
Abstract
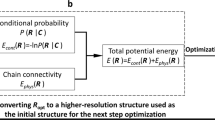
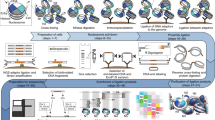
Chromosome conformation capture technologies such as Hi-C are widely used to investigate the spatial organization of genomes. Because genome structures can vary considerably between individual cells of a population, interpreting ensemble-averaged Hi-C data can be challenging, in particular for long-range and interchromosomal interactions. We pioneered a probabilistic approach for the generation of a population of distinct diploid 3D genome structures consistent with all the chromatin–chromatin interaction probabilities from Hi-C experiments. Each structure in the population is a physical model of the genome in 3D. Analysis of these models yields new insights into the causes and the functional properties of the genome's organization in space and time. We provide a user-friendly software package, called PGS, which runs on local machines (for practice runs) and high-performance computing platforms. PGS takes a genome-wide Hi-C contact frequency matrix, along with information about genome segmentation, and produces an ensemble of 3D genome structures entirely consistent with the input. The software automatically generates an analysis report, and provides tools to extract and analyze the 3D coordinates of specific domains. Basic Linux command-line knowledge is sufficient for using this software. A typical running time of the pipeline is ∼3 d with 300 cores on a computer cluster to generate a population of 1,000 diploid genome structures at topological-associated domain (TAD)-level resolution.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Dekker, J. et al. Capturing chromosome conformation. Science 295, 1306–1311 (2002).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Duan, Z. et al. A three-dimensional model of the yeast genome. Nature 465, 363–367 (2010).
Dixon, J.R. et al. Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015).
Sexton, T. et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472 (2012).
Ay, F. et al. Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression. Genome Res. 24, 974–988 (2014).
Rao, S.S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F. & Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 30, 90–8 (2012).
Nagano, T. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013).
Kind, J. et al. Single-cell dynamics of genome-nuclear lamina interactions. Cell 153, 178–192 (2013).
Beliveau, B.J. et al. Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat. Commun. 6, 7147 (2015).
Buenrostro, J.D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–90 (2015).
Stevens, T.J. et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017).
Junier, I., Dale, R.K., Hou, C., Kepes, F. & Dean, A. CTCF-mediated transcriptional regulation through cell type-specific chromosome organization in the -globin locus. Nucleic Acids Res. 40, 7718–7727 (2012).
Barbieri, M. et al. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. USA 109, 16173–16178 (2012).
Meluzzi, D. & Arya, G. Recovering ensembles of chromatin conformations from contact probabilities. Nucleic Acids Res. 41, 63–75 (2013).
Giorgetti, L. et al. Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell 157, 950–963 (2014).
Zhang, B. & Wolynes, P.G. Topology, structures, and energy landscapes of human chromosomes. Proc. Natl. Acad. Sci. USA 112, 6062–6067 (2015).
Tjong, H. et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proc. Natl. Acad. Sci. USA 113, E1663–E1672 (2016).
Dai, C. et al. Mining 3D genome structure populations identifies major factors governing the stability of regulatory communities. Nat. Commun. 7, 11549 (2016).
Li, Q. et al. The three-dimensional genome organization of Drosophila melanogaster through data integration. Genome Biol. 18, 145 (2017).
Serra, F. et al. Restraint-based three-dimensional modeling of genomes and genomic domains. FEBS Lett. 589, 2987–2995 (2015).
Peng, C. et al. The sequencing bias relaxed characteristics of Hi-C derived data and implications for chromatin 3D modeling. Nucleic Acids Res. 41, e183 (2013).
Zhang, Z., Li, G., Toh, K.-C. & Sung, W.-K. 3D chromosome modeling with semi-definite programming and Hi-C data. J. Comput. Biol. 20, 831–46 (2013).
Varoquaux, N., Ay, F., Noble, W.S. & Vert, J.-P. A statistical approach for inferring the 3D structure of the genome. Bioinformatics 30, i26–i33 (2014).
Lesne, A., Riposo, J., Roger, P., Cournac, A. & Mozziconacci, J. 3D genome reconstruction from chromosomal contacts. Nat Methods 11, 1141–1143 (2014).
Hu, M. et al. Bayesian inference of spatial organizations of chromosomes. PLoS Comput. Biol. 9, e1002893 (2013).
Zou, C., Zhang, Y. & Ouyang, Z. HSA: integrating multi-track Hi-C data for genome-scale reconstruction of 3D chromatin structure. Genome Biol. 17, 40 (2016).
Baù, D. & Marti-Renom, M.A. Genome structure determination via 3C-based data integration by the integrative modeling platform. Methods 58, 300–306 (2012).
Rousseau, M., Fraser, J., Ferraiuolo, M.A., Dostie, J. & Blanchette, M. Three-dimensional modeling of chromatin structure from interaction frequency data using Markov chain Monte Carlo sampling. BMC Bioinformatics 12, 414 (2011).
Russel, D. et al. Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 10, e1001244 (2012).
Alber, F. et al. Determining the architectures of macromolecular assemblies. Nature 450, 683–694 (2007).
Dostie, J. et al. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 16, 1299–1309 (2006).
Wang, S., Xu, J. & Zeng, J. Inferential modeling of 3D chromatin structure. Nucleic Acids Res. 43, e54 (2015).
Trieu, T. & Cheng, J. Large-scale reconstruction of 3D structures of human chromosomes from chromosomal contact data. Nucleic Acids Res. 42, e52 (2014).
Trieu, T. & Cheng, J. MOGEN: a tool for reconstructing 3D models of genomes from chromosomal conformation capturing data. Bioinformatics 32, 1286–1292 (2016).
Paulsen, J. et al. Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol. 1–15 (2017).
Gehlen, L.R. et al. Chromosome positioning and the clustering of functionally related loci in yeast is driven by chromosomal interactions. Nucleus 3, 370–83 (2012).
Kirkpatrick, S., Gelatt, C.D. & Vecchi, M.P. Optimization by simulated annealing. Science 220, 671–680 (1983).
Hestenes, M.R. & Stiefel, E. Methods of conjugate gradients for solving linear systems. J. Res. Nat. Bur. Stand. 49, 409–436 (1952).
Imakaev, M. et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012).
Knight, P.A. & Ruiz, D. A fast algorithm for matrix balancing. IMA J. Numer. Anal. 33, 1029–1047 (2013).
Szalaj, P. et al. 3D-GNOME: an integrated web service for structural modeling of the 3D genome. Nucleic Acids Res. 44, W288–W293 (2016).
Di Pierro, M., Zhang, B., Aiden, E.L., Wolynes, P.G. & Onuchic, J.N. Transferable model for chromosome architecture. Proc. Natl. Acad. Sci. USA 113, 12168–12173 (2016).
Durand, N.C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Acknowledgements
This work was supported by the Arnold and Mabel Beckman Foundation (BYI program) (to F.A.), the National Institutes of Health (grant U54DK107981 to F.A. and X.J.Z., grant P41 GM109824 to F.A., and a National Heart, Lung, and Blood Institute (NHLBI) grant (MAP-GEN U01HL108634) to X.J.Z.), and an NSF CAREER grant (1150287 to F.A.). F.A. is a Pew Scholar in Biomedical Sciences, supported by the Pew Charitable Trusts. We also thank W. Li for his contributions and discussions.
Author information
Authors and Affiliations
Contributions
H.T., K.G., and F.A. developed the method with the help of N.H.; N.H., H.T., and H.S. worked on the software design and implementation. N.H. and H.T. carried out analysis and developed tools included in the package. N.H., H.T. H.S., X.J.Z., and F.A. wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Data
Technical details of PGS. (PDF 221 kb)
Rights and permissions
About this article

Cite this article
Hua, N., Tjong, H., Shin, H. et al. Producing genome structure populations with the dynamic and automated PGS software. Nat Protoc 13, 915–926 (2018). https://doi.org/10.1038/nprot.2018.008
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2018.008
This article is cited by
-
Strong interactions between highly dynamic lamina-associated domains and the nuclear envelope stabilize the 3D architecture of Drosophila interphase chromatin
Epigenetics & Chromatin (2023)
-
A maximum-entropy model to predict 3D structural ensembles of chromatin from pairwise distances with applications to interphase chromosomes and structural variants
Nature Communications (2023)
-
Predicting scale-dependent chromatin polymer properties from systematic coarse-graining
Nature Communications (2023)
-
Evaluating the role of the nuclear microenvironment in gene function by population-based modeling
Nature Structural & Molecular Biology (2023)
-
Integrative genome modeling platform reveals essentiality of rare contact events in 3D genome organizations
Nature Methods (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
