
Abstract
Loss of seed dispersal is a key agronomical trait targeted by ancient human selection and has been regarded as a milestone of crop domestication. In this study, in the legume crop soybean Glycine max (L.) Merr. which provides vegetable oils and proteins for humans, we show that the key cellular feature of the shattering-resistant trait lies in the excessively lignified fibre cap cells (FCC) with the abscission layer unchanged in the pod ventral suture. We demonstrate that a NAC (NAM, ATAF1/2 and CUC2) gene SHATTERING1-5 (SHAT1-5) functionally activates secondary wall biosynthesis and promotes the significant thickening of FCC secondary walls by expression at 15-fold the level of the wild allele, which is attributed to functional disruption of the upstream repressor. We show that strong artificial selection of SHAT1-5 has caused a severe selective sweep across ~116 kb on chromosome 16. This locus and regulation mechanism could be applicable to legume crop improvement.
Similar content being viewed by others

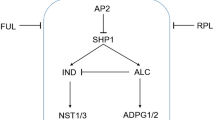
Introduction
A critical turning point in human history is the advent of Neolithic Revolution (~10,000 years ago) that resulted in a broad array of domesticated plant and animal species from their respective wild ancestors1,2,3. Loss of seed dispersal or fruit dehiscence, an agronomically important trait, was frequently targeted by human selection to enable more efficient cultivation during most crop domestication2,3. In cereal crops, the loss of seed shattering has been revealed to be mostly associated with abscission layer (AL) elimination or modification at the junction between the pedicel and the lemma4,5,6,7,8,9,10. However, anatomical differences between the fruit structures of monocot (such as cereals) and eudicot crops (such as legumes) imply fundamentally different mechanisms underlying seed shattering and pod shattering6,11,12. It has been widely accepted that soybean was domesticated from its annual wild relative, Glycine soja Sieb. and Zucc., in East Asia ~5,000 years ago1,13. Agricultural and biological scientists have continued to try to find the tissue contributing to pod shattering resistance and its underlying genetic mechanism in cultivated soybeans11,12. Quantitative trait locus (QTL) mapping has proven to be an effective approach in dissecting the gene (s) responsible for the domestication syndrome in several crop species14. In rice (Oryza sativa), maize (Zea mays) and tomato (Lycopersicon esculentum), QTL mapping has enabled the successful isolation and characterization of genes that govern plant architecture (TEOSINTE BRANCHED1), seed shattering (sh4) and fruit size (fw2.2)7,15,16. In the past 15 years, QTL mapping in soybean has identified a series of genomic regions related to pod shattering scattered among several linkage groups17,18,19,20,21. qPDH1 is a recently identified and fine-mapped pod shattering QTL on linkage group J (chromosome 16) with 10 putative candidate genes20,21. Unfortunately, no single gene of the 10 candidate genes has been implicated as the regulator for pod shattering resistance. In addition, no observable difference between shattering-resistant and shattering-susceptible phenotypes of near-isogenic lines was found to be associated with pod shattering resistance20. Therefore, it was suggested that pod shattering resistance might have been achieved through undetectable subtle morphological changes in fruit structure, or associated with other factors rather than morphology20. Although extensive work has been done to find the structures and controlling genes that contribute to pod shattering resistance in soybean (for example, see the study by Saxe et al.17, Bailey et al.18, Liu et al.19 and Suzuki et al.20), the exact cellular and genetic mechanisms underlying this trait have yet to be determined. Identification of the exact structure and the genes controlling shattering that were targeted by artificial selection would not only decipher the genetic mechanisms underlying the fixation of such a trait in domesticated soybeans but also offer insights into the evolution of complex morphological traits widely existing in nature4,22,23.
In this study, we employed multiple experimental approaches to explore the genetic mechanism underlying the evolution of shattering-resistant phenotype in domesticated soybeans. We found that the excessively lignified fibre cap cells (FCC) endowed the domesticated soybean with pod shattering-resistance phenotype and were promoted by a NAC gene SHAT1-5 by expression at 15-fold the level of the wild allele through repressor disruption. This regulatory change is correlative with strong artificial selection of GmSHAT1-5 during soybean domestication with hitchhiking effect on closely linked loci across ~116 kb in chromosome 16 of the soybean genome. This mechanism is distinct from the one underlying grain shattering resistance of domesticated cereals.
Results
FCC are involved in pod shattering resistance
HEINONG44 (G. max) is an elite cultivar from Heilongjiang province in Northeast China, which is resistant to pod shattering, and ZYD00755 (G. soja) is a wild soybean from Northeast China, which is shattering susceptible (Fig. 1a–c). We confirmed the shattering difference between HEINONG44 and ZYD00755 by shattering index analysis (5% for HEINONG44 versus 100% for ZYD00755) and mechanical force test (13.6 N for HEINONG44 versus 2.5 N for ZYD00755) (Methods) (Supplementary Fig. 1).

(a) Plant architecture and pod shattering character of cultivated soybean HEINONG44 and wild soybean ZYD00755. (b) Magnified views of mature pod of HEINONG44 (~50 DPA). (c) Magnified views of mature pod of ZYD00755 (~50 DPA). (d) Semi-thin cross sections (500 nm) of the HEINONG44 ventral suture of pods showing FCC located at the junctions between two vascular bundle valves with the AL below. (e) Magnified views of (d), showing the secondary walls of FCC were heavily thickened with more secondary deposition in the upper part than lower part in HEINONG44. (f) Semi-thin cross sections (500 nm) of the ZYD00755 ventral suture of pods. (g) Magnified views of (f), showing the secondary walls of FCC were poorly developed with only moderate deposition in the upper part in ZYD00755. vb, vascular bundle. Scale bars in (a), 10 cm; (b,c), 1 cm; (d,f), 200 μm; (e,g), 80 μm.
Cross sections of mature fruit (30 days past anthesis, DPA) of HEINONG44 and ZYD00755 showed that both fruits had a lignified endocarp and respective vascular bundle in dorsal and ventral sutures (Supplementary Fig. 2a–d), as previously described11,12. At the ventral suture, where pod shattering first took place, the two vascular bundle valves in both species were jointed along the fused line of the carpel with parenchymal AL delimitating the vascular bundle valve (Supplementary Fig. 2e,f). It is obvious that the two ventral vascular bundle valves were separated more easily in ZYD00755 than in HEINONG44 as fruit gradually become dried (Supplementary Fig. 2a,b). We next focused our attention on the elaborate structure of the ventral suture and attempted to find the cellular difference between HEINONG44 and ZYD00755 by detailed anatomical examination of semi-thin sections of the pod ventral sutures. We found that the secondary wall of the FCC in the ventral suture of the pod was heavily thickened in G. max but poorly developed in G. soja (Fig. 1e,g). It is also evident in the dried fruit of HEINONG44 that has two valves of the vascular bundle connected by only the FCC (Supplementary Fig. 3). In contrast, there were no differences detected in the AL cells between HEINONG44 and ZYD00755 (Fig. 1d–g). A previous study demonstrates that an endopolygalacturonase (Endo-PG), which is expressed in the fruit AL, is necessary for cell wall disintegration in cultivated soybean12. The effective breakdown of the AL in both HEINONG44 and ZYD00755 suggests that these homologous cells were not remarkably changed during soybean domestication. The AL, which consists of parenchyma cells and facilitates separations of organs, has been the recurrent target of selection during the crop domestication process24. In rice, sorghum (Sorghum bicolor) and tomato, the disruption of a functional AL is responsible for the loss of seed shattering and fruit shedding7,10,25. Our results herein are indicative of the excessive secondary wall thickening in the FCC rather than the modification of the AL that endows the pod with shattering resistance in cultivated soybeans.
Candidate genes associated with pod shattering resistance
Upon comparison and homologue analyses, the FCC with a lower AL in soybean pods is likely homologous to the lignified valve margin cells with a lateral AL in Arabidopsis siliques in structure and dehiscence function11,12,26. We therefore cloned 11 soybean orthologues of the Arabidopsis SHP1/2, IND, NST1/2, ALC, which were involved in the regulatory network to establish the identity of the valve margin cells and AL (Supplementary Fig. 4). GlycineEndo-PG, which encodes an Endo-PG and is expressed in the fruit AL, was also cloned12. We then examined the noncoding nucleotide diversity of these genes and compared the value with three previously characterized neutrally evolving genes (Glyma13g07220, Glyma13g17830 and Glyma19g33520) (Hyten et al.27; Guo et al.28) from a panel of cultivated and wild soybean accessions from diverse geographical locations in China (Supplementary Table 1). For the three neutral genes, sequence analysis revealed that the cultivated soybean (π ranging from 0.00042 to 0.00176) preserved about 50% of the variation found in wild soybean populations (π ranging from 0.00094 to 0.00347), and the relative genetic diversity between cultivated and wild soybean (πGm/πGs) is 45–53% (Table 1), suggesting a bottleneck effect during soybean domestication as previously reported27,28,29. Ten of the twelve candidate genes in cultivated soybeans retained 35~91% of the genetic diversity present in G. soja (Table 1). In addition, Tajama’s D and Fu and Li’s D* test for non-neutral evolution indicated that these 13 genes (including three neutral markers) evolved neutrally in both wild and cultivated soybeans (Supplementary Table 2). Thus, it seems unlikely that these genes were targeted by artificial selection and evolved in a non-neutral manner during the domestication process. However, Glyma04g39210 and Glyma16g02200 exhibited a dramatic reduction in diversity such that all soybean accessions share the same haplotype. All single-nucleotide polymorphism (SNP) variation was lost in cultivated soybean populations (Table 1). By scanning the soybean QTL map (Grant et al.30), we further confirmed that Glyma16g02200 was localized within an identified QTL region associated with pod dehiscence (pod dehis 1–5), which was repeatedly identified in several independent mapping populations17,18,19. Meanwhile, Glyma04g39210 resided in a genomic segment with QTLs unrelated to pod dehiscence. Glyma16g02200 encodes a NAC domain transcription factor with a putative function in secondary cell wall thickening and is the homologue of AtNST1/2 in Arabidopsis (Supplementary Fig. 4). The putative function of Glyma16g02200 is consistent with the anatomical difference in the FCC between HEINONG44 and ZYD00755. In addition, evidence shows that the function of NAC domain super family in activating secondary cell wall thickening is widely conserved between monocots and dicots31,32. Given the highly conserved function of the NST1-like gene in transcription regulation of secondary cell walls, we believe that Glyma16g02200 is the most likely underlying gene for pod dehis 1–5 QTL. We here designated this gene as SHATTERING1-5 (SHAT1-5) according to the name (pod dehis 1–5) given by the soybean QTL database30.
Expression and functional analyses of SHAT1-5
Real-time qPCR was conducted to determine the expression pattern of SHAT1-5 gene. SHAT1-5 was expressed at relatively low levels in tissues that have a low content of sclerenchymatous cells (leaf, young root, flower and inflorescence), and at high levels in stems and fruits, which contain cells with heavily secondary cell wall thickening in both HEINONG44 and ZYD00755 (Supplementary Fig. 5a; expression of GsSHAT1-5, also known as GsNST1B, in ZYD00755 was recently reported in Dong et al.33). During fruit development, SHAT1-5 remained at a fairly low expression level before stage-5 (13 DPA); thereafter SHAT1-5 exhibited a sharp increase in expression at stage-6 fruits (18 DPA) and adopted a significant drop at the final developmental stage (24 DPA) in both HEINONG44 and ZYD00755 (Supplementary Fig. 5b,c). To determine the expression localization of SHAT1-5 in fruits, we prepared fruit sections of two different stages (stage-5, 13 DPA; stage-6, 18 DPA) and detected SHAT1-5 proteins using a SHAT1-5 specific antibody. In stage-5 fruit (13 DPA), GmSHAT1-5 proteins were evidently localized specifically in the cells that were differentiating into FCC in the pod ventral suture (Fig. 2a), and were intensely accumulated in the FCC of the pod ventral suture in stage-6 (18 DPA) fruits (Fig. 2b). We next used a laser microdissection system to isolate the FCC and AL tissue in stage-6 fruits (18 DPA) from HEINONG44 and ZYD00755, respectively, to examine whether SHAT1-5 had differential expression between them (Supplementary Fig. 6). Real-time qPCR results showed that GmSHAT1-5 was expressed in HEINONG44 at 15-fold the level of GsSHAT1-5 in ZYD00755 (Fig. 2d), while other genes (GlycineSHP-A/B, GlycineALC-A/B, GlycineNST1, GlycineNST2, GlycineIND-A/B/C/D, GlycineEndo-PG) had no obvious difference in expression between the wild and cultivated orthologues (Fig. 2d,e). The highly increased expression of GmSHAT1-5 is strongly correlated with the heavily thickened FCC secondary walls in cultivated soybeans. These results suggest that the heavily thickened FCC in cultivated soybean is attributed to the over-expression of the SHAT1-5 gene.

(a) GmSHAT1-5 protein localized in developing FCC of stage-5 fruit (13 DPA) (red arrow). (b) GmSHAT1-5 protein intensely accumulated in FCC of stage-6 fruit (18 DPA) with signal stronger in the upper part. (c) Negative control with empty primary antibody generating no signal. (d) Expression analysis of all orthologues in the FCC. (e) Expression analysis of all orthologues in the AL. Note: GmSHAT1-5 expressed at 15.02-fold the level of GsSHAT1-5 in FCC, while other genes expressed in HEINONG44 0.6~4-fold over their counterparts in ZYD00755. Error bars in (d,e) indicated s.d. of four biological replicates. Scale bars in (a–c), 70 μm.
To validate the biological function of SHAT1-5, we conducted functional complementary tests in an Arabidopsis nst1-1;nst3-1 mutant (Methods) with pendent stem and indehiscent fruit phenotypes as a result of simultaneous loss of lignified cells in stems and fruits34,35. The expressions of GmSHAT1-5 and GsSHAT1-5 coding sequence with respective Arabidopsis promoters (pAtSND1 and pAtNST1) all rescued the mutant phenotype to the wild type with erect stems (Fig. 3a,b, red triangles and Supplementary Fig. 7a) and dehiscent fruits (Fig. 3c,d, red triangles and Supplementary Fig. 7b). We further found that expression of either GmSHAT1-5 or GsSHAT1-5 can effectively restore the expression of a number of transcription factors, which are involved in the secondary cell wall thickening regulation (Supplementary Fig. 8). These results suggest that both GmSHAT1-5 and GsSHAT1-5 have a conserved function to activate secondary cell wall biosynthesis in Arabidopsis. With regard to fruit shattering, it is interesting to note that NST1-like genes are involved in a similar pathway of secondary cell wall biosynthesis in soybean and Arabidopsis, but each employs a distinct mechanism to shatter the fruit because of different fruit structure between them12,26,35.

(a) Expression of GmSHAT1-5/GsSHAT1-5 under control of AtSND1 promoter effectively restores mutants to erect stem. (b) Anatomical analysis reveals that the specific expression of GmSHAT1-5/GsSHAT1-5 under control of AtSND1 promoter can activate secondary cell wall deposition in the stem interfascicular fibres (red triangles) of nst1-1;nst3-1 mutant. (c) Expression of GmSHAT1-5/GsSHAT1-5 under control of AtNST1 promoter effectively restores the indehiscent fruit phenotype of nst1-1;nst3-1 mutant (red triangles). (d) Anatomical analysis reveals that the specific expression of GmSHAT1-5/GsSHAT1-5 under control of AtNST1 promoter rescues the indehiscent fruit by activating secondary wall thickening in the valve margins and endocarps (red triangles) of nst1-1;nst3-1 mutant. Scale bars in (a), 10 cm; (b), 70 μm; (c), 500 mm; (d), 40 μm.
To determine the genetic effect of SHAT1-5 on pod shattering, we further conducted artificial hybridization between HEINONG44 and ZYD00755, in which F1 plants were characterized by fruits of partial shattering resistance (shattering index, 22.6%) as compared with its full-shattering parental line ZYD00755 (shattering index, 100%). In the F2 plants, we found that the GmSHAT1-5 allele, as determined by the G/T SNP 509 bp downstream of the stop codon in the 3′ flanking sequence, was invariably co-segregated with the heavily thickened FCC (Fig. 4a–c). The presence of highly lignified FCC can effectively prevented the pod from being shattering in the heterozygotes (shattering index <40%) (Fig. 4a–c; Supplementary Fig. 9). Anatomical observation on dry fruits of homozygote GmSHAT1-5 and heterozygote SHAT1-5 (GmSHAT1-5/GsSHAT1-5) showed that, as in HEINONG44, the shattering resistance was realized through the connection of two valve bundles by highly thickened FCC (Supplementary Fig. 10). Furthermore, in the FCC of F2 homozygote of GmSHAT1-5 and GsSHAT1-5 allele, we found that GmSHAT1-5 expressed at ~13-fold the level of GsSHAT1-5 (Fig. 4d). Collectively, all the observed data presented above provide compelling evidence that the GmSHAT1-5 effectively controls the shattering-resistant trait by much over-expression in the FFC in cultivated soybeans.

(a) Semi-thin cross sections of ventral sutures in pods of F2 plant with GmSHAT1-5/GmSHAT1-5 homozygotic genotype. (b) Semi-thin cross sections of ventral sutures in pods of F2 plant with GmSHAT1-5/GsSHAT1-5 heterozygotic genotype. (c) Semi-thin cross sections of ventral sutures in pods of F2 plant with GsSHAT1-5/GsSHAT1-5 homozygotic genotype. The FCC is indicated by a double-headed arrow. Upper panels of (a–c) show GlycineSHAT1-5 genotyped with a SNP (G versus T). (d) Comparative expression analysis of GlycineNST1 and GlycineSHAT1-5 in the Fcc of F2 homozygotic plants with GmSHAT1-5 and GsSHAT1-5 allele. Note: GmSHAT1-5 expressed at ~13.34-fold the level of GsSHAT1-5 while GmNST1 ~1.72-fold of GsNST1 in the FCC of F2 segregants, GlycineNST1 and GlycineSHAT1-5 denote the two gene loci, respectively. Error bars in (c,d) indicate s.d. of four biological replicates. Scale bars in (a–c), 70 μm.
SHAT1-5 locus is a target of artificial selection
Since expression and genetic analyses combined with functional investigations suggest SHAT1-5 as the key regulator in the domestication of pod shattering resistance, it strongly implies that SHAT1-5 might have been the target of artificial selection during soybean domestication. To evaluate the impact of artificial selection on SHAT1-5 locus, we first analysed the DNA polymorphism in the ~5.3 kb genomic region of SHAT1-5. Severe reduction in nucleotide diversity was found in SHAT1-5 with all soybean landraces sharing a uniform haplotype including two SNP substitutions fixed at the 3′ regulatory region, indicative of intense selection removing rare haplotypes at this locus36,37 (Fig. 5a,b). In a simple model of selection, a single favoured haplotype that contributed to the morphological adaptation has always been the target of artificial selection and become fixed during domestication. In the phylogenetic analyses, domesticated sequences would be expected to differentiate from the wild ancestor sequences to form a single clade38,39. To ascertain the evolution pattern of SHAT1-5 gene during soybean domestication, we conducted phylogenetic analysis based on the 5,244 bp genomic region of SHAT1-5. Given that Glyma07g05660 (GlycineNST1), the paralogue of SHAT1-5, has evolved in a neutral manner during domestication (Table 1; Supplementary Table 2), we also cloned it as a neutral locus for comparison. In the Glyma07g05660 tree, the soybean landraces were dispersed into multiple branches with six landraces embedded in G. soja populations in a typical neutral manner due to incomplete lineage sorting among these sequences38 (Supplementary Fig. 11a). In contrast, all soybean landraces were gathered together in a well supported clade in the SHAT1-5 tree (Supplementary Fig. 11b). These results suggest that cultivated soybean sequences of SHAT1-5 locus were derived from the same haplotype that distinct from all other wild soybean populations, indicating that this locus has experienced severe artificial selection likely in a single domestication event28.

(a) Nucleotide diversity (π) for G. max (purple line) and G. soja (green line) across the ~5.3 kb genomic region of GlycineSHAT1-5 locus. (b) Haplotype variation in the ~5.3 kb GlycineSHAT1-5 genomic region. The position of the polymorphism site is indicated by the number above. The G. max and G. soja specific nucleotides and haplotypes are indicated by blue and yellow shade, respectively. (c) Values of genetic diversity of 17 individual genes in G. max (red rhombus) and G. soja (blue rhombus) across a ~365 kb genomic region in chromosome 16. (d) Relative genetic diversity of G. max to G. soja. Note that five genes (4, 5, 6, 8 and 9) closely linked to GlycineSHAT1-5 (7, red bar) exhibit a severe reduction in genetic diversity in the domesticated soybean population (c,d). The relative genomic location of the 17 genes is indicated by solid bars. The ~116 kb selective sweep is indicated by the double-headed red arrow. The identity of the 17 genes is listed in Supplementary Table 3.
We further examined the DNA polymorphism ranging from 290 to 936 bp of 17 genes in an ~365 kb segment of chromosome 16 flanking the SHAT1-5 locus in cultivated and wild soybean accessions (Fig. 5c and Supplementary Table 3). We observed a genomic region around SHAT1-5 locus with π=0 across ~116 kb from −63 kb to 53 kb of the SHAT1-5 start codon with five linked genes involved (Glyma16g02140, Glyma16g02150, Glyma16g02190, Glyma16g02220 and Glyma16g02240) (Fig. 5c,d). In the G. soja populations, however, these five loci exhibited considerable genetic diversity (Fig. 5c and Supplementary Table 3). A previous study has shown that the domesticated soybean genome has exceptional high LD value29. To seek further evidence of selection on SHAT1-5 locus, we calculated the r2 value of both intralocus and interlocus sequence for all pairwise comparisons of segregating sites, a measurement of LD, in the aforementioned five genes in the ~116 kb region. The lack of sufficient polymorphism site hampers our ability to assess r2 value in the cultivated soybean population. However, in the wild soybean population, we found a low LD pattern across this genomic region with r2 intralocus comparison ranging from 0.026 to 0.335 (Supplementary Table 4). For interlocus comparison, the r2 values are typically 0.2–0.4, suggesting that there is non-random associations of these gene loci in wild soybean genome (Fig. 6). On the basis of the above findings, the appropriate interpretation of this result is that severe artificial selection might have extended the GmSHAT1-5 haplotype to the flanking loci as a result of ‘hitchhiking’ effect36,37,40. In addition, because of the stringent cleistogamic nature of the cultivated soybean, the low recombination rate across the genome is also expected to aggravate the genomic effect of artificial selection.

The average r2 value is typically lower than 0.6 in both intralocus and interlocus comparisons. The SHAT1-5 locus is indicated by a red ball and other gene loci are indicated by black balls on the chromosome segment (1.59M–1.77M). N.C. denote no pairwise comparison conducted due to lack of polymorphism sites.
A repressive element upstream of GmSHAT1-5 alters expression
The origin of morphological novelties is usually associated with expression alteration of functionally conserved developmental genes through mutations in cis-regulatory elements41,42,43. Given that both GmSHAT1-5 and GsSHAT1-5 can functionally complement the Arabidopsis mutant, we speculated that the driving force behind the GmSHAT1-5 upregulation in the FCC of G. max might lie in the regulatory region. We thus cloned the 5.0 kb upstream regulatory sequence from HEINONG44 and ZYD00755, respectively, and identified a number of SNPs and a 20 bp InDel between the two conserved sequences (sequence similarity is 98.61%). We used PlantProm (Shahmuradov et al.44) to predict the putative cis-element in these SNPs and the InDel. The 20 bp deletion in −4.0 kb of GmSHAT1-5 promoter region interested us, as it destroys the integrity of a GARP protein binding site of AGAT (Hosoda et al.45) with unknown exact function (Fig. 7a). We use transient assays in Arabidopsis mesophyll protoplast to test the effect of this cis-element on gene expression. Quartal replicates of this element (WT) and mutation of the core binding site (MU) were cloned into reporter construct upstream of the minimal cauliflower mosaic virus promoter (mpCaMV), respectively. With WT, it was obvious that this element repressed the expression of the GUS reporter as compared with mpCaMV (Fig. 7b,c). The constructs with mutation of the core binding sites (from AGAT to ACTA) increased expression of the GUS reporter fourfold relative to the WT construct (Fig. 7b,c). We next deleted the AGAT core motif (DE) to mimic the cultivated soybean promoter. Quartal replicates of the sequence fused to mpCaMV greatly boosted expression of the GUS reporter to 43-fold relative to the WT control construct (Fig. 7b,c), almost reminiscent of differential expression of GmSHAT1-5 and GsSHAT1-5. These results strongly suggest the AGAT element represses GUS expression, and by inference GsSHAT1-5 expression in vivo. Thus, the disruption of this functional repressive element can causatively, at least partially, explain the upregulation of GmSHAT1-5 in domesticated soybeans.

(a) The 20 bp deletion in the upstream regulatory region of GmSHAT1-5 in HEINONG44. The GARP-biding site is underlined with the core-biding site indicated by red letters. (b) Diagrams of the reporter constructs used for transient expression analysis. WT, tetramer of the wild type 20 bp InDel sequence; MU, tetramer of mutant sequence (the mutated core GARP-binding site was indicated by lower case); DE, tetramer of deletion sequence with core GARP-binding site deleted; mpCaMV, minimal promoter of the CaMV 35S promoter. (c) Transient expression analysis of GARP-binding site in Arabidopsis protoplast. Error bars in (c) indicate s.d. of three biological replicates.
Discussion
The candidate gene approach has been widely used to identify the genes controlling domestication traits and has been successful in a limited number of crops4,22. Here we employed multiple approaches to try to uncover the cellular and genetic basis behind pod shattering resistance in domesticated soybeans. The key domesticated cellular feature responsible for the shattering resistance lies in the FCC, rather than the AL, in the ventral suture of the pod controlled by SHAT1-5 functionally activating secondary cell wall biosynthesis. The FCC with heavily thickened secondary cell walls in domesticated soybean prevents the pod from shattering, which is attributed to the over-expression of GmSHAT1-5 gene in the FCC, at 15-folds the level of the wild allele. We further found evidence that disruption of a repressive element greatly enhances the specific expression of GmSHAT1-5 in the FCC, promoting an excessive deposition of secondary cell walls. Correlatively, severe artificial selection exerted on SHAT1-5 has caused a ~116 kb selection sweep around this locus. The series of causatively related events finally brought about the pod shattering-resistant phenotype in domesticated soybeans (Fig. 8).

In domesticated soybeans, the pod shattering resistance is caused by the excessive deposition of secondary cell walls in FCC. In the wild progenitor G. soja, the FCC is characteristic of only 2~3 layer of cells with secondary walls moderately thickened (pink). In G. max, on the contrary, the FCC is composed of multiple layer of cells with secondary walls heavily thickened (violet). The AL (blue) and the vascular bundle (red) perform normal function in both G. max and G. soja with no detectable change between them. A NAC (NAM, ATAF1/2 and CUC2) gene, SHATTERING1-5 (SHAT1-5) functionally activating secondary wall biosynthesis, promotes the heavily thickening of FCC secondary walls by expression at 15-fold the level of the wild allele, which is attributed to functional disruption of the upstream repressor. Note: the repressive element in GsSHAT1-5 promoter (red star) is indicated by bold letters and the core motif ‘GAT’ is indicated by red bold letters. VB, vascular bundle. Scale bars in the fruit figures represent 1 cm.
Generally, gene upregulation could be realized by recruiting an enhance element or excision of a repressive element in the regulatory region42,43. In maize, the upregulation of TEOSINTE BRANCHED1, which encodes a growth repressor in the axillary shoot to bring about apical dominance in domesticated maize, is recently found to be attributed to a transponsable element (Hopscotch) acting as an enhancer inserted in ~60 kb promoter region46,47. It is suggested that artificial selection seems to preferentially target the stable highly expressed genes across the maize genome48. The changes of fruit colour in grape (Vitis vinifera) and orange (Citrus sinensis) are demonstrated to be caused by repressive element excision in a MYB transcription factor during their respective crop improvements49,50. Taken together, our findings herein highlight the important role of cis-regulatory changes in upregulating gene expression, which in turn brings about phenotypic variation, targeted by human selection during the crop domestication process. Even though the disruption of a repressive element is evidently the most promising mechanism underlying the upregulation of SHAT1-5 in cultivated soybeans, we cannot exclude the roles of other possible mutations in the more remote regulatory regions presently unchecked.
A recent study has identified that a 113 bp InDel in the promoter of Glyma16g25600 encodes a bZIP transcription factor and is located in a major QTL related to pod shattering (qPDH1)51. It is noted that a previous transcriptome investigation of gene expression in Arabidopsis related to stem bolting indicates that members of the bZIP family have a high frequency of upregulation during secondary cell wall thickening52. In rice, loss of seed shattering is caused by the modification of the AL as the result of impaired function of two genes, Sh4 and qSH1, which encode Myb3-like and homeodomain transcription factors, respectively7,53. Interestingly, qSH1 gene expression is completely abolished in sh4-2 mutant background, suggesting these two genes act in the same genetic pathway in directing AL identity54. It would be possible that SHAT1-5 and Glyma16g25600 may be involved in the same or similar pathways to regulate secondary cell wall thickening and shattering resistance in domesticated soybeans. Re-examination of qPDH1 near-isogenic lines fruits at the cellular level combined with functional investigation of Glyma16g25600 would help to test this assumption.
During crop domestication, loss of seed dispersal or fruit shattering has been known to be associated with AL disruptions consequent upon related gene function alterations4,5,6. In eudicots, the loss of fruit dehiscence is a derived morphologically adaptive character in many lineages, but very little is known about the genetic basis underlying the evolution and diversification of such an important trait. In Arabidopsis and its relatives in Brassicaceae, the mutants or natural variants with indehiscent fruit are frequently related to the loss-of-function or loss-of-expression of genes involved in the regulation of cell identity of lignified valve margin and ALs26,55,56. In the legume plant Medicago, a coding change in SHP orthologs is recently found to be correlative with the origin of the specialized coiled morphology of the indehiscent fruit, but the exact cellular and genetic basis of such fruit indehiscence is still unknown57. Our findings represent a causative mechanism for pod shattering resistance, particularly applicable to this eudicot crop with fundamentally different fruit structures from both cereals and Arabidopsis. These results also suggest that GmSHAT1-5 is a promising locus potentially applicable to transgenic breeding for improving soybean yield due to its direct effect on pod shattering resistance and possible involvement in stem lodging resistance.
Methods
Plant materials and growth conditions
Glycine max (HEINONG44), Glycine soja (ZYD00755) and various cultivated soybean landraces and wild soybean accessions from diverse geographical locations covering China (Supplementary Table 1) were obtained from the seed bank of the Chinese Academy of Agricultural Sciences (Beijing). The seedlings were transplanted in the greenhouse of the State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, the Chinese Academy of Sciences.
Arabidopsis thaliana nst1-1;nst3-1 mutant (ecotype Columbia-0) was generated by crossing nst1-1 mutant (T-DNA-tagged line SALK_120377) with nst3-1 mutant (T-DNA-tagged line SALK_149909) (Mitsuda et al.34) and provided by Professor Masaru Ohme-Takagi. The mutants were germinated on 1/2 Murashige and Skoog medium at 23 °C. The seedlings were transplanted in the soil under 16-h-light (200 μmol m−2 s−1, 23 °C) and 8-h-dark (20 °C) conditions.
To generate the G. max and G. soja hybrids, we pollinated castrated HEINONG44 flower with pollen from ZYD00755. The resultant hybrids and segregants were genotyped using the G. max and G. soja specific SNP (G versus T) in the 3′ flanking regulatory region of GlycineSHAT1-5 gene.
Molecular population genetic analyses
We amplified PCR product from G. max and G. soja genomic DNA using TaKaRa ExTaq (TaKaRa, China). Sequences were analysed using CHROMAS software, and aligned with Clustal X software58, the matrix was adjusted and refined manually using BioEdit software59. The matrix was then imported into Dnasp 5.10 software60. Values of genetic diversity per base pair (π) were estimated for the cultivated soybean group and wild soybean group, respectively. Watterson’s estimator of θ per base pair was calculated based on the total number of polymorphism site. Sliding window analysis of genetic diversity (π) was calculated using a 500-bp sliding window with a 25-bp step using the 5,244 bp SHAT1-5 full length genomic sequence with average pairwise difference per base pair between sequences61. For a pairwise comparison of LD, as determined by the value of r2, only SNP sites in the respective sequence were included for calculation; significant comparisons were determined by the Fisher’s exact test using Dnasp 5.1060. Tajima’s D-value and Fu and Li’s D*-value were calculated as the probability of the sequence departure of a neutrally evolved model using Dnasp 5.1060. Phylogenetic trees were constructed using the ~5.3 kb genomic sequence of Glyma07g05660 and SHAT1-5, respectively, based on the Neighbor-Joining method62 with MEGA 5.0 software63. Bootstrap values were estimated (with 1,000 replications) based on Kimura’s two-parameter model64.
Shattering index evaluation and microscopy
For evaluating the shattering index of G. max and G. soja, two different conditions were applied. Under natural conditions (in the field, without artificial disturbance), the number of shattering pods of HEINONG44 and ZYD00755 was counted from 100 pods, respectively, after pod maturity in two consecutive years (2009 and 2010). Under experimental conditions, 100 brown fruits each (~40 DPA) were collected and kept in an oven at 37 °C for 4 days and then the number of shattering pods was counted, respectively. The statistics were an average of three biological replicates.
Pod shattering mechanical force was determined as the minimum force necessary to break the fruit into two parts using a digital mechanical force gauge (HANDPI, China). The statistics were an average of three replicates.
The fruit structure of HEINONG44 and ZYD00755 was observed with hand-cut sections. Sections (~100 μm) of mature fruits (30 DPA) were stained with 2% phloroglucinol (m/v), then mounted with 30% HCl (v/v) and analysed using Zeiss Axio Imager A1 microscopy (Carl Zeiss, Germany). The secondary cell walls were examined using sections of 500 nm cut from a diamond knife on a Leica Ultra-Cut microtome (Leica, Germany) and observed with a Zeiss LSM 510 META confocal microscope (Carl Zeiss). Dried fruits (~45 days after anthesis) of HEINONG44 were collected and rehydrated, 10 μm sections were dissected using a rotary microtome (MICROME, Germany) and analysed using Zeiss Axio Imager A1 microscopy (Carl Zeiss) to observe FCC in the ventral suture.
RNA extraction and real-time qPCR
Total RNA of roots, shoots, young stems (node 1 from shoot), mature stems (node 2 from shoot), inflorescences, leaves, flowers and fruit shells were extracted from HEINONG44 and ZYD00755, respectively, using SV Total RNA Isolation System (Promega, USA). The cDNA was synthesized using a RevertAid H Minus First-Strand cDNA Synthesis Kit (Thermo Scientific, USA).
For real-time qPCR, probes of 100–150 nt length were generated using gene-specific primers for GlycineSHP-A/B, GlycineIND-A/B/C/D, GlycineNST1, GlycineSHAT1-5, GlycineNST2, GlycineALC-A/B, GlycineEndo-PG and ACTIN (Supplementary Table 5). The SYBR Premix ExTaq (TaKaRa, China) was used to perform real-time qPCR with ROX as a reference dye on Stratagene Mx3000P system (Agilent Technologies, USA). The relative expression level of individual gene was determined using ratio=2−ΔΔCT method65. For each gene, a total of four biological replicates were conducted.
Laser microdissections and expression analyses
Stage-6 fruits (18 days after anthesis) were fixed and infiltrated using a vacuum pump. 8-μm sections were dissected using a rotary microtome (MICROME) and placed on PEN Membrane glass slides (Life Technology, USA). To dissolve the paraffin, the slides were exposed to xylene for 1 h and repeated three times, then processed into a series of xylene/ethanol solutions and dried.
Laser capture microdissection of FCC and AL was performed on Arcturus Laser Capture Microdissection system (Life Technology) at the Institute of High Energy Physics (CAS). The target tissues were collected on Macro LCM Caps (Life Technology), then dissolved in the collection tubes (Life Technology). Total RNA was extracted using miRNeasy FFPE kit (Qiagen, Germany). The cDNA was synthesized using a RevertAid H Minus First-Strand cDNA Synthesis Kit (Thermo Scientific), and real-time qPCR was conducted as mentioned above. For gene relative expression levels, both in FCC and AL, all HEINONG44 individual orthologous genes were standardized to ZYD00755, respectively. For each gene, four biological replicates were conducted.
Recombinant proteins and immunocytochemistry
The C terminus of the GlycineSHAT1-5 gene was isolated using primers GlycineNST1/SHAT1-5-EX-F/R (Supplementary Table 5) from ZYD00755 cDNA. The PCR product was digested with BamH I and Hind III, inserted into pET30a vector (Merck, Germany), and introduced into BL21 Escherichia coli cells. The His tagged recombinant proteins were purified from the soluble fraction of the cell lysate using Ni2+ sepharose (GE Healthcare, USA). The respective proteins were injected into rabbits, and the primary antibody was purified (Beijing Protein Innovation, China).
For immunocytochemical localization, the process was followed as previously described66. Briefly, stage-5 (13DPA) and stage-6 (18DPA) of the ventral sutures of HEINONG44 were fixed with 4% paraformaldehyde and embedded with paraffin, 10-μm sections were dissected using a rotary microtome (MICROME) on poly-L-lysine coated glass slides (Sigma-Aldrich, USA). The sections were dewaxed, rehydrated and treated with infiltration solution (3% H2O2 in 1 × PBS) to deactivated the endogenous proteinase. The sections were blocked with 2% BSA for 30 min and then incubated with primary antibody solutions (1:100 diluted with 2% BSA in 1 × PBS). After removing the non-specific primary antibody, FITC-conjugated secondary antibody (1:200 diluted with 2% BSA in 1 × PBS) was mounted on the sections. The signal was observed with a Zeiss LSM 510 META confocal microscope (Carl Zeiss) after removing the non-specific secondary antibody. We conducted control experiment on sections with no primary antibody supplied.
Complementation of Arabidopsis nst1-1;nst3-1 mutant
We first replaced the CaMV 35S promoter of pCAMBIA1301 vector with ~3 Kb of Arabidopsis SND1 and Arabidopsis NST1 promoters, respectively, using BamH I and Hind III to generate pAtSND1-GUS and pAtNST1-GUS vectors. The full length of GlycineSHAT1-5 coding sequence from HEINONG44 and ZYD00755 cDNA was, respectively, inserted into Hind III/Bgl II sites of pAtSND1-GUS and pAtNST1-GUS vector and then the construct was delivered into the nst1-1;nst3-1 mutant. At least 10 independent T1 transgenic plants with obvious complemented phenotype were selfed to generate T3 plants. The analyses were performed on T3 populations.
For evaluating the shattering index of wild-type, nst1-1;nst3-1 and transgenic plants, a random impact experiment was conducted with a modification of previously described method67. Briefly, a total number of 100 mature siliques of WT, nst1-1;nst3-1 or transgenic lines were harvested and kept in an oven at 37 °C for 2 days. The siliques were transferred into a petri dish with five 2-mm steel balls (weighing ~300 mg) and then attached the petri dish to an eppendorf shaker (Eppendorf, Germany). The petri dish was agitated for 60 s, the total number of shattering siliques was recorded for each genotype. The statistics were average of three biological replicates. The stem mechanical force was recorded as the minimum force necessary to break the inflorescence stem into two parts using a digital mechanical force gauge (HANDPI). For each genotype, three biological replicates were conducted. Real-time qPCR were conducted using cDNA from inflorescence stem of representative transgenic lines using primers listed in the Supplementary Table 5. For real-time qPCR, three biological replicates were conducted.
Transient gene expression assay
An oligonucleotide extending from −46 bp containing the minimal promoter of the cauliflower mosaic virus 35S promoter (CaMV) was synthesized and inserted into the EcoR I and Bgl II sites of binary pCAMBIA1301 vectors to create the mpCaMV-GUS construct. The plasmid was sequenced to ensure the proper insertion of the sequence. Then a double-stranded synthetic oligonucleotide comprising a tetramer of the wild-type InDel sequence (WT) (5′-ATTAAAAAAATAAATAAGATATT-3′), mutated core GARP-binding site (MU) (5′-ATTAAAAAAATAAATAACTAATT-3′) or deleted core GARP-binding site (DE) (5′-ATTAAAAAAATAAA-3′) was inserted into EcoR I and BamH I sites upstream of mpCaMV to generate the respective reporter construct. The plasmid was sequenced to ensure the proper insertion of the elements. The resultant constructs were introduced into Arabidopsis mesophyll protoplast by polyethylene glycol-mediated transformation method. The pCAMBIA1302 vectors were used to co-transfect into the protoplast as control to evaluate the transformation efficiency. The protoplast was cultured at room temperature for 6 h in the dark. The relative expression of GUS was monitored using real-time PCR based on total RNA extracted from the protoplast. Three biological replicate was conducted.
Additional information
Accession Codes: Gene sequences from G. max and G. soja have been deposited in the DDBJ/EMBL/GenBank database under accession codes KJ173520 to KJ173523.
How to cite this article: Dong, Y. et al. Pod shattering resistance associated with domestication is mediated by a NAC gene in soybean. Nat. Commun. 5:3352 doi: 10.1038/ncomms4352 (2014).
References
Harlan, J. R. Crops and Man American Society of Agronomy (1975).
Diamond, J. Evolution, consequences and future of plant and animal domestication. Nature 418, 700–707 (2002).
Purugganan, M. D. & Fuller, D. Q. The nature of selection during plant domestication. Nature 457, 843–848 (2009).
Olsen, K. M. & Wendel, J. F. A bountiful harvest: genomic insight in to crop domestication phenotypes. Annu. Rev. Plant Biol. 64, 47–70 (2013).
Doebley, J. F., Gaut, B. S. & Smith, B. D. The molecular genetics of crop domestication. Cell 127, 1309–1321 (2006).
Lenser, T. & Theißen, G. Molecular mechanisms involved in convergent crop domestication. Trend Plant Sci. 18, 704–714 (2013).
Li, C., Zhou, A. & Sang, T. Rice domestication by reducing shattering. Science 311, 1936–1939 (2006).
Simons, K. J. et al. Molecular characterization of the major wheat domestication gene Q. Genetics 172, 547–555 (2005).
Taketa, S. et al. Barley grain with adhering hulls is controlled by an ERF family transcription factor gene regulating a lipid biosynthesis pathway. Proc. Natl Acad. Sci. USA 105, 4062–4067 (2008).
Lin, Z. et al. Parallel domestication of the Shattering1 genes in cereals. Nat. Genet. 44, 720–724 (2012).
Tiwari, V. S. & Bhatia, V. S. Character of pod anatomy associated with resistance to pod shattering in soybean. Ann. Bot. 76, 483–485 (1995).
Christiansen, L. C., Dal Degan, F., Ulvskov, P. & Borkhardt, B. Examination of the dehiscence zone in soybean pods and isolation of a dehiscence-related endopolygalacturonase gene. Plant Cell Environ. 25, 479–490 (2002).
Hymowitz, T. On the domestication of soybean. Econ. Bot. 24, 408–421 (1970).
Mauricio, R. Mapping quantitative trait loci in plants: uses and caveats for evolutionary biology. Nat. Rev. Genet. 2, 370–381 (2001).
Doebley, J., Stec, A. & Hubbard, L. The evolution of apical dominance in maize. Nature 386, 485–488 (1997).
Frary, A. et al. fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289, 85–88 (2000).
Saxe, L. A., Clark, C., Lin, S. F. & Lumpkin, T. A. Mapping the pod shattering trait in soybean. Soybean Genet. Newsl. 23, 250–253 (1996).
Bailey, M. A., Mian, M. A. R., Carter, T. E., Ashley, D. A. & Boerma, H. R. Pod dehiscence of soybean: identification of quantitative trait loci. J. Hered. 88, 152–154 (1997).
Liu, B. et al. QTL mapping of domestication-related traits in soybean (Glycine max). Ann. Bot. 100, 1027–1038 (2007).
Suzuki, M., Fujino, K. & Funatsuki, H. A major soybean QTL, qPDH1, controls pod dehiscence without marked morphological change. Plant Prod. Sci. 12, 217–223 (2009).
Suzuki, M., Fujino, K., Nakamoto, Y., Ishimoto, M. & Funatsuki, H. Fine mapping and development of DNA markers for the qPDH1 locus associated with pod dehiscence in soybean. Mol. Breeding 25, 407–418 (2010).
Ross-Ibarra, J., Morrell, P. L. & Gaut, B. S. Plant domestication, a unique opportunity to identify the genetic basis of adaptation. Proc. Natl Acad. Sci. USA 104, 8641–8648 (2007).
Reznick, D. N. & Ricklefs, R. E. Darwin’s bridge between microevolution and macroevolution. Nature 457, 837–842 (2009).
Roberts, J. A., Elliott, K. A. & Gonzalez-Carranza, Z. Abscission, dehiscence, and other cell separation processes. Annu. Rev. Plant Biol. 53, 131–158 (2002).
Mao, L. et al. JOINTLESS is aMADS-box gene controlling tomato flower abscission zone development. Nature 406, 910–913 (2000).
Østergaard, L. Don’t ‘leaf’ now. The making of a fruit. Curr. Opin. Plant Biol. 12, 36–41 (2009).
Hyten, D. L. et al. Impacts of genetic bottlenecks on soybean genome diversity. Proc. Natl Acad. Sci. USA 103, 16666–16671 (2006).
Guo, J. et al. A single origin and moderate bottleneck during domestication of soybean (Glycine max): implications from microsatellites and nucleotide sequences. Ann. Bot. 106, 505–514 (2010).
Lam, H. M. et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059 (2010).
Grant, D., Nelson, R. T., Cannon, S. B. & Shoemaker, R. C. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 38, 843–846 (2010).
Zhong, R., Lee, C. & Ye, Z. Evolutionary conservation of the transcriptional network regulating secondary cell wall biosynthesis. Trend Plant Sci. 15, 625–632 (2010).
Zhao, Q. & Dixon, R. A. Transcriptional networks for lignin biosynthesis: more complex than we thought? Trend Plant Sci. 16, 227–233 (2011).
Dong, Y., Wang, B. H. & Wang, Y. Z. Functional characterization of the orthologs of AtNST1/2 in Glycine soja (Fabaceae) and the evolutionary implications. J. Syst. Evol. 51, 693–703 (2013).
Mitsuda, N. et al. NAC transcription factors, NST1 and NST3, are key regulators of the formation of secondary walls in woody tissues of Arabidopsis. Plant Cell 19, 270–280 (2007).
Mitsuda, N. & Ohme-Takagi, M. NAC transcription factors NST1 and NST3 regulate pod shattering in a partially redundant manner by promoting secondary wall formation after the establishment of tissue identity. Plant J. 56, 768–778 (2008).
Innan, H. & Kim, Y. Pattern of polymorphism after strong artificial selection in a domestication event. Proc. Natl Acad. Sci. USA 101, 10667–10672 (2004).
Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 39, 197–218 (2005).
Wang, R. L., Stec, A., Hey, J., Lukens, L. & Doebley, J. F. The limits of selection during maize domestication. Nature 398, 236–239 (1999).
Clark, R. M., Linton, E., Messing, J. & Doebley, J. F. Pattern of diversity in the genomic region near the maize domestication gene tb1. Proc. Natl Acad. Sci. USA 101, 700–707 (2004).
Smith, J. M. & Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 23, 23–25 (1974).
Carroll, S. B. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134, 25–36 (2008).
Wittkopp, P. J. & Kalay, G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 13, 59–69 (2012).
Wray, G. A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 8, 206–216 (2007).
Shahmuradov, I. A. et al. PlantProm: a database of plant promoter sequences. Nucleic Acids Res. 31, 114–117 (2003).
Hosoda, K. et al. Molecular structure of the GARP family of plant Myb-related DNA binding motifs of the Arabidopsis response regulators. Plant Cell 14, 2015–2029 (2002).
Clark, R. M., Wagler, T. N., Quijada, P. & Doebley, J. A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nat. Genet. 38, 594–597 (2006).
Studer, A., Zhao, Q., Ross-Ibarra, J. & Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 43, 1160–1163 (2011).
Hufford, M. B. et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 44, 808–811 (2012).
Kobayashi, S., Goto-Yamamoto, N. & Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 304, 982 (2004).
Butelli, E. et al. Retrotransposons control fruit specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 24, 1242–1255 (2012).
Gao, M. & Zhu, H. Fine mapping of a major quantitative trait locus that regulates pod shattering in soybean. Mol. Breeding 32, 485–491 (2013).
Ehlting, J. et al. Global transcript profiling of primary stems from Arabidopsis thaliana identifies candidate genes for missing links in lignin biosynthesis and transcriptional regulators of fiber differentiation. Plant J. 42, 618–640 (2005).
Konishi, S. et al. An SNP caused loss of seed shattering during rice domestication. Science 312, 1392–1396 (2006).
Zhou, Y. et al. Genetic control of seed shattering in rice by the APETALA2 transcription factor shattering abortion1. Plant Cell 24, 1034–1048 (2012).
Lenser, T. & Theißen, G. Conservation of fruit dehiscence pathways between Lepidium campestre and Arabidopsis thaliana sheds light on the regulation of INDEHISCENT. Plant J. 76, 545–556 (2013).
Mühlhausen, A., Lenser, T., Mummenhoff, K. & Theißen, G. Evidence that an evolutionary transition from dehiscent to indehiscent fruits in Lepidium (Brassicaceae) was caused by a change in the control of valve margin identity genes. Plant J. 73, 824–835 (2013).
Fourquin, C. et al. A change in SHATTERPROOF protein lies at the origin of a fruit morphological novelty and a new strategy for seed dispersal in Medicago genus. Plant Physiol. 162, 907–917 (2013).
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Nei, M. & Li, W. H. Mathematical-model for studying genetic-variation in terms of restriction endonucleases. Proc. Natl Acad. Sci. USA 76, 5269–5273 (1979).
Saitou, N. & Nei, M. The neighbor-joining method-a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide-sequences. J. Mol. Evol. 16, 111–120 (1980).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, 2002–2007 (2001).
Paciorek, T., Sauer, M., Balla, J., Wiśniewska, J. & Friml, J. Immunocytochemical technique for protein localization in sections of plant tissues. Nat. Protoc. 1, 104–107 (2006).
Arnaud, N. et al. Gibberellins control fruit patterning in Arabidopsis thaliana. Gen. Dev. 24, 2127–2132 (2010).
Acknowledgements
We thank Song Ge, Tao Sang, James F. Smith, Chaoying He, Qingyin Zeng and Yalong Guo for their constructive comments and language improvements on this article. We thank Lijuan Qiu and Hui Zhang (ICS-CAAS, Beijing) and the National Infrastructure of Crop Germplasm Resource for providing us the wild soybean and domesticated soybean germplasms. We also thank Masaru Ohme-Takagi (AIST, Tsukuba) for providing the nst1-1;nst3-1 mutant, Fengqing Dong (IB-CAS, Beijing) for her technical assistance of resin sections, Bing Wang (IHEP-CAS, Beijing) and Haisheng Liu (Life Technology, Beijing) for their assistance of laser capture microdissections. This work was funded by the State Major Basic Research Foundation of China (2007FY110500 to Y.Z. Wang) and National Natural Science Foundation of China (Grants 30990240 and 31170198 to Y.Z. Wang).
Author information
Authors and Affiliations
Contributions
Y.-Z.W. initiated, designed and supervised the research and wrote the article. Y.D. designed and performed the research, analysed the data and wrote the article. X.Y. and J.L. were involved in DNA sequence isolation and analyses. B.-H.W. and B.-L.L. were involved in expression and transgene experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-11, Supplementary Tables 1-5 and Supplementary References (PDF 1013 kb)
Rights and permissions
About this article
Cite this article
Dong, Y., Yang, X., Liu, J. et al. Pod shattering resistance associated with domestication is mediated by a NAC gene in soybean. Nat Commun 5, 3352 (2014). https://doi.org/10.1038/ncomms4352
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms4352
This article is cited by
-
Comparative transcriptome and co-expression network analysis revealed the genes associated with senescence and polygalacturonase activity involved in pod shattering of rapeseed
Biotechnology for Biofuels and Bioproducts (2023)
-
Accelerating crop domestication through genome editing for sustainable agriculture
Journal of Plant Biochemistry and Biotechnology (2023)
-
Major genetic locus with pleiotropism determined seed-related traits in cultivated and wild soybeans
Theoretical and Applied Genetics (2023)
-
Genome-wide association study for biomass accumulation traits in soybean
Molecular Breeding (2023)
-
Current overview on the genetic basis of key genes involved in soybean domestication
aBIOTECH (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
