not exhibit an exclusively binary expression pattern, but are often expressed at multiple different levels. Given the binary nature of our input spatial reference map, our initial models require the mean and variance parameters in each bin to take one of only two possible parameter values across the embryo. Second, assuming independent expression of the landmark genes is not well-justified. Many of these genes are likely to be co-regulated and to exhibit correlated expression even within a bin, particularly as Seurat considers imputed gene expression values.
To extend our initial models we wished to remove the assumption of independence between genes, and to estimate mean and variance parameters separately for each cell and each bin. In order to estimate a valid covariance matrix for n landmark genes for an individual bin, we needed data from at least n cells (we use 2n in practice) that were representative of that bin. We reasoned that we could use our initial mapping of 851 spatial centroids (above) to identify these cells, and therefore to estimate these parameters.
We implemented the following procedure for each bin. (i) We calculated the L2 distance for each of the 851 spatial centroids to the center of the specified bin. (ii) We selected the 2n cells that had the lowest L2 distance (where n is the number of landmark genes) and were likely to be close in space to the bin. (iii) We used these cells to estimate a vector of n means as well as an n × n covariance matrix specific to each bin. At the end of this procedure, we had constructed 64 new multivariate normal models. As before, we then used Seurat to calculate the likelihood that a cell originated from each of these bins, given these updated models.
We note that the cells selected as ‘closest’ to each bin may not be unique for each of the 64 bins, and some cells are therefore likely to contribute to the estimated models for multiple bins. This represents a smoothing across the embryo that is similar to a sliding window, particularly when the number of landmark genes is high. For example, computing covariance matrices for the 47 landmark genes in our original reference map would require 100 cells per bin, which is not available in our current scale of data. Thus, we aimed to most efficiently choose a new set of landmark genes. Notably, as we already had a preliminary spatial mapping, we were no longer restricted to choosing genes that were part of the original spatial reference set. We reasoned that the most informative genes would be those that had the strongest loadings in a PCA, and therefore selected an n of 18 genes (three genes with the highest and lowest loadings for each of three principal components) to use as input for this analysis.
Evaluating Seurat's performance.
We examined a control experiment consisting of cells from a modified dissociation and dissection procedure, which strongly depletes cells from the animal cap and therefore enriches for the embryonic margin. We calculated the centroid (defined as the center of mass of Seurat's inferred probability mapping), of both enriched and nonenriched cells, and compared the percentage of cells that mapped to each tier-bin (1–8)—calculating the enrichment (fold-change) for the margin-enriched compared to the randomly dissociated cells.
We further tested Seurat's performance by examining 28 ‘reference cells’ which were manually isolated under a dissection microscope, and whose spatial origin is approximately known. For each reference cell, we compared Seurat's inferred origin with its experimentally measured spatial location. As Seurat assigns cells to an origin probabilistically, we wanted to reflect any uncertainty in our error measurements. For every reference cell, we examined all bins where Seurat assigned the cell with nonzero posterior probability and constructed a posterior probability-weighted distance metric, weighting the distance between the inferred bin and the measured bin by the posterior probability of assignment.
Additionally, we used Seurat to infer spatial patterns for genes with known expression patterns (i.e., the landmark genes). Specifically, for each landmark gene G, we performed the following process.
a. We removed G from the spatial reference map, leaving a total of 46 genes remaining in the map.
b. We reran Seurat on the input data. In this case, the localization of G was not known to Seurat, and could not influence downstream inferences. For all bins Bi, i = 1.64, and cells Cj, j = 1.851, Seurat calculates the posterior probability:
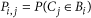
c. We ‘inferred’ the spatial localization of G given Seurat's posterior inferences. Our inferred in situ represents a probability-weighted estimate of gene expression across the entire embryo. Specifically for all bins Bi, i = 1.64, we calculated the expression level of gene G in the bin as:

where M is the measured expression matrix and M(G,Cj) represents the nonlogged expression level of gene G in cell Cj. Note that for the evaluation of all final in situ patterns, we used the measured (nonimputed) estimates of gene expression in this inference. Thus, the imputed values help to guide individual cells to their correct spatial origin (i.e., to calculate Pi,j), but the inferred in situ patterns in Figures 3f, 4b, 5d and Supplementary Figure 5 use the measured (nonimputed) values for M.
d. We tested whether our inferred expression levels were sufficient to correctly classify the binarized landmark in situ pattern. We assayed the accuracy of this classification using an ROC curve.
Determination of archetypal patterns of gene expression.
Seurat's spatial inferences enable us to not only re-infer spatial patterns for the landmark genes, but also to create computational in situ patterns for any gene detected in our RNA-seq data. We therefore inferred spatial patterns for all genes that were likely to exhibit spatially restricted expression patterns across our data set. Specifically, we took all genes that displayed weak evidence of being variable across our single cells (see ‘Identification of highly variable genes’, though here we applied a z-score cutoff of 1 instead of 2) and added all genes exhibiting ‘significant’ loadings of the first five principal components using a Bonferroni-corrected P-value of 0.01. Finally, we removed genes that were detected in less than 20 cells in the overall data set, as they may present strong spatial patterns that are simply the result of aberrant expression in a very small number of cells, leaving us with 2,190 remaining genes. We then inferred the spatial localization patterns of all these genes. As our goal at this stage was to identify broad clusters of spatial gene patterning, we inferred spatial patterns using imputed measurements for each of these genes to ameliorate technical noise.
We further examined these 2,190 patterns to search for genes whose expression patterns exhibited significant spatial variability across the embryo. To accomplish this, we calculated a ‘spatial CV’ for each gene, by calculating the coefficient of variation of its expression levels across all 64 bins. We identified 290 genes with a CV greater than 0.25, implying spatial heterogeneity. We chose this cutoff because it excluded known housekeeping genes (e.g., ribosomal proteins) from further analysis, as these genes are unlikely to be heavily spatially patterned.
We next performed k-means clustering on the remaining 290 patterns. Specifically, the input for the k-means clustering was a 290 × 64 matrix, containing the expression level of all 290 genes in each of the 64 spatial bins. We used a k = 9, as this was the largest value of k for which we observed distinct and nonoverlapping clusters9. The nine clusters represent ‘archetypes’ of gene expression, namely, broad spatial patterns representing clusters of similarly localized genes.
Downsampling (power) analyses.
In order to test the number of landmark genes required for a spatial reference map, we remapped cells using only subsets of the landmark gene set in our study. Our baseline was remapping the cells using 46 of the 47 landmarks that were included in our archetypal analysis, without the optional quantitative refinement step. We then randomly selected 2, 4, 6 or all 9 of the archetypes and selected evenly across the chosen archetypes to produce a final set of 2–45 landmark genes. Although we selected evenly from the different archetypes, some archetypes had fewer landmarks than others; thus, in many cases, there are fewer genes from one archetype than another, if we had selected all of the landmarks present from a given archetype. After choosing the limited set of archetypes, we remapped cells and compared the mean change in centroid position, as Euclidian distance, in terms of bins, and the sum of the change in posterior probabilities.
In order to assess the effect of redundant landmarks more directly, we chose two sets of 4 landmarks that had identical binary input patterns. One set (osr1, mixl1, ndr1 and ndr2) were expressed narrowly around the embryo in the two bins closest to the margin, and the other set (ta, ism1, sebox and tbx16) were expressed more broadly around the embryo in the four bins closest to the margin. Thus, these patterns defined a total of three bins (one defined by the absence of genes from either set, another defined by the expression of the broader set but absence of the narrower set, and a final bin defined by the expression of both the broader and narrower set). As a baseline, we calculated the posterior probabilities using the full set of 47 landmarks, without quantitative refinement, and then summed probabilities from the smaller bins to create the three large bins. We then remapped cells using every combination of one, two, three or four landmarks from each set and measured the total change in posterior probabilities.
Identification and characterization of embryonic subpopulations.
We used a combination of supervised and unsupervised analyses to identify rare and functionally coherent subpopulations in the developing zebrafish embryo. To identify cells representing progenitors for the prechordal plate and the endoderm, we first used Seurat's spatial mappings to identify all cells in the three (out of eight) spatial tiers closest to the embryonic margin, corresponding to a total of 252 cells. We then performed an unbiased PCA of these cells, and performed k-means clustering for genes that were significantly associated with the three principal components (jack straw, P < 10−5). We identified two distinct clusters of cells from the k-means analysis. Cells from these clusters were also clearly distinguished by the second and third principal components, as well as by the expression patterns of known marker genes (e.g., gsc and sox32), enabling us to identify these cells as either prechordal plate or endoderm progenitors. A similar PCA analysis, conducted over all 851 cells in the data set, identified a separate subpopulation strongly distinguished by the fourth principal component (‘Apoptotic-like’ cells, Supplementary Fig. 6f).
To identify markers that were enriched for the prechordal plate progenitors and the ‘apoptotic-like’ cells, we implemented a likelihood-ratio test (LRT) for single-cell differential expression49. Importantly, this test is designed to simultaneously test for changes in both the percentage of cells expressing a gene, as well as the quantitative RNA levels with these cells. All differential expression testing was performed on measured (nonimputed) values.
For Gene Ontology analysis of ‘apoptotic-like’ markers, we took all genes with a Bonferroni-corrected LRT P-value < 0.01 and used this list for Gene Set Enrichment Analysis50.
Seurat package.
Seurat is available as an open-source software package in R, and is available as the Supplementary Code. In addition, clear documentation (R markdown files) showing the commands and output for the analysis of this data set are included in the Supplementary Note.
Accession codes.
GEO: GSE66688.
Accession codes
References
Graveley, B.R. et al. The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479 (2011).
Gerstein, M.B. et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330, 1775–1787 (2010).
Schier, A.F. Genomics: zebrafish earns its stripes. Nature 496, 443–444 (2013).
Islam, S. et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 21, 1160–1167 (2011).
Ramsköld, D. et al. Full-length mRNA-seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777–782 (2012).
Jaitin, D.A. et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014).
Pollen, A.A. et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 32, 1053–1058 (2014).
Shalek, A.K. et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240 (2013).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Treutlein, B. et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014).
Hashimshony, T., Wagner, F., Sher, N. & Yanai, I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Reports 2, 666–673 (2012).
Deng, Q., Ramsköld, D., Reinius, B. & Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196 (2014).
Lovatt, D. et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat. Methods 11, 190–196 (2014).
Lee, J.-H. et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014).
Ho, R. & Kimmel, C. Commitment of cell fate in the early zebrafish embryo. Science 261, 109–111 (1993).
Schier, A.F. & Talbot, W.S. Molecular genetics of axis formation in zebrafish. Annu. Rev. Genet. 39, 561–613 (2005).
Kimmel, C.B., Warga, R.M. & Schilling, T.F. Origin and organization of the zebrafish fate map. Development 108, 581–594 (1990).
Roussigné, M., Blader, P. & Wilson, S.W. Breaking symmetry: the zebrafish as a model for understanding left-right asymmetry in the developing brain. Dev. Neurobiol. 72, 269–281 (2012).
Solnica-Krezel, L. & Sepich, D.S. Gastrulation: making and shaping germ layers. Annu. Rev. Cell Dev. Biol. 28, 687–717 (2012).
Durruthy-Durruthy, R. et al. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell 157, 964–978 (2014).
Brennecke, P. et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 10, 1093–1095 (2013).
Kharchenko, P.V., Silberstein, L. & Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 11, 740–742 (2014).
Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Series B Stat. Methodol. 58, 267–288 (1996).
Howe, D.G. et al. ZFIN, the Zebrafish Model Organism Database: increased support for mutants and transgenics. Nucleic Acids Res. 41, D854–D860 (2013).
Kudoh, T. et al. A gene expression screen in zebrafish embryogenesis. Genome Res. 11, 1979–1987 (2001).
Thisse, B. et al. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 77, 505–519 (2004).
Soumillon, M., Cacchiarelli, D., Semrau, S., van Oudenaarden, A. & Mikkelsen, T. Characterization of directed differentiation by high-throughput single-cell RNA-seq. Preprint at http://biorxiv.org/content/early/2014/03/05/003236 (2014).
Grün, D., Kester, L. & van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat. Methods 11, 637–640 (2014).
Shalek, A.K. et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369 (2014).
Kimmel, C.B., Ballard, W.W., Kimmel, S.R., Ullmann, B. & Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 (1995).
Le Guellec, D., Morvan-Dubois, G. & Sire, J.-Y. Skin development in bony fish with particular emphasis on collagen deposition in the dermis of the zebrafish (Danio rerio). Int. J. Dev. Biol. 48, 217–231 (2004).
Kikuchi, Y. Casanova encodes a novel Sox-related protein necessary and sufficient for early endoderm formation in zebrafish. Genes Dev. 15, 1493–1505 (2001).
Yoon, C., Kawakami, K. & Hopkins, N. Zebrafish vasa homologue RNA is localized to the cleavage planes of 2- and 4-cell-stage embryos and is expressed in the primordial germ cells. Development 124, 3157–3165 (1997).
Stachel, S.E., Grunwald, D.J. & Myers, P.Z. Lithium perturbation and goosecoid expression identify a dorsal specification pathway in the pregastrula zebrafish. Development 117, 1261–1274 (1993).
Fürthauer, M., Thisse, B. & Thisse, C. Three different noggin genes antagonize the activity of bone morphogenetic proteins in the zebrafish embryo. Dev. Biol. 214, 181–196 (1999).
Kawahara, A., Wilm, T., Solnica-Krezel, L. & Dawid, I.B. Antagonistic role of vega1 and bozozok/dharma homeobox genes in organizer formation. Proc. Natl. Acad. Sci. USA 97, 12121–12126 (2000).
Seo, H.-C., Drivenes, Ø., Ellingsen, S. & Fjose, A. Expression of two zebrafish homologues of the murine Six3 gene demarcates the initial eye primordia. Mech. Dev. 73, 45–57 (1998).
Fodor, E. et al. Full transcriptome analysis of early dorsoventral patterning in zebrafish. PLoS ONE 8, e70053 (2013).
Pauli, A. et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science 343, 1248636 (2014).
Achim, K. et al. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol. 33, doi:10.1038/nbt.3209 (13 April 2015).
Combs, P.A. & Eisen, M.B. Sequencing mRNA from cryo-sliced Drosophila embryos to determine genome-wide spatial patterns of gene expression. PLoS ONE 8, e71820 (2013).
Junker, J.P. et al. Genome-wide RNA tomography in the zebrafish embryo. Cell 159, 662–675 (2014).
Pauli, A. et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 22, 577–591 (2012).
Bennett, J.T. et al. Nodal signaling activates differentiation genes during zebrafish gastrulation. Dev. Biol. 304, 525–540 (2007).
Thisse, C., Thisse, B., Schilling, T.F. & Postlethwait, J.H. Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119, 1203–1215 (1993).
Clay, H. & Ramakrishnan, L. Multiplex fluorescent in situ hybridization in zebrafish embryos using tyramide signal amplification. Zebrafish 2, 105–111 (2005).
Chung, N.C. & Storey, J.D. Statistical significance of variables driving systematic variation in high-dimensional data. Bioinformatics 31, 545–554 (2015).
Benaglia, T., Chauveau, D., Hunter, D. & Young, D. Mixtools: an R package for analyzing finite mixture models. J. Stat. Softw. 32, 1–29 (2009).
McDavid, A. et al. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics 29, 461–467 (2013).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Acknowledgements
We thank members of the Regev and Schier laboratories for helpful discussion, O. Wurtzel for assistance with the R markdown scripts, A. Pauli for sharing the aplnrb plasmid, K. Rogers for donating superior, unpublished in situ hybridizations. This work was supported by F32 HD075541 (R.S.), the Jane Coffin Childs Memorial Fund for Medical Research (J.A.F.), the NIH (A.F.S.), National Human Genome Research Institute, Center of Excellence in Genome Science 1P50HG006193, the Klarman Cell Observatory and Howard Hughes Medical Institute (A.R.).
Author information
Authors and Affiliations
Contributions
R.S., J.A.F., A.F.S. and A.R. conceived the research. R.S. and J.A.F. wrote the Seurat package and performed computational analysis of the RNA-seq data. J.A.F., D.G. and R.S. performed experimental work. J.A.F. and R.S. analyzed the data and produced figures. J.A.F., R.S., D.G., A.F.S. and A.R. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.R. is a consultant to the Driver Group, which analyzes tumor samples, a potential sample fodder for spatial analysis.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7, Supplementary Tables 1 and 2, Supplementary Text and Supplementary Note (PDF 6126 kb)
Supplementary Code
Seurat source code (ZIP 28 kb)
41587_2015_BFnbt3192_MOESM13_ESM.mov
Animal cap depletion and dissection of margin-enriched cell population from zebrafish embryos for dissociation (MOV 9690 kb)
Rights and permissions
About this article

Cite this article
Satija, R., Farrell, J., Gennert, D. et al. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502 (2015). https://doi.org/10.1038/nbt.3192
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.3192
This article is cited by
-
Tissue-specific RNA Polymerase II promoter-proximal pause release and burst kinetics in a Drosophila embryonic patterning network
Genome Biology (2024)
-
SuperCellCyto: enabling efficient analysis of large scale cytometry datasets
Genome Biology (2024)
-
Dynamics of host immune responses and a potential function of Trem2hi interstitial macrophages in Pneumocystis pneumonia
Respiratory Research (2024)
-
Multi-omics and immunogenomics analysis revealed PFKFB3 as a targetable hallmark and mediates sunitinib resistance in papillary renal cell carcinoma: in silico study with laboratory verification
European Journal of Medical Research (2024)
-
Identification of female-enriched and disease-associated microglia (FDAMic) contributes to sexual dimorphism in late-onset Alzheimer’s disease
Journal of Neuroinflammation (2024)
