
Abstract
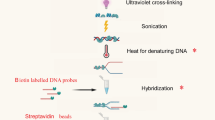
Chromatin immunoprecipitation (ChIP) is a powerful technique to study interactions between transcription factors (TFs) and DNA in vivo. For genome-wide de novo discovery of TF-binding sites, the DNA that is obtained in ChIP experiments needs to be processed for sequence identification. The sequences can be identified by direct sequencing (ChIP-SEQ) or hybridization to microarrays (ChIP-CHIP). Given the small amounts of DNA that are usually obtained in ChIP experiments, successful and reproducible sample processing is challenging. Here we provide a detailed procedure for ChIP of plant TFs, as well as protocols for sample preparation for ChIP-SEQ and for ChIP-CHIP. Our ChIP procedure is optimized for high signal-to-noise ratio starting with tissue fixation, followed by nuclei isolation, immunoprecipitation, DNA amplification and purification. We also provide a guide for primary data analysis of ChIP-SEQ data. The complete protocol for ChIP-SEQ/ChIP-CHIP sample preparation starting from plant harvest takes ∼7 d.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

References
Kuo, M.H. & Allis, C.D. In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19, 425–433 (1999).
de Folter, S., Urbanus, S.L., van Zuijlen, L.G., Kaufmann, K. & Angenent, G.C. Tagging of MADS domain proteins for chromatin immunoprecipitation. BMC Plant Biol. 7, 47 (2007).
Margueron, R., Trojer, P. & Reinberg, D. The key to development: interpreting the histone code? Curr. Opin. Genet. Dev. 15, 163–176 (2005).
Pfluger, J. & Wagner, D. Histone modifications and dynamic regulation of genome accessibility in plants. Curr. Opin. Plant Biol. 10, 645–652 (2007).
Tariq, M. & Paszkowski, J. DNA and histone methylation in plants. Trends Genet. 20, 244–251 (2004).
Kim, T.H. & Ren, B. Genome-wide analysis of protein-DNA interactions. Annu. Rev. Genomics Hum. Genet. 7, 81–102 (2006).
Massie, C.E. & Mills, I.G. Chromatin immunoprecipitation (ChIP) methodology and readouts. Methods Mol. Biol. 505, 123–137 (2009).
Benhamed, M. et al. Genome-scale Arabidopsis promoter array identifies targets of the histone acetyltransferase GCN5. Plant J. 56, 493–504 (2008).
Bernatavichute, Y.V., Zhang, X., Cokus, S., Pellegrini, M. & Jacobsen, S.E. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS ONE 3, e3156 (2008).
Turck, F. et al. Arabidopsis TFL2/LHP1 specifically associates with genes marked by trimethylation of histone H3 lysine 27. PLoS Genet. 3, e86 (2007).
Lee, J. et al. Analysis of transcription factor HY5 genomic binding sites revealed its hierarchical role in light regulation of development. Plant Cell 19, 731–749 (2007).
Oh, E. et al. Genome-wide analysis of genes targeted by PHYTOCHROME INTERACTING FACTOR 3-LIKE5 during seed germination in Arabidopsis. Plant Cell 21, 403–419 (2009).
Morohashi, K. & Grotewold, E. A systems approach reveals regulatory circuitry for Arabidopsis trichome initiation by the GL3 and GL1 selectors. PLoS Genet. 5, e1000396 (2009).
Kaufmann, K. et al. Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biol. 7, e1000090 (2009).
Gomez-Mena, C., de Folter, S., Costa, M.M., Angenent, G.C. & Sablowski, R. Transcriptional program controlled by the floral homeotic gene AGAMOUS during early organogenesis. Development 132, 429–438 (2005).
Ito, T., Takahashi, N., Shimura, Y. & Okada, K. A serine/threonine protein kinase gene isolated by an in vivo binding procedure using the Arabidopsis floral homeotic gene product, AGAMOUS. Plant Cell Physiol. 38, 248–258 (1997).
Leibfried, A. et al. WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature 438, 1172–1175 (2005).
Wang, H., Tang, W., Zhu, C. & Perry, S.E. A chromatin immunoprecipitation (ChIP) approach to isolate genes regulated by AGL15, a MADS domain protein that preferentially accumulates in embryos. Plant J. 32, 831–843 (2002).
Rozowsky, J. et al. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 27, 66–75 (2009).
Quail, M.A. et al. A large genome center′s improvements to the Illumina sequencing system. Nat. Methods 5, 1005–1010 (2008).
Nowak, D.E., Tian, B. & Brasier, A.R. Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques 39, 715–725 (2005).
Zeng, P.Y., Vakoc, C.R., Chen, Z.C., Blobel, G.A. & Berger, S.L. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. Biotechniques 41, 694,696, 698 (2006).
Turck, F., Zhou, A. & Somssich, I.E. Stimulus-dependent, promoter-specific binding of transcription factor WRKY1 to Its native promoter and the defense-related gene PcPR1-1 in Parsley. Plant Cell 16, 2573–2585 (2004).
Bowler, C. et al. Chromatin techniques for plant cells. Plant J. 39, 776–789 (2004).
Xie, Z. & Grotewold, E. Serial ChIP as a tool to investigate the co-localization or exclusion of proteins on plant genes. Plant Methods 4, 25 (2008).
Schmidt, D., Stark, R., Wilson, M.D., Brown, G.D. & Odom, D.T. Genome-scale validation of deep-sequencing libraries. PLoS ONE 3, e3713 (2008).
Katou, Y., Kaneshiro, K., Aburatani, H. & Shirahige, K. Genomic approach for the understanding of dynamic aspect of chromosome behavior. Methods Enzymol 409, 389–410 (2006).
Johnson, D.S. et al. Systematic evaluation of variability in ChIP-chip experiments using predefined DNA targets. Genome Res. 18, 393–403 (2008).
Park, P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680 (2009).
Li, H., Ruan, J. & Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–1858 (2008).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714 (2008).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Hillier, L.W. et al. Whole-genome sequencing and variant discovery in C. elegans. Nat. Methods 5, 183–188 (2008).
Dohm, J.C., Lottaz, C., Borodina, T. & Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 36, e105 (2008).
Arabidopis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Kozarewa, I. et al. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat. Methods 6, 291–295 (2009).
Spyrou, C., Stark, R., Lynch, A.G. & Tavare, S. BayesPeak: Bayesian analysis of ChIP-seq data. BMC Bioinformatics 10, 299 (2009).
Robertson, G. et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4, 651–657 (2007).
Bentley, D.R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008).
Ni, Z. et al. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 457, 327–331 (2009).
Acknowledgements
We thank S. de Folter and F. Turck for their initial advice on the ChIP experiments and W. Busch for advice on sample processing. Some parts of the computations were done at the Poznań Supercomputing and Networking Center.
Author information
Authors and Affiliations
Contributions
K.K. designed and carried out experiments and wrote the paper; J.M.M. analyzed data, developed computational tools and edited the manuscript; M.O. designed and carried out experiments; L.F. supervised experiments; P.K. supervised the data analysis and development of computational tools; and G.C.A. supervised the experiments and analysis and edited the manuscript.
Corresponding author
Supplementary information
Supplementary Figure 1 | ChIP-seq and ChIP-chip coverage comparison.
Solexa reads were randomly generated from the Arabidopsis genome using MAQ1 and uniquely mapped using SOAP2. Affymetrix 1.0R tiling probes were uniquely mapped using SOAP with the same parameters. The number of Affymetrix probes and Solexa reads representing non-overlapping genomic windows of size 30432 bp was calculated. Different number of solexa reads were simulated, the same number as unique Affymetrix probes (black), 3 times more (green), 3 times less (red). For illustrative reasons only chromosome 1 is shown. ChIP-seq has similar coverage, except near centromeric regions where the coverage is higher. (PDF 90 kb)
(1) Li, H., Ruan, J. & Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18, 1851-8 (2008).
(2) Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713-4 (2008).
Supplementary Figure 2 | Flowchart of different steps in the ChIP-SEQ data analyses, with examples for different general available software packages.
*For some algorithms, the reads are not extended. (PDF 528 kb)
The references associated with this figure are:
(1) Li, H., Ruan, J. & Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18, 1851-8 (2008).
(2) Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713-4 (2008).
(3) Kao, W.C., Stevens, K. & Song, Y.S. BayesCall: A model-based base-calling algorithm for high-throughput short-read sequencing. Genome Res 19, 1884-95 (2009).
(4) Rougemont, J. et al. Probabilistic base calling of Solexa sequencing data. BMC Bioinformatics 9, 431 (2008).
(5) Langmead, B., Trapnell, C., Pop, M. & Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009).
(6.) Spyrou, C., Stark, R., Lynch, A.G. & Tavare, S. BayesPeak: Bayesian analysis of ChIP-seq data. BMC Bioinformatics 10, 299 (2009).
(7) Johnson, D.S., Mortazavi, A., Myers, R.M. & Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497-502 (2007).
(8) Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008).
(9) Valouev, A. et al. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 5, 829-34 (2008).
(10) Ji, H. et al. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol 26, 1293-300 (2008).
(11) Bailey, T.L., Williams, N., Misleh, C. & Li, W.W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34, W369-73 (2006).
(12) Foat, B.C., Morozov, A.V. & Bussemaker, H.J. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics 22, e141-9 (2006).
(13.) Kel, A., Voss, N., Jauregui, R., Kel-Margoulis, O. & Wingender, E. Beyond microarrays: Finding key transcription factors controlling signal transduction pathways. BMC Bioinformatics 7 Suppl 2, S13 (2006).
(14) Bellora, N., Farre, D. & Mar Alba, M. PEAKS: identification of regulatory motifs by their position in DNA sequences. Bioinformatics 23, 243-4 (2007).
(15) Hestand, M.S. et al. CORE_TF: a user-friendly interface to identify evolutionary conserved transcription factor binding sites in sets of co-regulated genes. BMC Bioinformatics 9, 495 (2008).
(16) Nicol, J.W., Helt, G.A., Blanchard, S.G., Jr., Raja, A. & Loraine, A.E. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25, 2730-1 (2009).
Rights and permissions
About this article
Cite this article
Kaufmann, K., Muiño, J., Østerås, M. et al. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat Protoc 5, 457–472 (2010). https://doi.org/10.1038/nprot.2009.244
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2009.244
This article is cited by
-
The Mediator kinase module enhances polymerase activity to regulate transcriptional memory after heat stress in Arabidopsis
The EMBO Journal (2024)
-
Genomic and epigenomic determinants of heat stress-induced transcriptional memory in Arabidopsis
Genome Biology (2023)
-
Dual specificity and target gene selection by the MADS-domain protein FRUITFULL
Nature Plants (2023)
-
PIF7 is a master regulator of thermomorphogenesis in shade
Nature Communications (2022)
-
Plasma membrane-nucleo-cytoplasmic coordination of a receptor-like cytoplasmic kinase promotes EDS1-dependent plant immunity
Nature Plants (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
