
Abstract
Despite the fact that binge alcohol drinking (intake resulting in blood alcohol concentrations (BACs) ⩾80 mg% within a 2-h period) is the most prevalent form of alcohol-use disorders (AUD), a large knowledge gap exists regarding how this form of AUD influences neural circuits mediating alcohol reinforcement. The present study employed integrative approaches to examine the functional relevance of binge drinking-induced changes in glutamate receptors, their associated scaffolding proteins and certain signaling molecules within the central nucleus of the amygdala (CeA). A 30-day history of binge alcohol drinking (for example, 4–5 g kg−1 per 2 h−1) elevated CeA levels of mGluR1, GluN2B, Homer2a/b and phospholipase C (PLC) β3, without significantly altering protein expression within the adjacent basolateral amygdala. An intra-CeA infusion of mGluR1, mGluR5 and PLC inhibitors all dose-dependently reduced binge intake, without influencing sucrose drinking. The effects of co-infusing mGluR1 and PLC inhibitors were additive, whereas those of coinhibiting mGluR5 and PLC were not, indicating that the efficacy of mGluR1 blockade to lower binge intake involves a pathway independent of PLC activation. The efficacy of mGluR1, mGluR5 and PLC inhibitors to reduce binge intake depended upon intact Homer2 expression as revealed through neuropharmacological studies of Homer2 null mutant mice. Collectively, these data indicate binge alcohol-induced increases in Group1 mGluR signaling within the CeA as a neuroadaptation maintaining excessive alcohol intake, which may contribute to the propensity to binge drink.
Similar content being viewed by others

INTRODUCTION
Binge drinking is defined as consuming enough alcohol to reach intoxication in a 2-h period, with intoxication defined as a blood alcohol concentration (BAC) ⩾80 mg% (National Institute on Alcohol Abuse and Alcoholism, 2004). As 22.6% of persons aged ⩾12 years participated in binge drinking in the United States in 2011 (Substance Abuse and Mental Health Services Administration, 2012), characterizing that the neurobiological impact of binge drinking is crucial for the development of relevant pharmacotherapies for the treatment of this form of alcohol-use disorder (AUD).
Research on AUD neurobiology has focused on the mesocorticolimbic system, including the ventral tegmental area and the basal forebrain (for example, Cassell et al, 1999; Koob, 2003; Pandey, 2003). Within the basal forebrain, a neural circuitry forms a separate entity termed the extended amygdala (Alheid and Heimer, 1988) composed of: the bed nucleus of the stria terminalis, the central nucleus of the amygdala (CeA), the shell of the nucleus accumbens (NACshell) and the sublenticular substantia innominata (Alheid and Heimer, 1988). Alcohol increases indices of glutamate neurotransmission within the NAC, CeA and bed nucleus of the stria terminalis (c.f., Gass and Olive, 2008; Siggins et al, 2003; see also Kash et al, 2009; Melendez et al, 2005; Obara et al, 2009; Roberto et al, 2004, 2006; Szumlinski et al, 2008; Wills et al, 2012). Of relevance to this report, repeated bouts of binge alcohol intake can sensitize glutamate release, as well as increase the expression of certain postsynaptic glutamate receptors and their intracellular scaffolding protein Homer2 and downstream effectors within the NACshell (Cozzoli et al, 2009, 2012; Szumlinski et al, 2007); however, very little experimental attention has focused on the role for glutamatergic neurotransmission within other extended amygdala regions vis-à-vis binge drinking. This being said, a chronic history of alcohol experience, produced by either continuous-access procedures or vapor inhalation, elevates CeA indices of both Group1 metabotropic glutamate receptor (mGluR1/5) and ionotropic NMDA receptor signaling (Obara et al, 2009; Roberto et al, 2004, 2006). Moreover, systemic pretreatment with an mGluR5 antagonist attenuates the reinstatement of alcohol seeking in an operant paradigm and reduces the concomitant elevation in biochemical indices of CeA activation (Schroeder et al, 2008). The above data, coupled with evidence that inhibiting Protein Kinase C epsilon, a downstream effector of mGluR5 (Conn and Pin, 1997), within the CeA attenuates binge alcohol intake (Lesscher et al, 2009), lead us to hypothesize that a history of binge alcohol drinking increases Group1 mGluR function within the CeA to maintain excessive alcohol intake.
Group1 mGluRs activate various intracellular signaling cascades, including the direct activation of phospholipase C (PLC) through αq signaling (Nakamura et al, 2004) and phosphatidylinositol 3-kinase (PI3K) through βγ signaling (Rong et al, 2003). Thus, we characterized the effects of a chronic history of binge alcohol drinking upon indices of Group1 mGluR function and then employed various neuropharmacological and transgenic approaches to determine the functional relevance of observed changes in CeA protein expression for the maintenance of excessive alcohol-drinking behavior.
MATERIALS AND METHODS
Subjects
The present studies employed adult (8 weeks old) male C57BL/6J (B6) mice (Jackson Laboratories, Bar Harbor, ME, USA) or adult (7–10 weeks old) male C57BL/6J X 129Xi/SvJ mice with null mutations of Homer2 and their wild-type (WT) counterparts (consistent with Cozzoli et al, 2009; 2012). Mice were housed in individual polyethylene cages under a 12-h-reverse light/dark cycle. Food and water were available ad libitum, except when water bottles were removed for the daily 2-h alcohol/sucrose bottle presentation. All experimental protocols were consistent with the guidelines of the National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals (NIH Publication No. 80–23, revised 1996) and approved by the IACUC of the University of California, Santa Barbara.
Drinking-In-the-Dark (DID) Procedures
The DID procedures to entice high alcohol intake (3.5–5.0 g kg−1 per 2 h) and produce BACs ⩾80 mg% were consistent with those employed previously (Cozzoli et al, 2012; Crabbe et al, 2009; Moore and Boehm, 2009; Rhodes et al, 2005). Briefly, 3 h after lights out, the home cage water bottle was replaced by a 50-ml sipper tube containing 20% alcohol (v/v) or 5% sucrose (w/v) in tap water. In all studies, mice were allowed to drink for a total of 2 h and then the home cage water bottle was returned. Control animals in the immunoblotting studies received a 50-ml sipper tube containing tap water in lieu of 20% alcohol.
Immunoblotting Procedures
For this study, B6 mice were subjected to 30 consecutive days of 20% alcohol or water (control) drinking under DID procedures. At 24 h withdrawal, animals were decapitated, brains were sectioned (1.0 mm thick) along the coronal plane at the level of the amygdala, and the CeA and adjacent basolateral amygdala were dissected out over ice using an 18-gauge needle. The 24-h time point was selected, as it corresponds approximately to the time when animals would receive their next alcohol presentation (that is, animals undergo 22-h periods of withdrawal daily) and to be consistent with the experimental design employed in our earlier immunoblotting studies on NAC tissue (for example, Cozzoli et al, 2009; 2012). The tissue homogenization, protein transfer, antibody incubation and developing procedures were identical to those reported previously (Cozzoli et al, 2009, 2012; Goulding et al, 2011). Bis-Tris gradient gels (4–12%) (Life Technologies, Grand Island, NY, USA) were used to separate Homers, PI3K and p-(Tyr458)PI3K, and Tris-Acetate gradient gels (3–8%) (Life Technologies) were used to separate the glutamate receptor proteins and PLCβ3. The following rabbit polyclonal antibodies were used: anti-Homer2a/b (Cosmo Bio USA, Carlsbad, CA, USA) and anti-Homer1b/c (GeneTex, Irvine, CA, USA), anti-mGluR5 (Upstate Cell Signaling Solutions, Lake Placid, NY, USA), anti-GluN2A and anti-GluN2B (Calbiochem, San Diego, CA, USA), anti-PI3K (Upstate Cell Signaling Solutions), anti-p(Tyr458)PI3K (Cell Signaling, Danvers, MA, USA) and anti-PLCβ3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). An anti-mGluR1a mouse polyclonal antibody (Upstate Cell Signaling Solutions) was also used and a rabbit anti-calnexin polyclonal primary antibody (Stressgen, Victoria, British Columbia, Canada) indexed protein loading and transfer. All primary antibodies were diluted 1:1000, with the exception of the anti-PLCβ3 and –p-PI3K, which were diluted 1:500 prior to incubation. The levels of immunoreactivity for all proteins, detected using standard chemiluminescent approaches, were quantified using Image J (NIH, Bethesda, MD, USA), and each protein of interest was first normalized to that of its appropriate calnexin signal to provide a protein/calnexin ratio. These ratios were then normalized to the mean ratios for each protein of the water control for each individual gel (n=6–7 per gel). The data were analyzed using t-tests (α=0.05).
Surgical Procedures
The surgical procedures to implant bilateral guide cannulae into the CeA were similar to those employed for studies of the NAC in previous reports (for example, Cozzoli et al, 2009, 2012; Goulding et al, 2011; Szumlinski et al, 2008). Under isoflurane anesthesia, guide cannulae (20 gauge, 10 mm long) were implanted 2 mm over the CeA of mice using the following coordinates from the mouse brain atlas of Paxinos and Franklin (2004): AP: −1.25; ML: ±2.70; DV: −2.70 mm from Bregma. To prevent continuous externalization, dummy cannulae (24 gauge; length equivalent to guide cannula) were placed inside the guide cannulae and only removed before testing.
Intracranial Drug-Infusion Procedures
To examine a role for mGluR1, mGluR5, PLC and PI3K within the CeA, a series of neuropharmacological studies was conducted. Bilaterally cannulated B6, Homer2 WT and/or Homer2 knockout (KO) mice were trained to consume 20% alcohol under DID procedures until stable intake was established (<10% variability across three consecutive presentations), which occurred typically within the first 4–6 days of drinking. Separate groups of mice received microinfusions of a maximum of 5 of the following drug doses: the mGluR1 antagonist JNJ-16259685 [(3,4-Dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycyclohexyl)-methanone] (0, 0.1, 0.5, 5 and 15 pg per side; Tocris Bioscience, Ellisville, MO, USA), the mGluR5 antagonist MTEP [3-((2-Methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride] (0, 0.3 and 3.0 μg per side; Tocris Bioscience), the PLC inhibitor U-73122 [1-[6-((17β-3-Methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1 H-pyrrole-2,5-dione] (0, 0.58, 5.8, 58, 580, 5800, 58 000 fg per side; EMD Millipore, Billerica, MA, USA), the highly selective, class 1 PI3K inhibitor GDC-0941 [2-(1 H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine] (0, 0.53 and 5.3 pg per side, Axon Medchem BV, Groningen, The Netherlands), the less selective PI3K antagonists LY 294002 [2-(4-morpholinyl)-8-phenyl-4 H-1-benzopyran-4-one hydrochloride] (0 and 0.17 ng per side, Tocris Bioscience) and wortmannin (0, 50 ng per side; Sigma-Aldrich, St Louis, MO, USA). The highly selective polo-like kinase-1 antagonist cyclapolin 9 [7-nitro-5-(trifluoromethyl)-2-benzo thiazolecarboxamide-3-oxide] (0, 38 pg per side, Tocris Bioscience) was also infused to control for one of the potential off-target effects of LY 294002 and wortmannin (Cozzoli et al, 2012). Microinfusions were delivered via a 33-gauge injector (12 mm long) at a rate of 0.25 μl min−1 for a total volume of 0.25 μl per side, and the injectors remained in place for an additional 60 s. All drugs were dissolved in sterile water with the exception of JNJ-16259685 and GDC-0941, which were dissolved in 0.1% dimethyl sulphoxide. The order of dosing either within drug (in the cases of dose–response studies) or across drugs (in the cases of co-infusion studies) was pseudo counterbalanced, and 2–4 days were allowed between microinfusions to re-establish baseline intake (see also Cozzoli et al, 2009; 2012). Immediately after infusion, mice were returned to their home cages and presented with the 20% alcohol-containing sipper tube for 2 h. To assess for nonspecific effects of intra-CeA antagonist infusion, an effective dose of a particular antagonist for reducing alcohol intake was then examined for effects upon 5% sucrose intake during a 2-h session. Standard cresyl violet histochemical procedures were used to verify injector cannulae localization in the CeA. As not all mice in the neuropharmacological experiments received all intracranial treatments as a result of experimental design (maximum of five microinjections) or loss of guide cannulae patency (maximum of two mice per experiment), the alcohol intake data for the drug studies were analyzed using between-subjects analysis of variances (ANOVAs), followed by LSD post-hoc tests when appropriate (α=0.05).
Small-Hairpin RNA (shRNA) Vector Infusion and Four-Bottle-Choice Procedures
To assay the functional relevance of CeA Homer2b for binge drinking, we infused a neurotropic AAV expressing a shRNA for the knockdown of Homer2b (shRNA-Homer2) directly into the CeA (at 0.1μl min−1 for 5 min). For visualization of transduced cells, the shRNA-Homer2 construct also included the renilla GFP (hrGFP) reporter gene under the control of the chicken-beta actin promoter, and control AAVs carried the hrGFP reporter gene only. Details of our shRNA and control vectors, as well as our infusion procedures, are provided in Cozzoli et al (2012). Three weeks following AAV infusion, mice were subjected to DID procedures. In all, two independent experiments were conducted. The first employed our standard DID procedures and assayed the effects of shRNA-Homer2b upon the intake of 20% alcohol (averaged over 1 week of drinking) and 5% sucrose (averaged over 1 week of drinking). The second employed a multiple bottle-choice version of the DID procedures, in which AAV-infused mice were presented with 5, 10, 20 and 40% (v/v) alcohol for 1 week, followed by 5, 10 and 15% (w/v) sucrose for 1 week. Spillage from the bottles containing the four different alcohol concentrations was estimated from that recorded by conducting the four-bottle-choice procedures on three empty cages during testing. The average spillage for each concentration was calculated and subtracted from the volume consumed by the animals prior to calculation of their g kg−1 alcohol intake. After behavioral testing, mice were perfused transcardially with PBS, followed by a 4% paraformaldehyde solution, and then their brains were sliced along the coronal plane for visualization of the GFP reporter using fluorescent microscopy. Light microscopy was employed to examine for gross signs of neurotoxicity, as conducted previously (for example, Cozzoli et al, 2009, 2012; Goulding et al, 2011; Szumlinski et al, 2008). The behavioral data from the AAV studies were analyzed using two-way ANOVA, with treatment as repeated measure (single-bottle DID), or a mixed-design ANOVA, with repeated measures of the concentration factor (multi-bottle DID; α=0.05).
RESULTS
Binge Drinking Elevates CeA Levels of Glutamate-Associated Proteins
The B6 mice in the immunoblotting study consumed on average 4.8±0.37 g kg−1 per 2 h over the entire 30-day drinking period, which is predicted, based on published results (for example, Rhodes et al, 2005) to result in BACs ⩾80 mg%. As illustrated in Figure 1b, we observed an increase in CeA levels of Homer2a/b (t22=2.356, P=0.03), mGluR1a (t22=2.491, P=0.02), GluN2B (t22=3.779, P=0.001) and PLCβ3 (t22=2.749, P=0.01) at 24 h withdrawal from binge drinking, whereas GluN2A levels were moderately reduced (t22=1.877, P=0.07). There were no observable changes in the total protein expression of Homer1b/c, mGluR5, p-PI3K or PI3K within the CeA (t-tests, all P-values >0.05). As shown in Figure 1c, the alcohol-induced increases in protein expression were not apparent within the basolateral amygdala. Thus, short-term withdrawal from a chronic history of binge drinking increases mGluR1 and GluN2B expression within the CeA, concomitant with increases in Homer2a/b and PLCβ3.

Thirty days of alcohol drinking under drinking-in-the-dark procedures upregulated indices of glutamate signaling within the central nucleus of the amygdala (CeA). (a) Representative immunoblots for the protein levels of Homer1b/c, Homer2a/b, mGluR1, mGluR5, GluN2A, GluN2B, PLC-β3, p-PI3K, PI3K and calnexin in the CeA of mice following a 24-h period of withdrawal after 30 days of 2-h access to 20% alcohol (a) or water (W). (b) Summary of the change in protein expression within the CeA at a 24-h withdrawal from 30 days of alcohol drinking under DID procedures, expressed as a percentage of the average protein expression of water-drinking controls (Water). Compared with water controls, binge alcohol drinking significantly increased levels of Homer2 (↑42%), mGluR1 (↑56%), GluN2B (↑63%) and PLC-β3 (↑29%). (c) A parallel study conducted on tissue from the basolateral amygdala failed to reveal any significant alterations in protein levels following binge alcohol drinking, when compared with water controls. Data represent the mean±s.e.m. *P<0.05 (t-tests).
mGluR1, mGluR5 and PLC within the CeA Maintain Binge Alcohol Consumption
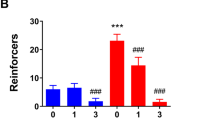
We next evaluated whether intact activation of mGluR1/5, PLC and PI3K signaling within the CeA maintained excessive alcohol consumption (Figure 2). Intra-CeA infusion of the mGluR1 antagonist JNJ-16259685 or the mGluR5 antagonist MTEP both reduced alcohol intake under DID procedures (JNJ: F(4,24)=5.46, P=0.006; MTEP: (2,14)=10.26, P=0.006), with significant reductions in intake produced by the 5 and 15 pg doses of JNJ-16259685 (Figure 2a) and the 3 μg dose of MTEP (Figure 2b) (post-hoc tests, P-values <0.05). PLC inhibition by U-73122 robustly decreased binge alcohol intake with significant attenuations observed at all doses tested (Figure 2c) (F(6,88)=5.06, P=0.0001; post-hoc tests, P<0.05). Importantly, intra-CeA infusion of 3.0 μg per side MTEP, 15 pg per side JNJ-16259685 or 0.58 fg per side U-73122 did not significantly affect sucrose intake under DID procedures, relative to vehicle, indicating a selective effect of these inhibitor doses on alcohol drinking (Figure 2d; P>0.05). Additionally, intra-CeA infusion of the selective Class I PI3K inhibitor GDC-0941 failed to alter binge drinking even when administered at a dose 10 times its reported in vitro IC50 for inhibiting p110α (for example, Folkes et al, 2008) (Figure 2e; P>0.05). Although intra-NAC infusion of the non-selective PI3K antagonists wortmannin and LY 294002 were demonstrated to reduce binge alcohol drinking previously by our group (Cozzoli et al, 2009, 2012), we failed to detect significant effects following infusion into the CeA. Additionally, an infusion of the polo-like kinase-1 inhibitor cyclapolin 9 (employed as a control for potential off-target effects of these inhibitors) failed to significantly alter alcohol intake (Figure 2f; P>0.05).

Blockade of CeA mGluR1, mGluR5 and PLC, but not PI3K, reduces binge alcohol drinking in B6 mice without having an impact on sucrose drinking. Summary of the effects of an intra-CeA infusion of the mGluR1 antagonist JNJ-16259685 (a), the mGluR5 antagonist MTEP (b) and the PLC antagonist U-73122 (c) upon 20% alcohol intake during DID procedures. (d) Summary of the effects of the intra-CeA infusion of JNJ, MTEP and U-73122 upon 5% sucrose intake during DID procedures. Summary of the effects of an intra-CeA infusion of the PI3K inhibitor GDC-0941 (e) as well as the PI3K inhibitors LY 294002 and wortmannin and the polo-like kinase-1 antagonist cyclapolin 9 (f) upon 20% alcohol intake during DID procedures. Data represent the mean±s.e.m. *P<0.05 versus respective vehicle (VEH) pretreatment.
To address whether or not the effects of mGluR1/5 and PLC inhibition were interdependent, we next assayed the effects of co-infusing effective doses of either MTEP (3.0 μg per side) or JNJ-16259685 (15 pg per side) with U-73122 (0.58 fg per side) on binge alcohol intake. All intracranial manipulations lowered alcohol intake, relative to vehicle-infused controls (Figure 3) (F(3,39)=12.46, P<0.0001) and post-hoc comparisons confirmed a reduction in alcohol intake by the very low dose of U-73122 (t(13)=2.63, P=0.02). Interestingly, the U-73122-induced attenuation of drinking was not influenced by MTEP co-infusion (t-test, P>0.05) but was accentuated by JNJ-16259685 co-infusion –t(13)=2.80, P=0.015). These data suggest that the attenuation of binge drinking by mGluR5 antagonism is dependent upon intact activation of PLC, whereas that produced by mGluR1 blockade may be independent of PLC activity.

Combined blockade of CeA mGluR1 and PLC, but not mGluR5 and PLC, further reduces binge alcohol drinking in B6 mice when compared with PLC blockade alone. Summary of the effects of an intra-CeA infusion of the PLC antagonist U-73122 (0.58 fg per side) alone or in combination with the mGluR5 antagonist MTEP (3.0 μg per side) or JNJ-16259685 (15 pg per side) upon 20% alcohol intake during DID procedures. Data represent the mean±s.e.m., *P<0.05 versus vehicle (VEH) pretreatment.
Homer2b within the CeA and Binge Alcohol Consumption
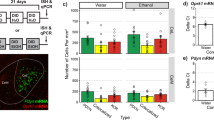
We next assessed the functional relevance of the Group1 mGluR-scaffolding protein Homer2 by infusing intra-CeA AAVs carrying shRNA-Homer2b or a control vector and by testing for effects upon the intake of 20% alcohol under DID procedures. However, despite histological verification of microinjector localization and neuronal transduction, as well as immunoblotting verification (n=4/AAV) of an ∼50% knockdown of protein expression within the CeA by our shRNA construct (Figures 4a–c), the results of two independent replicates of this experiment failed to indicate group differences for the intake of either 20% alcohol (GFP: 3.52±0.34 g kg−1, n=16; shRNA: 4.23±0.33 g kg−1, n=21) or 5% sucrose (GFP: 7.84±0.43 g kg−1, n=16; shRNA: 8.64±0.92 g kg−1, n=21) (P>0.05). We next conducted a third replicate of the study to address the possibility that Homer2b knockdown in the CeA altered alcohol/sucrose sensitivity, in which the mice were presented simultaneously with 5, 10, 20 and 40% alcohol or with 5, 10 and 15% sucrose for 2 h daily over the course of 7 days. Using this dose–response approach, we observed a significant effect of CeA Homer2b knockdown upon total alcohol intake (Figures 4d) (t(25)=3.10, P=0.005), which reflected reduced consumption of 40% alcohol but not other alcohol concentrations (Figures 4e) (AAV × concentration: F(3,75)=5.52, P=0.002; 5, 10 and 20% post-hoc tests: P>0.05; 40%: P<0.01). As blood-sampling procedures can interfere with subsequent fluid intake, we opted not to collect blood samples from our AAV-infused mice and conducted a parallel study of binge drinking under our four-bottle-choice procedures in manipulation-naive B6 mice. The alcohol intakes of these B6 mice were approximately equivalent to that exhibited by our shRNA-infused animals (mean total intake =9.90±1.91; n=19), and the average BACs obtained at the end of the 2-h drinking session were well above the NIAAA criterion for binge drinking (mean BAC =126.7±7.93 mg%). Thus, reducing Homer2b levels within the CeA significantly lowers alcohol intake under the four-bottle-choice procedures but does not eliminate binge alcohol intake under these or the one-bottle conditions. However, no effect of CeA Homer2b knockdown was apparent for total sucrose intake when we employed a dose–response approach (GFP: 20.46±2.49 g kg−1 versus shRNA: 15.83±0.89 g kg−1; t-test, P>0.05).

CeA knockdown of Homer2b decreases total alcohol intake and intake of 40% alcohol in a four-bottle-choice version of the DID procedure. (a) Histological verification of microinjector localization into the CeA using light microscopy. The black arrow indicates the tip of the guide cannula and the white arrow indicates the tip of the microinjector within the CeA. (b) Visualization of the green fluorescent protein (GFP) tag using fluorescent microscopy indicating neuronal transduction within the CeA (B at × 10 and B’ at × 20 magnification). (c) Immunoblotting verification of Homer2 knockdown by shRNA within punches taken from the CeA using anti-Homer2 (KO=Homer2 KO animal). (d) Homer2b knockdown produced by shRNA-Homer2b infusion in the CeA of B6 mice (shRNA) significantly reduced total alcohol intake in a four-bottle-choice DID paradigm, relative to AAV-GFP-infused controls (GFP), which reflected a reduced consumption of 40% alcohol but not other concentrations (e). Data represent the mean±s.e.m., *P<0.05 versus GFP.
Homer2 is Necessary for the Inhibition of Binge Drinking by mGluR1, mGluR5 and PLC Antagonists
To determine whether or not the effects of mGluR1, mGluR5 and PLC blockades require Homer2 expression, we conducted a neuropharmacological study in Homer2 WT and KO mice. A comparison of alcohol intake under DID procedures failed to indicate genotypic differences in the absence of any intracranial manipulation (WT=4.2±0.48 versus KO=3.56±0.39 g kg−1; t-test, P=0.33). However, there was a clear genotypic difference in the capacity of effective doses of JNJ-16259685, MTEP and U-73122 to reduce alcohol intake under DID procedures (Figure 5) (Genotype × drug: F(4,80)=3.69, P=0.02). Consistent with the data for baseline drinking, genotypic differences were not apparent for vehicle-infused mice (t-test, P=0.25)). However, in WT mice, intra-CeA drug infusions significantly reduced alcohol intake (F(4,42)=12.46, P<0.0001), with all treatments significantly lowering alcohol drinking below that exhibited by vehicle-infused animals (post-hoc tests, all P-values <0.05). In contrast, none of the compounds altered alcohol intake when infused intra-CeA in KO animals (one-way ANOVA, P>0.05).

Homer2 is necessary for the attenuating effects of CeA mGluR1, mGluR5 and PLC blockade. Summary of the effects of an intra-CeA infusion of effective doses of the mGluR1 antagonist JNJ-16259685 (15 pg per side), the mGluR5 antagonist MTEP (3 μg per side) and the PLC antagonist U-73122 (0.58 fg per side) upon 20% alcohol intake during DID procedures by Homer2 WT and KO mice. The data represent the mean±s.e.m., *P<0.05 versus vehicle (VEH) pretreatment.
DISCUSSION
The present results fill a void in our knowledge concerning the neurobiology of binge drinking by directly examining the role for glutamate receptor activation within the CeA. When combined with earlier work (Cozzoli et al, 2009, 2012), the present data provide compelling evidence that binge drinking upregulates Group1 mGluR signaling throughout the extended amygdala, and the activation of both mGluR1 and mGluR5 signaling within the CeA is relevant for alcohol intake. However, we also show in vivo distinctions in the neuropharmacology of these two receptor subtypes within the CeA as it relates to binge drinking, in particular with respect to the activation of PLC. Importantly, from a medication-development perspective, herein we show that PLC inhibition was effective at reducing binge drinking and was devoid of effects upon the consumption of a non-alcoholic, palatable liquid. In these ways, the results of the present study advance our understanding of the neurocircuitry, neurochemistry and molecular biology of binge drinking.
Binge Alcohol Drinking Elevates CeA Indices of Glutamate Signaling
Consistent with studies conducted in alcohol vapor-exposed (Roberto et al, 2006) or chronic, continuous alcohol access-exposed rats (Obara et al, 2009), a month-long history of binge alcohol consumption by B6 mice increased CeA levels of Homer2, mGluR1, GluN2B and PLCβ3 proteins. Thus, the capacity of ingested alcohol to elevate CeA mGluR1 expression does not appear to depend highly upon the species studied, the scheduling of alcohol availability or the chronicity of drinking. Moreover and consistent with earlier results (Obara et al, 2009), we failed to detect alcohol-induced changes in protein expression within the adjacent basolateral amygdala. Thus, both the NACshell (Cozzoli et al, 2009, 2012) and CeA glutamate systems appear to be particularly sensitive to the effects of binge alcohol. As the CeA and NACshell share cytoarchitectural and functional anatomical similarities as components of the extended amygdala (Alheid and Heimer, 1988), it is suggested from the collection of data to date that short-term withdrawal from binge drinking under limited-access procedures is sufficient to augment excitability within this circuit highly implicated in the negative affective state accompanying alcohol withdrawal (Koob, 2013). Future studies should examine the longevity of these receptor adaptations and examine for changes in other receptor systems within the CeA that is also implicated in alcohol dependence (for example, ionotropic glutamate, GABA and serotonin receptors; for example, Bell et al, 2006; Koob, 2013).
mGluR5-PLC, not -PI3K, Signaling in the CeA Maintains Excessive Drinking
Group1 mGluRs transduce glutamate signals through a number of intracellular pathways (Conn and Pin, 1997). Within the NACshell, mGluR5-Homer2-PI3K signaling, mediated presumably by the activation of βγ subunits (Rong et al, 2003), maintains binge alcohol consumption (Cozzoli et al, 2009, 2012). As Group1 mGluR stimulation of PLC can result in the activation of PKCs and as recent evidence implicates Protein Kinase C epsilon activity within the CeA in the manifestation of binge drinking (Lesscher et al, 2009), we probed signaling through mGluR1/5 to PLC and PI3K via Homer2 within the CeA. The average alcohol consumption of the mice in the present neuropharmacological studies ranged from 3.8 g kg−1 per 2 h (GDC-0941 study; Figure 5e) to 4.9 g kg−1 per 2 h (for example, U-73122 study; Figure 5c). These alcohol intakes are predicted from correlational analyses to elevate BACs above 80 mg% (for example, Rhodes et al, 2005) and, thus, constitute binge alcohol consumption. Despite no alcohol-induced change in protein expression, intra-CeA mGluR5 blockade reduced alcohol intake in both B6 mice and B6-129 hybrid mice, suggesting an increase in CeA mGluR5 function by a month-long history of binge drinking. The increase in mGluR5 function likely relates to the alcohol-induced rise in CeA Homer2a/b expression, as (1) Homers are well characterized to regulate Group1 mGluR expression, trafficking and signaling in vivo (Ary et al, 2013; c.f., Szumlinski et al, 2008); (2) Homer2 knockdown is sufficient to lower binge alcohol intake (albeit only at a high concentration); and (3) the attenuating effects of intra-CeA mGluR5 antagonists are absent in the Homer2 KO mice. Thus, as reported for the NACshell (Cozzoli et al, 2009; 2012), mGluR/Homer2 signaling in the CeA maintains excessive alcohol drinking, although it remains to be determined whether or not this signaling contributes to the initiation of binge alcohol intake.
Akin to the effects of CeA mGluR5 antagonism, intra-CeA infusion of a potent and soluble mGluR1 antagonist significantly reduced alcohol drinking in both B6 and B6-129 hybrid mice; moreover, this effect depended upon the intact Homer2 expression as determined by studies in KO mice. To the best of our knowledge, this is the first neuropharmacological study in the alcohol field to confirm a role for CeA mGluR1 in behavior. Intriguingly, although both mGluR5 and mGluR1 are well characterized to stimulate PLC (Conn and Pin, 1997), the two receptor subtypes diverged with respect to the effects of antagonist co-infusion with a PLC inhibitor. Demonstrating interdependence in signaling, the effects of co-inhibition of mGluR5 and PLC were not additive, whereas co-inhibition of mGluR1 and PLC produced a greater reduction in binge intake than PLC alone. These results suggest that, at least in the CeA, mGluR1 signaling through an alternate, PLC-independent pathway is involved in maintaining binge drinking. Whereas the specific pathway involved cannot be discerned from the present studies, it clearly does not involve PI3K as none of the three antagonists tested were effective. These data are in contrast to our reports for the NAC, where binge drinking under either DID or SHAC (Scheduled High Alcohol Consumption) procedures enhanced indices of PI3K activity and PI3K inhibition significantly lowered binge drinking in behavioral studies (Cozzoli et al, 2009, 2012). However, the present study also failed to detect drinking-induced changes in amygdalar PI3K activity. Such data strongly argue against a major role for PI3K signaling within the CeA in the neurobiology of binge alcohol drinking.
CeA Homer2 Contributes, in Part, to High-Dose Alcohol Intake
Homer2 regulates Group1 mGluR and NMDA receptor trafficking and function (c.f., Shiraishi-Yamaguchi and Furichi, 2007; Szumlinski et al, 2008). Thus, we examined whether there is a critical role for Homer2 within the CeA for binge alcohol consumption. As observed in our previous studies of binge alcohol intake (Cozzoli et al, 2009, 2012), constitutive deletion of Homer2 did not have an impact on alcohol intake under DID procedures. This finding continues to intrigue our group as Homer2 KO mice are reported to exhibit a very clear alcohol-aversive phenotype when assayed for voluntary alcohol consumption under free-access and operant self-administration procedures, as well in alcohol-induced place-conditioning studies (Szumlinski et al, 2005). Although we have argued in the past that developmental compensations secondary to Homer2 deletion or redundancy in function across Homer1, Homer2 and Homer3 isoforms might contribute to the failure to observe effects of Homer2 deletion upon binge alcohol drinking (Cozzoli et al, 2009; 2012), the results of a recent meta-analysis of studies of behavior conducted across 37 distinct mutant mouse lines indicate that voluntary alcohol consumption under limited-access ‘binge’ models (that is, SHAC and DID) are less-sensitive paradigms for detecting genotypic differences in alcohol consumption versus conventional, continuous-access, two-bottle-choice procedures. Moreover, voluntary alcohol intake under limited-access procedures (notably the SHAC procedure) is unrelated to that observed under continuous-access conditions. Thus, our failure to detect an effect of Homer2 deletion upon binge alcohol intake using SHAC or DID procedures could very well relate to the apparent ‘resistance to mutation’ of these models (see Blednov et al, 2012). The question as to why limited-access binge models are less sensitive to genotypic differences in voluntary alcohol consumption is one that is beyond the scope of the present report but, in all likelihood, relates to the scheduling and limiting of alcohol availability to the period of the circadian cycle in which mice innately consume the majority of their fluid intake for the day, which may be less subject to influence by constitutive gene deletion.
Despite the fact that Homer2 deletion does not impinge upon a binge drinking phenotype, previous studies employing AAV-mediated Homer2b knockdown within the NACshell established that Homer2b in this region was critical for a binge drinking phenotype (Cozzoli et al, 2009, 2012). As observed in the NAC (Cozzoli et al, 2009, 2012), Homer2a/b levels were increased in the CeA by binge drinking, suggesting that alcohol upregulates Homer2 expression throughout the extended amygdala to maintain excessive alcohol drinking. Thus, it was hypothesized that CeA Homer2b knockdown would reduce alcohol intake. However, when conventional, single-bottle DID procedures were employed, CeA Homer2b knockdown had no effect. As Homer2 deletion produces a rightward and downward shift in the dose–response functions for alcohol preference and intake under continuous-access conditions (Szumlinski et al, 2005), although NAC Homer2b over-expression in B6 mice augments selectively the intake of higher alcohol concentrations (Szumlinski et al, 2008), we offered shRNA-infused mice concurrent access to multiple alcohol concentrations for the 2-h drinking period. Under these conditions, alcohol intake was very high in both control and shRNA-infused animals. Whereas CeA Homer2b knockdown significantly reduced alcohol consumption (particularly at the 40% concentration), the total alcohol intake exhibited by shRNA-infused mice is estimated to result in BACs well above the 80 mg% NIAAA criterion for binge drinking. As Homer2 deletion produces an alcohol-intolerant and -adverse animal (Szumlinski et al, 2005), the possibility exists that Homer2, particularly within the CeA, may facilitate the development of tolerance to the aversive properties of alcohol, although the mechanisms through which this occurs require comprehensive investigation. One possibility might relate to effects of our shRNA-Homer2b construct upon the expression of glutamate receptors, as we have recently reported a coincident reduction in Homer2b and mGluR5 (but not mGluR1) upon intraprefrontal cortex infusion of our shRNA-Homer2b AAV (Ary et al, 2013). Nevertheless, the current studies showing the necessary role of Homer2 for the attenuating effects of intra-CeA mGluR1, mGluR5 and PLC antagonism demonstrates the importance of Homer2 for mGluR1 and mGluR5-PLC signaling within the CeA for the maintenance of an excessive drinking phenotype.
CONCLUSIONS
Using a combination of immunoblotting, neuropharmacological and behavioral genetic approaches, we show for the first time that two different mGluR1/5-Homer2 signaling cascades within the CeA underpin binge alcohol drinking in a murine model. Such data pose alcohol-induced plasticity within CeA Group1 mGluR signaling pathways in the etiology and treatment of this prevalent form of AUD.
FUNDING AND DISCLOSURE
All authors declare no conflict of interest in relation to the work described. This research was supported by NIH grants AA016650 (KKS), DA00266 and DA011742 (PFW), AA016981 (DAF), and a grant from the Department of Veterans Affairs (DAF). Funding to MK was provided by UNSW, as well as the Australian Research Council Future Fellowship. DKC was supported by T32 AA007468. The following organizations have financially compensated for professional services to Debra K Cozzoli and Deborah A Finn (Department of Behavioral Neuroscience, Oregon Health & Science University and VA Medical Research); Justin Courson, Melissa G Wroten, Andrew B Thompson, Dan Maliniak, and Karen K Szumlinski (Department of Psychological and Brain Sciences, University of California Santa Barbara); Georg Jonquieres and Matthias Klugmann (Translational Neuroscience Facility, School of Medical Sciences, University of New South Wales); and Paul F Worley (Department of Neuroscience, Johns Hopkins University School of Medicine). Daniel I Greentree, Emily N Lum, and Rianne R Campbell received academic, not financial, compensation for their participation in this research project from the Departments of Psychological and Brain Sciences (DIG, RRC) or Molecular, Cellular and Developmental Biology (ENL), University of California, Santa Barbara.
References
Alheid GF, Heimer L (1988). New perspectives in basal forebrain organization of special relevance for neuropsychiatric disorders: the striatopallidal, amygdaloid, and cotricopetal components of substantia innominata. Neuroscience 27: 1–39.
Ary AW, Lominac KD, Wroten MG, Williams AR, Campbell RR, Ben-Shahar O et al (2013). Imbalances in prefrontal cortex CC-Homer1 versus CC-Homer2 expression promote cocaine preference. J Neurosci 33: 8101–8113.
Bell RL, Rodd ZA, Lumeng L, Murphy JM, McBride WJ (2006). The alcohol-preferring P rat and animal models of excessive alcohol drinking. Addict Biol 11: 270–288.
Blednov YA, Mayfield RD, Belknap J, Harris RA (2012). Behavioral actions of alcohol: phenotypic relations from multivariate analysis of mutant mouse data. Genes Brain Behav 11: 424–435.
Cassell MD, Freedman LJ, Shi C (1999). The intrinsic organization of the central extended amygdala. Ann N Y Acad Sci 877: 217–241.
Conn PJ, Pin JP (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205–237.
Cozzoli DK, Courson J, Caruana AL, Miller BW, Greentree DI, Thompson AB et al (2012). Nucleus accumbens mGluR5-associated signaling regulates binge alcohol drinking under drinking-in-the-dark procedures. Alcohol Clin Exp Res 36: 1623–1633.
Cozzoli DK, Goulding SP, Zhang PW, Xiao B, Hu JH, Ary AW et al (2009). Binge drinking upregulates accumbens mGluR5-Homer2-PI3K signaling: functional implications for alcoholism. J Neurosci 29: 8655–8668.
Crabbe JC, Metten P, Rhodes JS, Yu CH, Brown LL, Phillips TJ et al (2009). A line of mice selected for high blood ethanol concentrations shows drinking in the dark to intoxication. Biol Psychiatry 65: 662–670.
Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G et al (2008). The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class 1 PI3 kinase for the treatment of cancer. J Med Chem 51: 5522–5532.
Gass JT, Olive MF (2008). Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol 75: 218–265.
Goulding SP, Obara I, Lominac KD, Gould AT, Miller BW, Klugmann M et al (2011). Accumbens Homer2-mediated signaling: a factor contributing to mouse strain differences in alcohol drinking? Genes Brain Behav 10: 111–126.
Kash TL, Baucum AJ 2nd, Conrad KL, Colbran RJ, Winder DG (2009). Alcohol exposure alters NMDAR function in the bed nucleus of the stria terminalis. Neuropsychopharmacology 34: 2420–2429.
Koob GF (2003). Alcoholism: allostasis and beyond. Alcohol Clin Exp Res 27: 232–243.
Koob GF (2013). Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci 13: 3–30.
Lesscher HM, Wallace MJ, Zheng L, Wang V, Deitchman JK, McMahon T et al (2009). Amygdala protein kinase C epsilon controls alcohol consumption. Genes Brain Behav 8: 493–499.
Melendez RI, Hicks MP, Cagle SS, Kalivas PW (2005). Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol Clin Exp Res 29: 326–333.
Moore EM, Boehm SL 2nd (2009). Site-specific microinjection of baclofen into the anterior ventral tegmental area reduces binge-like ethanol intake in male C57BL/6J mice. Behav Neurosci 123: 555–563.
Nakamura M, Sato K, Fukaya M, Araishi K, Aiba A, Kano M et al (2004). Signaling complex formation of phospholipase Cbeta4 with metabotropic glutamate receptor type 1alpha and 1,4,5-triphosphate receptor at the perisynapse and endoplasmic reticulum in the mouse brain. Eur J Neurosci 20: 2929–2944.
National Institute on Alcohol Abuse and Alcoholism (2004). NIAA Newsletter http://pubs.niaaa.nih.gov/publications/Newsletter/winter2004/Newsletter_Number3.pdf.
Obara I, Bell RL, Goulding SP, Reyes CM, Larson LA, Ary AW et al (2009). Differential effects of chronic ethanol consumption and withdrawal on homer/glutamate receptor expression in subregions of the accumbens and amygdale of P rats. Alcohol Clin Exp Res 33: 1924–1934.
Pandey SC (2003). Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol Sci 24: 456–460.
Paxinos G, Franklin KBJ (2004) The mouse brain in stereotaxic coordinates. Academic Press: Maryland Heights, MO.
Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC (2005). Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav 84: 53–63.
Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR (2006). Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology 31: 988–996.
Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004). Acute and chronic ethanol alter glutamatergic transmission in rat central amygdale: an in vitro and in vivo analysis. J Neurosci 24: 1594–1603.
Rong R, Ahn J-Y, Huang H, Nagata E, Kalman D, Kapp JA et al (2003). PI3 kinase enhancer-Homer complex couples mGluR1 to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci 6: 1153–1161.
Schroeder JP, Spanos M, Stevenson JR, Besheer J, Salling M, Hodge CW (2008). Cue-induced reinstatement of alcohol-seeking behavior is associated with increased ERK1/2 phosphorylation in specific limbic brain regions: blockade by the mGluR5 antagonist MPEP. Neuropharmacology 55: 546–554.
Shiraishi-Yamaguchi Y, Furuichi T (2007). The Homer family proteins. Genome Biol 8 8: 206–212.
Siggins GR, Martin G, Roberto M, Nie Z, Madamba S, De Lecea L (2003). Glutamatergic transmission in opiate and alcohol dependence. Ann N Y Acad Sci 1003: 196–211.
Substance Abuse and Mental Health Services Administration (2012) Results from the 2011 National Survey on Drug Use and Health: Summary of National Findings. Substance Abuse and Mental Health Services Administration: Rockville, MD, USA.
Szumlinski KK, Ary AW, Lominac KD, Klugmann M, Kippin TE (2008). Accumbens Homer2 overexpression facilitates alcohol-induced neuroplasticity in C57BL/6J mice. Neuropsychopharmacology 33: 1365–1378.
Szumlinski KK, Diab ME, Friedman R, Henze LM, Lominac KD, Bowers MS (2007). Accumbens neurochemical adaptations produced by binge-like alcohol consumption. Psychopharmacology (Berl) 190: 415–431.
Szumlinski KK, Lominac KD, Oleson EB, Walker JK, Mason A, Dehoff MH et al (2005). Homer2 is necessary for EtOH-induced neuroplasticity. J Neurosci 25: 7054–7061.
Wills TA, Klug JR, Silberman Y, Baucum AJ, Weitlauf C, Colbran RJ et al (2012). GluN2B subunit deletion reveals key role in acute and chronic ethanol sensitivity of glutamate synapses in bed nucleus of the stria terminalis. Proc Natl Acad Sci USA 109: E278–E287.
Acknowledgements
We would like to thank Dr Bo Xiao and Mr Marlin Dehoff (Johns Hopkins University School of Medicine) for their efforts in developing the Homer2 knockout mice.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cozzoli, D., Courson, J., Wroten, M. et al. Binge Alcohol Drinking by Mice Requires Intact Group1 Metabotropic Glutamate Receptor Signaling Within the Central Nucleus of the Amygdale. Neuropsychopharmacol 39, 435–444 (2014). https://doi.org/10.1038/npp.2013.214
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.214
Keywords
This article is cited by
-
Matrix Metalloproteinase-9 Overexpression Regulates Hippocampal Synaptic Plasticity and Decreases Alcohol Consumption and Preference in Mice
Neurochemical Research (2020)
-
NMDA receptor subunits change in the prefrontal cortex of pure-opioid and multi-drug abusers: a post-mortem study
European Archives of Psychiatry and Clinical Neuroscience (2019)
-
Metabotropic glutamate receptor 5 binding in male patients with alcohol use disorder
Translational Psychiatry (2018)
-
mGlu1 receptor as a drug target for treatment of substance use disorders: time to gather stones together?
Psychopharmacology (2017)
-
Frequency of alcohol consumption in humans; the role of metabotropic glutamate receptors and downstream signaling pathways
Translational Psychiatry (2015)
