
Abstract
Major depressive disorder topped ischemic heart disease as the number one cause of disability worldwide in 2012, and women have twice the risk of men. Further, the comorbidity of depression and cardiometabolic disorders will be one of the primary causes of disability worldwide by 2020, with women at twice the risk. Thus, understanding the sex-dependent comorbidities has public health consequences worldwide. We propose here that sex differences in MDD-cardiometabolic comorbidity originate, in part, from pathogenic processes initiated in fetal development that involve sex differences in shared pathophysiology between the brain, the vascular system, the CNS control of the heart and associated hormonal, immune, and metabolic physiology. Pathways implicate neurotrophic and angiogenic growth factors, gonadal hormone receptors, and neurotransmitters such as gamma amino butyric acid (GABA) on neuronal and vascular development of HPA axis regions, such as the paraventricular nucleus (PVN), in addition to blood pressure, in part through the renin–angiotensin system, and insulin and glucose metabolism. We show that the same prenatal exposures have consequences for sex differences across multiple organ systems that, in part, share common pathophysiology. Thus, we believe that applying a sex differences lens to understanding shared biologic substrates underlying these comorbidities will provide novel insights into the development of sex-dependent therapeutics. Further, taking a lifespan perspective beginning in fetal development provides the opportunity to target abnormalities early in the natural history of these disorders in a sex-dependent way.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD) topped ischemic heart disease as the number one cause of disability worldwide in 2012 [1], and women have twice the risk of men [2,3,4]. MDD has been associated with multiple general medical illnesses, the number one being cardiovascular disease (CVD). In fact, the comorbidity of MDD and CVD, with a substantial prevalence [5,6,7,8,9,10,11], will be one of the primary causes of disability worldwide by 2020, with women at twice the risk [12,13,14]. MDD is an independent risk factor for the development and progression of coronary artery disease [5, 7, 10, 15, 16]. Although the risk for CVD alone is higher in men [17], this difference attenuates after menopause in women, and heart disease is the number one cause of death of women in the U.S. and in most middle-income countries. Given that we have no effective treatments for the comorbidity of MDD and CVD, one reason for the high death rate for CVD in women may be due, in part, to unrecognized and untreated MDD. In fact, numerous prospective studies demonstrated significantly elevated risks of coronary heart disease, myocardial infarction, or cardiac death among participants with depression [11, 18,19,20,21,22]. Depression predicts first cardiovascular events even among otherwise healthy people [21], and particularly in women [19, 21], with a risk of 1.5–6-fold.
Although the incidence of sex differences in MDD has been known for many years, even recent studies of brain circuitry and genes associated with mood dysregulation and MDD per se do not design studies to investigate sex effects nor incorporate even current sex-dependent knowledge into development of therapeutics. MDD is associated with abnormalities in stress response circuitry, i.e., brain circuitry regulating the hypothalamic pituitary adrenal (HPA) axis [23,24,25,26,27,28], including hypothalamus (HYPO), amygdala (AMYG), hippocampus (HIPP), anterior cingulate cortex (ACC), and ventromedial prefrontal and orbitofrontal cortices (vmPFC, OFC) [26, 27, 29,30,31,32], areas that are among the most sexually dimorphic in the brain, i.e., areas that develop in sex-dependent ways and function differently across the lifespan. HIPP, HYPO, AMYG, and PFC are dense in sex steroid and glucocorticoid receptors (GRs) [33,34,35] coupled with cytokine receptors [36,37,38,39], in particular, TNF-α, IL-1β, IL-6, the major co-activators of the HPA axis [36, 37, 40, 41]. In fact, activity in these areas has been associated with cortisol response [27, 42,43,44,45,46], autonomic dysfunction characterized by loss of parasympathetic cardiac tone [47, 48], and immune responses [36,37,38,39], brain activity, and hormonal physiology that we previously showed varied by sex [26, 49].
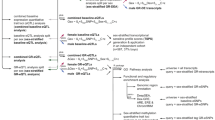
A number of studies demonstrated fetal risk factors for MDD [14, 50,51,52], with final common pathways involving abnormalities in maternal stress responses that are associated with fetal abnormalities in HPA axis development, or prenatal stress models of MDD. The HPA axis modulates inflammatory responses and microglial activation across the lifespan [53, 54], and neuroinflammation is associated with mood, autonomic nervous system (ANS), and vascular and metabolic disturbances. These HPA-immune connections have given rise to so-called prenatal stress-immune models of MDD and cardiometabolic disorders. We demonstrated that if these fetal stress and immune risk factors occur during a critical period of the sexual differentiation of the brain (2nd–3rd trimester), they impact highly sexually dimorphic brain regions [24] and the vasculature [55,56,57] in sex-dependent ways. We also argue here (see Fig. 1) that maternal prenatal glucocorticoid (GC) excess and immune pathway abnormalities beginning in fetal development are associated with sex-dependent impacts on the offspring’s brain circuitry that regulates mood, autonomic cardiac regulation, blood pressure, and metabolism, resulting in lifelong recurrent MDD, shared with dysregulation of hormone and immune responses to stress, autonomic dysfunction, and cardiometabolic dysregulation later in life [34, 58,59,60].

Prenatal stress and inflammation programs the fetal brain to increase the risk for depression and cardiometabolic disorders in adulthood. Diagrammatic representation of programming factors that can permanently affect the developing brain. Accordingly, prenatal stressors and immune responses can act through elevations in maternal glucocorticoids; This can involve changes in expression of a number of molecular targets such as brain-derived neurotrophic factor (BDNF), insulin-like growth factor 1 (IGF1), gamma amino butyric acid (GABA) signaling pathways that can act on neurons, glia, or vasculature to permanently alter neural circuitry and physiology. Of importance, there are sex biases in this mechanism which may represent interactions between glucocorticoid receptor (GR), estrogen receptor (ER), and androgen receptor (AR) signaling
Although population-level studies have demonstrated substantial sex differences in comorbidities with major public health implications worldwide, the pathways to explaining comorbidity are unclear. In part, this is due to a lack of focus on sex differences in diseases. However, beyond that, typically investigators studying the brain rarely think about the heart and associated vascular system or adiposity and vice versa. Moreover, studies focused on CVD and MDD generally begin in adulthood with the notion that either MDD causes CVD or metabolic syndrome (MetS), or CVD–MetS results in MDD. Given the substantial comorbidity worldwide, common fetal causes will have important implications for prevention or attenuation of disability in many countries [61], particularly in women.
Importance of considering comorbidity and sex
In medicine, disorders are traditionally thought of as separate entities and development of therapeutics are treated as such. Even the funding of research is predicated on a unitary approach to the study of disorders, i.e., divisions of funding streams through separate institutes at the National Institute of Health. However, we argue that more severe, long-term disability and greater expense to the healthcare system lies in the comorbidity of one or more disorders that share symptomatology and pathophysiology. Further, we argue here that a focus on sex differences in this shared pathophysiology will provide critical insights into understanding this comorbidity that will lead to the development of sex-dependent therapeutics with greater efficacy and implications that cross organ systems.
We propose that there are shared developmental origins implicating prenatal stress and immune pathways, suggesting a shared biological substrate as the basis for an increased likelihood of several disorders arising in the same individual. The conceptualization of prenatal maternal stress [62, 63] can include multiple kinds of maternal gestational exposures (both external or internally driven), such as obstetric conditions (e.g., preeclampsia, fetal growth restriction), maternal over- or undernutrition, or other environmental and psychological exposures. These exposures have been associated with abnormal immune responses, such as increased pro-inflammatory cytokines, that necessitate a maternal anti-inflammatory GC response in the in utero environment, thus the name, prenatal stress-immune models of disease vulnerability in the offspring. The pathways through which these stressors impact fetal growth and brain development in the offspring are not well understood, but likely involve genetic and epigenetic mechanisms mediated by hormones, growth factors, and/or markers of immune function that cross the placenta. Shared biological substrates can be envisioned on at least three levels (anatomical, molecular/biochemical, and genetic) that produce comorbid risk for these disorders with fetal origins that are sex dependent. Previous critical reviews by our team supported this case for MDD–CVD risk [24, 56] with respect to prenatal stress defined exclusively by excess GC exposure during development. Here, we expand this argument to mechanisms that implicate sex differences in the vasculature itself, blood pressure, metabolism, and the shared impact of the immune system.
Anatomy of stress and role of paraventricular nucleus
It is first important to understand the neuroanatomy of stress response circuitry in order to have a better understanding of the brain regions that are affected during development and why they have such widespread implications for disease vulnerability across the lifespan, and in particular, the key role that is coordinated by the paraventricular nucleus (PVN) in the HYPO. Historically, emotions and visceral function have been intimately associated, the latter of which is controlled by the ANS, coordinated to a large extent by the HYPO [64, 65]. Subsequently, a more complex network has been implicated including highly sexually dimorphic brain regions [66,67,68,69,70,71], such as AMYG [72, 73], HIPP, cingulate cortex [65, 74], medial PFC, and periacqueductal gray [75]. MDD has been characterized as maladaptive stress-induced neuroplastic alterations in the medial prefrontal cortico-amygdalo/hippocampo-hypothalamo-brainstem circuits [76, 77]. We and others previously demonstrated sex differences in this circuitry in healthy adults and deficits in MDD [26, 28, 49, 78]. Our findings are consistent with other studies of negative-high arousal stimuli [79,80,81,82], and sex differences to aversive cues [83,84,85,86]. Further, we demonstrated that some of these brain activity deficits in stress circuitry were significantly associated with loss of parasympathetic control in MDD women, including the HYPO [47, 48]. This is consistent with the fact that dysregulation of the nucleus of the solitary tract (which directly connects to the PVN), by the vagus nerve, and parasympathetic motor nuclei in the brainstem (innervated by preautonomic neurons in the PVN), can affect heart and cardiovascular function [16, 64, 87].
The PVN is the key brain region central to HPA axis function, implicated in MDD [88] and one of the most highly vascularized regions in the brain [89, 90]. It lies at the dorsal limit of the classical HYPO flanking the top of the third ventricle and has been implicated in a broad array of homeostatic and behavioral functions ranging from neuroendocrine and cardiovascular functions to affective, ingestive, and defensive behaviors [91, 92]. In rodents, its development can be regulated by GCs and gamma aminobutyric acid (GABA), with females being particularly vulnerable to GABA disruption in PVN neuronal [93, 94] and vascular development [95]. Previous human postmortem studies demonstrated that corticotropin releasing hormone (CRH)-producing neurons in PVN were increased in hypertension [96] in the same region as in MDD and regulated by estradiol [88], also suggesting a link for understanding sex differences in brain abnormalities (i.e., PVN) associated with MDD and cardiometabolic risk. This is not surprising since the PVN has been implicated in MDD and CVD in postmortem studies [88, 96, 97] and, in our work, in in vivo human structural [66] and functional imaging studies of sex differences in stress response circuitry in healthy adults [26, 66, 98], MDD subjects [28, 99], and ANS function [47, 48]. Further, we and others have demonstrated sex-dependent effects of GC excess in rats and mice on PVN and AMYG morphology [93, 100,101,102] and on expression of growth factors that regulate PVN and hippocampal function [103,104,105,106,107]. Thus, the PVN is a critical key relay station for understanding comorbidities among depression, CVD, and related MetSs.
Neurons of the PVN express receptors for almost all steroid hormones and from this arises the potential for sex differences in PVN function that could be driven by circulating sex steroid hormones. Moreover, steroid hormone receptors are among the markers that indicate different zones within the PVN. For example, cell groups reportedly contain immunoreactive estrogen receptors-α (ir-ERα), ERβ, androgen receptors (ARs), or GRs [108, 109]. Defects in the healthy development of the PVN provide an anatomical basis to predict shared comorbidity for disorders related to the functions of neurons normally located in or around the PVN. A number of mechanisms may lead to long-term alterations in PVN function that may or may not be sex dependent. These include changes in gene expression, cell death, connectivity, neuronal phenotypes, or positions thereof, or relationships to unique vasculature. Further, we and others have demonstrated environmental perturbations during development (e.g., obstetric complications or chronic social/psychological stress) cause changes in gene expression within the neurons of the PVN, in response to a common circulating factor (such as GCs), and perhaps as a function of epigenetic marks that are placed on DNA in response to such stimuli [110, 111]. For example, perinatal exposure of rats to the synthetic GC (sGC), dexamethasone (DEX), changed the methylation state of the brain-derived neurotrophic factor (BDNF) gene [110, 112] and/or levels of prepro-thyrotropin releasing hormone (TRH) [111]. Similarly, levels of maternal behavior altered the adult expression of GRs [113] and estrogen receptors [114]. In many cases, such influences have been shown to be sex dependent.
The vast majority of approaches to brain development examine neuronal, glial, and vascular factors independently, with notably less emphasis on vascular factors. The vascularization of the PVN is extensive compared to the surrounding tissue [115] and this density develops late in development [95, 116]. Therefore, alterations in PVN vasculature may provide another anatomical substrate to help explain shared comorbidities. In mice, PVN vascularization at birth is similar to the rest of the forebrain. Increased vascularization, however, becomes readily discernible by the second postnatal week [95]. Whatever drives the increased vascularity does so over a period of time when the PVN receives similar blood flow as the rest of the HYPO. Postnatal angiogenesis in the mouse PVN that would be later during prenatal human development may be driven by neural signals as suggested by the term “angioneurins” [117], but intrinsic to the unique environment and components of the PVN. Interestingly, neural activity may impact the development of the vasculature, since GABAB receptor signaling caused a 20% decrease in vascular characteristics of length or branch points [95].
The effectiveness of the blood brain barrier (BBB) may be a variable with significant impact in highly vascularized regions like the PVN. Disruption of BBB function in general might make the PVN appear selectively vulnerable because of the dense vascularization. In development, there is debate as to when the BBB “closes” or begins to regulate the flow of macromolecules into and out of the brain parenchyma. Our previous results suggested that perinatal GABAergic [95] or GC [118] treatments influence BBB development. Evidence currently exists for both sex-dependent GC [119] and reproductive hormone [120] influences on some aspects of BBB function, such as permeability and expression of molecular pumps.
Puberty and PVN
Puberty has been considered as a legitimate hormone-sensitive critical period in development rivaling that of early periods of brain differentiation [121]. Stress at puberty may unmask neuropsychiatric and neuropathological consequences of fetal programming [122]. The key question is what the nature of such programming is that the effect is delayed until another stimulus at puberty. At the molecular level, one can speculate epigenetic programming early in development may render genetic controls different at later points in development, such as shown with maternal grooming on GR methylation [113]. At a cellular level, one might look toward changes in immune cells such as microglia where long-term changes in cell numbers might result from early stimuli [123, 124]. Another group of cells to consider are those involved in neurovascular units. Maternal treatment with DEX caused BBB disruption in offspring prior to puberty independent of sex [57]. Lack of BBB competence was not apparent after puberty. However, astrocytes and pericyte components of the BBB differed as a function of fetal exposure, but dependent on sex. Disruption in BBB components in offspring from DEX-treated mothers may affect BBB function under specific physiological challenges, resulting in altered neuronal signaling. For example, spontaneously hypertensive rats have a breakdown of the BBB within the PVN, causing a feed-forward loop that increases blood pressure [125]. Although there may be previous permeability during a critical period, it would be detrimental to an organism to have a prolonged loss of BBB integrity, especially in a brain region like the PVN. Therefore, a repair process that maintains the ultimate BBB competency, but with underlying sex-dependent alterations in components, may result in susceptibility to further challenges. The emergence of sex differences in BBB components after puberty may be a critical substrate for sex-selective responses to stress after puberty. Thus, importantly, prenatal processes involved in sexual differentiation of the brain in regions regulating HPA axis and stress-immune responses in the offspring may be potentiated and/or enhanced after puberty.
Prenatal maternal programming of HPA axis in offspring development
There is a long history of work characterizing the HPA axis as central to understanding the development of MDD. Studies in humans and model animals demonstrated consistent HPA axis dysregulation associated with MDD, most notably elevated plasma cortisol levels, high cerebrospinal fluid CRH levels, and high cortisol secretion on DEX suppression test, although in some studies, hypocortisolemia [126,127,128,129,130,131]. These findings were not due to medication [132, 133]. Postmortem work has reported substantial decrease in the density of GR mRNA in MDD in frontal cortex, dentate gyrus, and subiculum, suggesting downregulation of GRs affecting the negative feedback system of the HPA axis and resulting in dysregulation of cortisol release [134].
Increasing evidence in rodent models has shown that perturbations in utero can cause long-term changes in the HPA physiology of the offspring, and, as seen in humans, they can increase disease risk in adulthood. These factors organize the HPA axis and ANS and their control of adult physiological functions and behaviors. A common signaling pathway between mother and fetus for many prenatal stressors includes rises in circulating GCs of maternal origin. Treatment with sGCs can mimic the prenatal stress-induced elevations in endogenous maternal GCs. Further, inhibition of 11β-hydroxysteroid dehydrogenase (11β-HSD), a placental enzyme that metabolizes GCs and acts as a buffer to maternal GCs [135], can also cause long-term changes in adult physiological parameters, including cardiac and metabolic function, neuroendocrine responses to stress and anxiety- and depressive-like behaviors in adulthood [136].
Studies in rodents show that prenatal stressors [137,138,139] can program the HPA axis causing elevated neuroendocrine responses to stress in adulthood. The effects of prenatal stress can be mimicked by prenatal exposure to GCs [140]. Of interest, the effects of prenatal stress on adult stress reactivity have been reported to be more profound in adult female offspring, depending on timing of the exposure [141,142,143]. Prenatal stressors elevate basal ACTH and CORT levels, increase the peak and extend the duration of the response to stressors in adulthood [140, 143]. Underlying these effects, are studies showing increased apoptosis in the PVN of fetal female, but not male, rats following chronic maternal immobilization [144]. The neuroendocrine changes in adulthood correlate with changes in anxiolytic behaviors [143]. Late gestation sGCs treatment resulted in increased anxiety and depressive-like behaviors in adult female, but not male, offspring [145]. This is consistent with clinical findings, since mood disorders have been characterized by a hyper-reactive HPA axis with disease risk greater in women than in men.
In contrast, postnatal treatment of rat pups with sGCS [146] blunted the stress-induced activation of the HPA axis in adulthood through elevated feedback inhibition, suggesting that the timing of GC exposure is important for directing the type of response observed and this may also impact sex-biased dysregulation. Further evidence of potential targets that are disrupted by prenatal stress and GCs comes from studies [147] that show the administration of 5-alpha-reduced neurosteroids to adult prenatally stressed rats reversed the effects of prenatal stress on the adult HPA axis, which occurred in a sex-dependent fashion. Given that sex-dependent changes can occur following prenatal perturbations, the importance of central changes in the factors regulating the HYPO and ANS stress-responsivity and how they impact long-term changes in cardiovascular, metabolic, and behavioral associated diseases are important future considerations. These topics will be addressed independently in the next sections of this review.
Prenatal programming of immune function
As discussed above, throughout the lifespan there is substantial bidirectional communication between the neuroendocrine stress pathways, ANS, and immune system [53, 54]. This crosstalk regulates both inflammation and mood. Many of the products of neuro-immune communication (cytokines, growth factors, neurotransmitters) can cross the placenta, such that immune activation experienced by the mother impacts the developing fetal immune system, brain, and vasculature, and we hypothesize perhaps programs future risk for neuro-immune diseases, including MDD (illustrated in Fig. 1).
Alterations in the inflammatory response of offspring may contribute to propensity to develop MDD, as depression has been proposed as an inflammatory disease. Evidence for an inflammatory basis for depression comes from several observations: (1) systemic immune responses are associated with a “sickness syndrome” of low mood, sleep disturbance, fatigue, anorexia; (2) pro-inflammatory cytokines, including IL-1β and IL-6, are elevated in the serum and plasma from patients with depression; and (3) anti-viral cytokine therapies used, such as interferons given to treat Hepatitis C, can induce depression [148]. Further, anti-inflammatory therapies including NSAIDs and biologics, ameliorate depressive symptoms and are under investigation in clinical trials as new antidepressants. Selective serotonin reuptake inhibitors (SSRIs) have been found to reduce levels of cytokines in MDD patients [149]. The immune dysregulation so common in MDD may, in part, be programmed by maternal immune dysregulation. Furthermore, there is a sex difference in the behavioral consequences of maternal immune programming that has been increasingly explored in preclinical models.
In general, investigations on how maternal immune responses impact development of offspring take one of two approaches. One approach models the lower levels of inflammation that are induced by activation of the HPA axis after a stress challenge. The other models mimic an over-exposure to bacterial or viral infection in pregnant mothers, for example, by injecting pregnant rodents with bacterial lipopolysaccharide (LPS) and/or the viral stimulus poly I:C, or both. This approach, termed “maternal immune activation (MIA)”, consistently results in neurodevelopmental changes in the offspring that have been associated with phenotypes related to sex differences in autism and schizophrenia [150]. Although depression and anxiety behaviors have not been well studied in this model, rats exposed to low dose LPS at prenatal day 10 had enhanced anxiety and depressive behaviors associated with reduced adult neurogenesis [151]. Further, prenatal LPS increased depression and anxiety-related behaviors in adult male offspring [152]. A priming effect in the MIA model has also been postulated such that the initial maternal immune exposure is considered a “first hit”, and a “second hit” at some point after birth amplifies the behavioral phenotype [153, 154]. This is consistent with recent work by our group demonstrating the impact of chronic social adversity on maternal prenatal immune activation abnormalities and neurodevelopment in the offspring [155].
Interestingly, based on the limited amount of data looking at sex differences in these models, the type of stress-immune challenge and level of inflammatory response experienced by the mother has been associated with sex differences in behavior of the offspring. However, timing of the exposure impacts whether the effect has greater consequences for male or female offspring. This may explain why in some studies higher inflammation (such as in the MIA models) led to greater anxiety or depressive (and neurodevelopmental) phenotypes in male offspring [152, 156], while work by our group and others [57, 110, 157, 158] resulted in increased anxiety and depression in female offspring.
Peripheral and central immune mechanisms can participate in how maternal immune dysregulation impacts the developing fetal immune system and brain, predisposing to increased risk for MDD in a sex-dependent manner. These mechanisms may include: (1) peripheral cytokines that act on brain, (2) microglial activation in the brain, (3) placental immune activation, (4) immune programming of the offspring immune system. First, prenatal and early life stress induces peripheral cytokines in the mother and fetus that change HPA axis function and are associated with sex-dependent risk for MDD [14, 52]. These prenatal studies are consistent with other studies associating mood disorders with innate immunity dysregulation [159,160,161,162]. The major cytokines studied include tumor necrosis alpha (TNF-α), interleukin- (IL)-1β, and IL-6 [163,164,165]. These cytokines are present in the maternal uterus, cross the placenta and BBB [166, 167], and are found in the fetal circulation and brain. Chronically elevated immune activation in mother and offspring as measured by cytokines predisposes to increased oxidative stress [168,169,170], the neural impact of which differs by sex [57, 110, 157, 158]. Exposure to cytokines and oxidative stress may lead to abnormalities in regions such as ACC, HIPP, and HYPO resulting in increased risk of negative stress response circuitry deficits and ANS dysregulation in the offspring.
Microglia
In addition to peripheral cytokines released by peripheral immune cells, central immune cells also may be important in fetal brain development. Microglia in particular are increasingly being recognized as being crucial in integrating environmental signals to regulate brain development. In addition, they exhibit vast sex differences in their response to these signals [171]; (see McCarthy et al., in this NPP edition). They express cytokine receptors as well as receptors for innate immune activators (i.e., TLR4, receptor for LPS). Although “activated” microglia are associated with neurobehavioral alterations and neuroinflammatory pathology, there is a significant sex difference in how microglia respond to the same immune challenge [154]. The microglia in females during LPS exposure reflect a completely distinct transcriptional signature from males, as determined by RNA-sequence profiling, with male microglia exhibiting a more mature phenotype after in vivo exposure [172]. In disorders such as chronic pain, T lymphocytes rather than microglia infiltrate and communicate directly with neurons in females [173]. This remains to be investigated in the context of prenatal immune activation and major depression.
The placenta is partially fetal, and therefore specific to the sex of the fetus. Although not typically considered an immune organ, the placenta also expresses cytokine and pattern recognition receptors and transmits information (through unclear pathways) to the fetal brain [174]. Some of this information may be through placental microbiome changes. Restraint stress led to microbiome changes in the mother, increased IL-1β in the placenta and brains of female (but not male fetal mice), decreased BDNF in the placenta and AMYG of adult female offspring, that was associated with increased anxiety like behaviors [175, 176].
Prenatal immune activation can also have significant effects in a sex-dependent way on the innate and adaptive immune response in the offspring, which again may contribute to the MDD risk in the adult offspring, although this type of immune programming is almost completely not understood. In a non-human primate study, pregnant rhesus macaques were injected with poly I:C LC to mimic viral infection [177]. The offspring exhibited increases in several cytokines, not just the expected pro-inflammatory cytokines, such as IL-1β, but a pronounced increase in allergic, Th2-mediated type inflammation. This observation is consistent with clinical studies in patients and preclinical studies in rodents, where maternal inflammation from either infection or non-infectious stress has been found to predispose to Th2-associated atopy (allergic diseases including asthma and eczema) in offspring of both sexes [178, 179]. The exception to this observation is when the mother has an autoimmune disease, for example, experimental autoimmune encephalitis (EAE) modeling of multiple sclerosis. In this case, the offspring of both sexes become more susceptible to induction of the autoimmune disease, associated with Th1-type inflammation [156]. Interestingly, in the non-EAE induced offspring, the males but not females showed increased depressive and anxious behaviors.
Prenatal programming of cardiovascular and autonomic function
Exposure to a variety of prenatal stressors including the administration of a sGC, such as DEX, dietary restriction, and inflammation can also program changes in cardiovascular function in adult offspring. While the ultimate phenotype of the cardiovascular dysfunction or pathology varies with the type and timing of prenatal insult, there are consistent changes that increase the risk of future CVD. In many cases, these can be tracked to changes in autonomic control of cardiovascular function with a key relay station being the PVN.
11β-HSD is normally expressed in the placenta and protects the fetus by buffering any elevations in GCs based on its ability to convert active GCs to inactive GCs. Prenatal GC exposure, resulting from increased endogenous transmission across the placental barrier secondary to reduced 11β-HSD expression, from elevations that can overwhelm placental 11 β-HSD, or after exogenous delivery of sGC to pregnant dams impact cardiovascular function in adult offspring in animal models of prenatal stress. Specifically, fetal increases in GCs can program changes in cardiovascular function in male and female rodent offspring, resulting in adulthood in increased vascular sensitivity to norepinephrine [180, 181], neuropeptide Y [182], electrical field stimulation [182], increased peripheral resistance, and reduced cardiac output [183]. With respect to blood pressure, both hypertension [184, 185] and hypotension [180, 186] have been reported. Moreover, adult offspring of prenatally stressed dams were more susceptible to elevations in blood pressure in response to high salt diet [187], angiotensin II (Ang II) [181], and restraint [180, 182, 188] suggesting the coincident programming of other physiological systems as well.
Excessive prenatal GC exposure has also been shown to result in male-specific impairment in sinoatrial node function and increased susceptibility to myocardial ischemic damage [189]. In addition, prenatal stressors have been shown to induce female-specific enhanced endothelin responsiveness [190] and impaired parasympathetic function (although males were not tested in the latter) [191]. Such results support the hypothesis that the in utero environment not only programs adult physiology, but it does so in a sexually dimorphic pattern, likely influenced by timing of exposure.
The effects of GC excess are thought to be mediated by central mechanisms, since GCs are readily found in neonatal rat brain following administration because of the immature BBB [192]. Data implicate the ANS as a mechanism that is targeted by prenatal stress or over-exposure to GCs or inflammatory molecules. In rats, O’Regan et al. [193] showed that prenatal DEX exposure caused a hypertensive phenotype in adult female offspring due to mild stressors, such as handling. These animals were also more sensitive to amphetamine, suggesting altered sympathetic responses, and further implicating coincident changes in mood dysregulation and anxiety-like behaviors.
Further support of underlying ANS dysfunction in prenatal DEX-exposed female offspring comes from studies showing that prenatal DEX treatment significantly reduced TRH and core body temperature (CBT), another marker of autonomic function, in female, but not male, offspring [194]. These have also been reported to be dysregulated in MDD. Of interest, there are no effects on overall activity in these studies, thereby ruling out activity as a source of the changes in arterial pressure or CBT. Together, these data show altered ANS activity in prenatal DEX-treated adult females that are consistent with findings regarding depressive and anxiety symptomatology and related to greater cardiovascular problems with age.
As in humans, prenatal GC exposures in rodents are coupled with inflammatory responses, known to be associated with hypertension and heart disease. Inflammation is an early mediator of hypertension and heart disease, as with MDD. Fetal exposure to an inflammatory environment can cause persistent negative consequences on the cardiovascular system of the adult offspring, as previously discussed for depressive symptomatology. For example, prenatal exposure to IL-6 resulted in hypertension, enhanced cardiovascular response to stress, and increased HPA axis activity in adult rats, with a greater effect observed in females [195]. This is interesting as prenatal IL-6 exposure has also been implicated in neurodevelopment and behavioral abnormalities associated with schizophrenia [150]. In addition, injecting the endotoxin LPS to pregnant dams produced diastolic dysfunction [196], associated with cardiac pathological remodeling in the heart involving hypertrophy, fibrosis [197], and apoptosis [196] in the adult offspring. The functional consequence of a prenatal inflammatory environment was impaired diastolic [196] and aortic dysfunction characterized by altered reactivity and connexin expression [198].
Prenatal stress alters the renin–angiotensin system (RAS)
The PVN receives afferent angiotensinergic projections from the subfornical organ (SFO), a circumventricular organ that is positioned outside the BBB and responds to circulating Ang II (see Fig. 2). Activation of angiotensin type 1 receptors (AT1R) within the SFO ultimately stimulates the sympathetic and inhibits parasympathetic neural pathways through relays in the PVN. Thus, abnormal development of the RAS in pre-ANS brain circuitry after prenatal GC excess may play an important role underlying increased risk of adult ANS imbalance. For example, prenatal [181] and perinatal [199] stress have been shown to increase the hypertensive response to Ang II infusion. Moreover, prenatal GC exposure has been shown to increase renal AT1R expression [185, 186, 200], renal Ang II production [201], and plasma renin activity [193]. However, the direction of RAS activity in male versus female offspring varied with the nature or timing of prenatal stress.

The paraventricular nucleus of the hypothalamus is a central player in the control of behavior, neuroendocrine function, and the autonomic nervous system. This figure shows a diagrammatic representation of some of the brain areas involved in regulating brainstem autonomic nuclei: NTS nucleus of the solitary tract; RVLM rostral ventral lateral medulla. Programming changes in the PVN can influence coincident changes in anxiety and depressive-like behaviors, stress-related neuroendocrine responses, and autonomic stress responses. The PVN also receives important input from circumventricular organs that have an incomplete blood brain barrier (BBB) such as the subfornical organ (SFO) which can respond to circulating peripheral signals such as angiotensin II (Ang II). PS parasympathetic, SYM sympathetic, AT1R angiotensin II receptor subtype 1. Motor X: motor nuclei of the vagus nerve (cranial nerve X). Dashed lines indicate principal projections of the PS nervous system, dotted lines indicate principal projections of the SYM nervous system
The role of enhanced RAS activity in cardiovascular control is further supported by studies showing that acute AT1R blockade lowered BP, partially normalized parasympathetic dysfunction, and fully normalized BP variability in adult male sheep that were prenatally exposed to sGC (females not tested) [202]. A maternal low-protein diet caused corticosterone-dependent increases in AT1b receptor expression coupled with reduced methylation of the AT1b receptor promoter [203]. Importantly, in addition to changes in angiotensin receptor expression, inhibition of corticosterone production in utero prevented hypertension in these offspring [203].
Prenatal stress has also been reported to alter the activity of the central RAS. Central infusion of losartan, an AT1R antagonist, reduced blood pressure and heart rate in the offspring of sGC-treated but not control male sheep (females not tested) [204]. Prenatal sGC exposure resulted in increased AT1R expression in the brainstem [204] and increased Ang II levels relative to the protective Ang(1-7) in the dorsal medulla [205] of adult offspring. While the mechanisms underlying increased RAS activity following prenatal stress are unknown, a cis-acting GC responsive element has been identified on the AT1aR promoter [206]. Together, these studies suggest prenatal stress alters the RAS through epigenetic programming of angiotensin receptor expression during gestation.
Prenatal programming of adult metabolic function
Animal studies have shown that prenatal stressors during late gestation predispose offspring to metabolic disorders in adulthood [207, 208]. Late gestation exposure of rodents to sGC increases gluconeogenesis in adult male, but not female offspring [193, 209], and alters insulin production by reducing the mass of pancreatic β-cells and their capacity to secrete insulin [210]. Prenatal GC exposure also increases circulating triglycerides, promotes storage of fat in the liver [211], and decreases fatty acid uptake in visceral adipose tissue [212] of adult offspring. Whereas, most studies have only examined adult male offspring, high fat diet-induced hepatosteatosis has been shown to be more profound in female offspring of DEX-treated dams [194]. This was coupled with female-biased decreases in adult circulating insulin-like growth factor 1 (IGF-1) [194].
GCs alter β-cell mass and function
During development and in adulthood, GCs have been shown to cause insulin resistance, hyperglycemia, and modulate β-cell proliferation to alter the total mass of pancreatic β-cells [213]. Support for this also comes from earlier studies that have shown that maternal food restriction can permanently decrease β-cell mass of neonates, secondary to elevated fetal corticosterone levels, regardless of post-weaning nutrition [214, 215]. In vitro studies showed that DEX treatment of embryonic pancreas caused a decrease in β-cell numbers and increase in acinar cells, whereas removal of GR signaling in acinar precursors increased β-cell mass [216]. Hence, a mechanism whereby DEX can modulate β-cell mass is by decreasing differentiation into β-cells. This differentiation step begins early in the third week of gestation in the rat (G14.5–15.5) [216]. In addition, GCs have been shown to be pro-apoptotic in a number of tissues including the pancreas where they activate the intrinsic mitochondrial cell death pathway, ultimately resulting in the activation of caspase-3 [217]. GCs have also been shown to program β-cell dysfunction through inhibition of pancreatic duodenal homeobox 1 (Pdx1), a transcription factor involved in β-cell maturation [216]. Impaired β-cell function and number have been reported following in utero DEX exposure during the last trimester [210, 218, 219], yet there are few studies examining sex differences in pancreas development or the effects of DEX on developing β-cells. Moreover, since normal pancreatic β-cell function is dependent upon an appropriate autonomic innervation [220, 221] in adulthood and during development [222], the same factors that underlie prenatal stress induced changes in cardiovascular function may similarly be involved in the programming of metabolism.
Growth hormone–IGF axis as target and mediator of fetal programming
Growth hormone (GH) and IGF-1 are independent and opposing risk factors for the development of metabolic disorders such as type II diabetes (T2D). GH, secreted by the anterior pituitary gland, promotes the expression and secretion of IGF-1 by the liver, but in mice lacking IGF-1, GH causes insulin resistance [223], whereas IGF-1 administration improves insulin sensitivity and contributes to glucose and FFA (free fatty acid) uptake [224]. Insulin and IGF-1 are closely related hormones controlling different aspects of growth and metabolism and they have tremendous overlap in function as each can bind to both the insulin receptor and the IGF-1 receptor [225]. During development, IGF-1 maintains β-cell mass [226] and the maturation of β-cells [227]. Prenatal DEX has been shown to cause growth restriction and reduced long-bone growth following weaning, with more severe growth restriction in females [194] and this correlated with decreases in circulating IGF-1. Insulin and IGF-1 appear to regulate overlapping responses. Thus, hyperinsulinemia produces early infant growth, whereas hypoinsulinemic babies were small [228]. Likewise, IGF-1 regulates glucose metabolism in states of insulin resistance. Administration of IGF-1 to Insulin Receptor knockout (IRKO) mice decreased plasma glucose levels by action through the IGF1R on skeletal muscle [229].
Prenatal stress and MetS in adulthood
Preclinical studies have shown that late gestation stressors predisposed offspring to MetS in adulthood [207, 208]. Correspondingly, late gestation exposure to sGC increased liver enzymes involved in gluconeogenesis in adult male, but not female offspring [193, 209], and altered production of insulin by reducing pancreatic β-cells and their capacity to secrete insulin [210]. Prenatal GCs also increased circulating triglycerides and storage of fat in the liver [211] and decreased fatty acid uptake in visceral adipose tissue [212]. Whereas, most of these studies were restricted to the examination of adult male offspring, we have previously demonstrated that high fat diet-induced hepatosteatosis was more profound in female offspring of DEX-treated dams which was coupled with sex-specific reductions in circulating IGF-1 [194]. Taken together, these studies highlight the role of prenatal GC exposure, whether from prenatal stress or treatment with sGC, on the impact of glucose homeostasis programming and risk for adult MetS.
The programming of MetS by prenatal stress may be initiated very early in gestation and have sex-selective effects expressed as different metabolic phenotypes in adult males and females. For example, O-linked N-acetyl glucosamine (O-GlcNAc) transferase (OGT) is expressed by the placenta and senses changes in maternal energy homeostasis to regulate epigenetic marks on chromatin [230]. Gestational stress during the first week of pregnancy in a mouse causes reductions in placental OGT which give rise to changes in offspring endocrine and hypothalamic mitochondrial function and body weight [231, 232]. Prenatal stress induced reductions in placental OGT were only found in male placentas, suggesting a pathway whereby early changes in placental OGT regulated sex-selective epigenetic modification of genes important for adult metabolism. Note that the earlier timing of stress induction resulted in greater impact on male than female placentas.
Gestational stress can also foster adult MetS by organizing hypothalamic nuclei controlling energy homeostasis [233]. A surge of circulating leptin occurs in development that peaks about postnatal day (PD) 9-12 in male and female rats [234]. Gestational malnutrition advanced the leptin surge, leading to hypothalamic leptin insensitivity in adult male offspring (females not tested) [235], while delaying the postnatal leptin surge led to diet-induced obesity in female rats (males not tested) [236]. Therefore, leptin is thought to be an important player in the development of both orexigenic and anorexigenic projections from the arcuate nucleus [237] and specifically acts during development to program the function of autonomic brainstem nuclei in adulthood [222]. Similarly, intrauterine growth restriction caused hyperleptinemia followed by leptin resistance by PD21 in female, but not male rats [238], while a leptin antagonist increased food intake in adult male, but not female offspring [239]. Together, these studies suggest sex-biased roles for the postnatal surge of leptin in the rodent, which can impact the development of hypothalamic regulation of energy balance in adulthood.
These studies are critical as we think about mechanisms to explain the high rate of the comorbidity of obesity and MetS with major depression. In fact, some have argued the importance of understanding distinctions between atypical and more classical depressive phenotypes, the former being more highly associated with overeating and weight gain and hypo- or normal cortisol levels than the classical melancholic depression, characterized by loss of appetite and weight and hypercortisolemia [240, 241].
Summary comment
The comorbidity of MDD and risk for CVD and associated MetS will be one of the primary causes of disability worldwide by 2020, and women are at twice the risk of men. In fact, these disorders are independent risk factors for memory decline and Alzheimer’s disease, which will overwhelm our economy without effective treatments. Thus, it is a major public health problem with substantial economic, social, and disease burden, particularly for women. Sex differences in MDD-cardiometabolic comorbidity originate, in part, from pathogenic processes initiated in fetal development that involve shared pathophysiology between the brain, the vascular system, the CNS control of the heart and associated hormonal, immune, and metabolic physiology. We hypothesize that prenatal stress and immune disruptions that occur during key gestational periods of the sexual differentiation of the brain will result in sex-dependent effects, dependent on exposure timing, on brain development in highly sexually dimorphic regions that regulate mood, stress, metabolism, and energy balance, the ANS, and the vasculature itself. Pathways implicate neurotrophic and angiogenic growth factors, gonadal hormone receptors, and neurotransmitters (such as GABA) on neuronal and vascular development of HPA axis regions, such as the PVN, in addition to blood pressure, in part through the renin angiotensin system, and insulin and glucose metabolism. The same prenatal exposures have consequences for sex differences across multiple organ systems that, in part, share common pathophysiology. Thus, we believe that applying a sex differences lens to understanding shared biologic substrates underlying these comorbidities will provide novel insights into the development of sex-dependent therapeutics. Further, taking a lifespan perspective beginning in fetal development provides the opportunity to target abnormalities early in the natural history of these disorders. By doing so, we believe this will increase the efficacy of these therapeutics even for one of the disorders and will move us closer to the ideals of precision medicine.
References
World Health Organization. Global health estimates 2014 summary tables: YLD by cause, age, and sex, 2000-2012. http://www.who.int/healthinfo/global_burden_disease/estimates/en/index2.html. Geneva, Switzerland.
Kendler K, Gatz M, Gardner C, Pedersen N. A Swedish national twin study of lifetime major depression. Am J Psychiatry. 2006;163:109–14.
Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289:3095–105.
Kessler RC, McGonagle KA, Swartz M, Blazer DG, Nelson CB. Sex and depression in the National Comorbidity Survey. I: lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29:85–96.
Barefoot JC, Helms MJ, Mark DB, Blumenthal JA, Califf RM, Haney TL, et al. Depression and long-term mortality risk in patients with coronary artery disease. Am J Cardiol. 1996;78:613–7.
Jones DJ, Bromberger JT, Sutton-Tyrrell K, Matthews KA. Lifetime history of depression and carotid atherosclerosis in middle-aged women. Arch Gen Psychiatry. 2003;60:153–60.
Kawachi I, Sparrow D, Vokonas PS, Weiss ST. Symptoms of anxiety and risk of coronary heart disease. the normative aging study. Circulation. 1994;90:2225–9.
Krishnan KR, Doraiswamy PM, Clary CM. Clinical and treatment response characteristics of late-life depression associated with vascular disease: a pooled analysis of two multicenter trials with sertraline. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:347–61.
Scherrer JF, Xian H, Bucholz KK, Eisen SA, Lyons MJ, Goldberg J, et al. A twin study of depression symptoms, hypertension, and heart disease in middle-aged men. Psychosom Med. 2003;65:548–57.
Everson SA, Kaplan GA, Goldberg DE, Salonen R, Salonen JT. Hopelessness and 4-year progression of carotid atherosclerosis; the Kuopio Ischemic Heart Disease Risk Factor Study. Arterioscler Thromb Vasc Biol. 1997;17:1490–5.
Glassman AH, Shapiro PA. Depression and the course of coronary artery disease. Am J Psychiatry. 1998;155:4–11.
Moller-Leimkuhler AM. Gender differences in cardiovascular disease and comorbid depression. Dialogues Clin Neurosci. 2007;9:71–83.
Naqvi TZ, Naqvi SS, Merz CN. Gender differences in the link between depression and cardiovascular disease. Psychosom Med. 2005;67 Suppl 1:S15–18.
Goldstein JM, Cherkerzian S, Buka SL, Fitzmaurice G, Hornig M, Gillman M, et al. Sex-specific impact of maternal-fetal risk factors on depression and cardiovascular risk 40 years later. J Dev Orig Health Dis. 2011;2:353–64.
Kawachi I, Colditz GA, Ascherio A, Rimm EB, Giovannucci E, Stampfer MJ, et al. Prospective study of phobic anxiety and risk of coronary heart disease in men. Circulation. 1994;89:1992–7.
Musselman DL, Evans DL, Nemeroff CB. The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry. 1998;55:580–92.
Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–215.
Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99:2192–217.
Rutledge T, Reis SE, Olson MB, Owens J, Kelsey SF, Pepine CJ, et al. Depression symptom severity and reported treatment history in the prediction of cardiac risk in women with suspected myocardial ischemia: the NHLBI-sponsored WISE study. Arch Gen Psychiatry. 2006;63:874–80.
Rutledge T, Reis VA, Linke SE, Greenberg BH, Mills PJ. Depression in heart failure a meta-analytic review of prevalence, intervention effects, and associations with clinical outcomes. J Am Coll Cardiol. 2006;48:1527–37.
Vaccarino V, McClure C, Johnson BD, Sheps DS, Bittner V, Rutledge T, et al. Depression, the metabolic syndrome and cardiovascular risk. Psychosom Med. 2008;70:40–48.
Van der Kooy K, van Hout H, Marwijk H, Marten H, Stehouwer C, Beekman A. Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int J Geriatr Psychiatry. 2007;22:613–26.
García-Gómez RG, López-Jaramillo P, Tomaz C. [The role played by the autonomic nervous system in the relation between depression and cardiovascular disease]. Rev Neurol. 2007;44:225–33.
Goldstein JM, Handa RJ, Tobet SA. Disruption of fetal hormonal programming (prenatal stress) implicates shared risk for sex differences in depression and cardiovascular disease. Front Neuroendocrinol. 2014;35:140–58.
Goldstein JM, Holsen L, Handa R, Tobet S. Fetal hormonal programming of sex differences in depression: linking women’s mental health with sex differences in the brain across the lifespan. Front Neurosci. 2014;8:247.
Goldstein JM, Jerram M, Abbs B, Whitfield-Gabrieli S, Makris N. Sex differences in stress response circuitry activation dependent on female hormonal cycle. J Neurosci. 2010;30:431–8.
Holsen LM, Lancaster K, Klibanski A, Whitfield-Gabrieli S, Cherkerzian S, Buka S, et al. HPA-axis hormone modulation of stress response circuitry activity in women with remitted major depression. Neuroscience. 2013;250:733–42.
Holsen LM, Spaeth SB, Lee JH, Ogden LA, Klibanski A, Whitfield-Gabrieli S, et al. Stress response circuitry hypoactivation related to hormonal dysfunction in women with major depression. J Affect Disord. 2011;1–3:379–87.
Mather M. The emotion paradox in the aging brain. Ann N Y Acad Sci. 2012;1251:33–49.
Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci. 1997;9:471–81.
Briceno EM, Rapport LJ, Kassel MT, Bieliauskas LA, Zubieta JK, Weisenbach SL, et al. Age and gender modulate the neural circuitry supporting facial emotion processing in adults with major depressive disorder. Am J Geriatr Psychiatry. 2015;23:304–13.
Dougherty D, Rauch SL. Neuroimaging and neurobiological models of depression. Harv Rev Psychiatry. 1997;5:138–59.
McEwen BS, De Kloet ER, Rostene W. Adrenal steroid receptors and actions in the nervous system. Physiol Rev. 1986;66:1121–88.
Pacak K, Palkovits M, Kopin IJ, Goldstein DS. Stress-induced norepinephrine release in the hypothalamic paraventricular nucleus and pituitary-adrenocortical and sympathoadrenal activity: in vivo microdialysis studies. Front Neuroendocrinol. 1995;16:89–150.
Tobet SA, Hanna IK. Ontogeny of sex differences in the mammalian hypothalamus and preoptic area. Cell Mol Neurobiol. 1997;17:565–601.
Schobitz B, de Kloet ER, Sutanto W, Holsboer F. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur J Neurosci. 1993;5:1426–35.
Anisman H, Merali Z. Cytokines, stress, and depressive illness. Brain Behav Immun. 2002;16:513–24.
Connor TJ, Leonard BE. Depression, stress and immunological activation: the role of cytokines in depressive disorders. Life Sci. 1998;62:583–606.
Harbuz MS, Stephanou A, Sarlis N, Lightman SL. The effects of recombinant human interleukin (IL)-1 alpha, IL-1 beta or IL-6 on hypothalamo-pituitary-adrenal axis activation. J Endocrinol. 1992;133:349–55.
Shintani F, Kanba S, Nakaki T, Nibuya M, Kinoshita N, Suzuki E, et al. Interleukin-1 beta augments release of norepinephrine, dopamine, and serotonin in the rat anterior hypothalamus. J Neurosci. 1993;13:3574–81.
Barbanel G, Ixart G, Szafarczyk A, Malaval F, Assenmacher I. Intrahypothalamic infusion of interleukin-1 beta increases the release of corticotropin-releasing hormone (CRH 41) and adrenocorticotropic hormone (ACTH) in free-moving rats bearing a push-pull cannula in the median eminence. Brain Res. 1990;516:31–6.
Kern S, Oakes TR, Stone CK, McAuliff EM, Kirschbaum C, Davidson RJ. Glucose metabolic changes in the prefrontal cortex are associated with HPA axis response to a psychosocial stressor. Psychoneuroendocrinology. 2008;33:517–29.
Liberzon I, King AP, Britton JC, Phan KL, Abelson JL, Taylor SF. Paralimbic and medial prefrontal cortical involvement in neuroendocrine responses to traumatic stimuli. Am J Psychiatry. 2007;164:1250–8.
Urry HL, Van Reekum CM, Johnstone T, Kalin NH, Thurow ME, Schaefer HS, et al. Amygdala and ventromedial prefrontal cortex are inversely coupled during regulation of negative affect and predict the diurnal pattern of cortisol secretion among older adults. J Neurosci. 2006;26:4415–25.
Veer IM, Oei NY, Spinhoven P, van Buchem MA, Elzinga BM, Rombouts SA. Endogenous cortisol is associated with functional connectivity between the amygdala and medial prefrontal cortex. Psychoneuroendocrinology. 2012;37:1039–47.
Cunningham-Bussel AC, Root JC, Butler T, Tuescher O, Pan H, Epstein J, et al. Diurnal cortisol amplitude and fronto-limbic activity in response to stressful stimuli. Psychoneuroendocrinology. 2009;34:694–704.
Garcia RG, Valenza G, Tomaz C, Barbieri R. Relationship between cardiac vagal activity and mood congruent memory bias in major depression. J Affect Disord. 2016;190:19–25.
Holsen LM, Lee JH, Spaeth SB, Ogden LA, Klibanski A, Whitfield-Gabrieli S, et al. Brain hypoactivation, autonomic nervous system dysregulation, and gonadal hormones in depression: a preliminary study. Neurosci Lett. 2012;514:57–61.
Mareckova K, Holsen LM, Admon R, Makris N, Seidman L, Buka S, et al. Brain activity and connectivity in response to negative affective stimuli: Impact of dysphoric mood and sex across diagnoses. Hum Brain Mapp. 2016;37:3733–44.
Jones PB, Tarrant CJ. Specificity of developmental precursors to schizophrenia and affective disorders. Schizophr Res. 1999;39:121–6.
van Os J, Jones P, Lewis G, Wadsworth M, Murray R. Developmental precursors of affective illness in a general population birth cohort. Arch Gen Psychiatry. 1997;54:625–31.
Gilman S, Cherkerzian S,MH, Walch J, Buka S, Goldstein J. Prenatal immune programming of sex-dependent risk for major depression. Transl Psychiatry. 2016;6:e822.
Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host. Nat Rev Immunol. 2006;6:318–28.
Talbot S, Foster SL, Woolf CJ. Neuroimmunity: physiology and pathology. Annu Rev Immunol. 2016;34:421–47.
Barker DJ. Intrauterine programming of adult disease. Mol Med Today. 1995;1:418–23.
Tobet S, Handa R, Goldstein JM. Sex-dependent pathophysiology as predictors of comorbidity of major depressive disorder and cardiovascular disease. Pflugers Arch. 2013;465:585–94.
Frahm KA, Tobet SA. Development of the blood-brain barrier within the paraventricular nucleus of the hypothalamus: influence of fetal glucocorticoid excess. Brain Struct Func. 2015;220:2225–34.
Beauregard M, Paquette V, Levesque J. Dysfunction in the neural circuitry of emotional self-regulation in major depressive disorder. Neuroreport. 2006;17:843–6.
Monroe SM, Harkness KL. Life stress, the “kindling” hypothesis, and the recurrence of depression: considerations from a life stress perspective. Psychol Rev. 2005;112:417–45.
Domes G, Schulze L, Bottger M, Grossmann A, Hauenstein K, Wirtz PH, et al. The neural correlates of sex differences in emotional reactivity and emotion regulation. Hum Brain Mapp. 2010;31:758–69.
Ustun TB, Ayuso-Mateos JL, Chatterji S, Mathers C, Murray CJ. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184:386–92.
Chan JC, Nugent BM, Bale TL. Parental advisory: maternal and paternal stress can impact offspring neurodevelopment. Biol Psychiatry. 2018;83:886–94.
Goldstein J. Impact of prenatal stress on understanding offspring psychopathology and comorbidity with general medicine. Biol Psychiatry. 2018, in press.
Lane RD, Wager TD. The new field of Brain-Body Medicine: what have we learned and where are we headed? Neuroimage. 2009;47:1135–40.
Papez JW. A proposed mechanism of emotion. J Neuropsychiatry Clin Neurosci. 1995;7:103–12.
Makris N, Swaab DF, van der Kouwe A, Abbs B, Boriel D, Handa R, et al. Volumetric parcellation methodology of the human hypothalamus in neuroimaging: normative data and sex differences. Neuroimage. 2012;69:1–10.
McEwen BS. Gonadal steroid influences on brain development and sexual differentiation. In: Greep R, editor. Reproductive physiology IV. 27. University Park: Baltimore; 1983. pp. 99–145.
Tobet S, Knoll JG, Hartshorn C, Aurand E, Stratton M, Kumar P, et al. Brain sex differences and hormone influences: a moving experience? J Neuroendocrinol. 2009;21:387–92.
Giedd JN, Vaituzis AC, Hamburger SD, Lange N, Rajapakse JC, Kaysen D, et al. Quantitative MRI of the temporal lobe, amygdala, and hippocampus in normal human development: ages 4-18 years. J Comp Neurol. 1996;366:223–30.
Goldstein JM, Seidman LJ, Horton NJ, Makris N, Kennedy DN, Caviness VS, et al. Normal sexual dimorphism of the adult human brain assessed by in vivo magnetic resonance imaging. Cereb Cortex. 2001;11:490–7.
Gorski RA. Sexual differentiation of the nervous system. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of neural science. 4th ed. New York: McGraw-Hill Health Professions Division; 2000. pp. 1131–46.
Kluver H, Bucy PC. Preliminary analysis of functions of the temporal lobes in monkeys. J Neuropsychiatry Clin Neurosci. 1997;9:606–20.
Zola-Morgan S, Squire LR, Alvarez-Royo P, Clower RP. Independence of memory functions and emotional behavior: separate contributions of the hippocampal formation and the amygdala. Hippocampus. 1991;1:207–20.
Mac LP. Psychosomatic disease and the visceral brain; recent developments bearing on the Papez theory of emotion. Psychosom Med. 1949;11:338–53.
Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35:192–216.
Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902.
Murray EA, Wise SP, Drevets WC. Localization of dysfunction in major depressive disorder: prefrontal cortex and amygdala. Biol Psychiatry. 2011;69:e43–54.
Jacobs EG, Holsen LM, Lancaster K, Makris N, Whitfield-Gabrieli S, Remington A, et al. 17beta-estradiol differentially regulates stress circuitry activity in healthy and depressed women. Neuropsychopharmacology. 2015;40:566–76.
Lang PJ, Bradley MM, Cuthbert MM. Motivated attention: affect, activation, and action. In: Lang PJ, Simons RF, Balaban MT, editors. Attention and orienting: sensory and motivational processes. Hillsdale, NJ: Erlbaum; 1997. pp. 97–135.
Perlstein W, Elbert T, Stenger VA. Dissociation in human prefrontal cortex of affective influences on working memory-related activity. Proc Natl Acad Sci USA. 2002;99:1736–41.
Taylor SF, Phan KL, Decker LR, Liberzon I. Subjective rating of emotionally salient stimuli modulates neural activity. Neuroimage. 2003;18:650–9.
Hariri AR, Mattay VS, Tessitore A, Fera F, Weinberger DR. Neocortical modulation of the amygdala response to fearful stimuli. Biol Psychiatry. 2003;53:494–501.
Lane RD, Reiman EM, Bradley MM, Lang PJ, Ahern GL, Davidson RJ, et al. Neuroanatomical correlates of pleasant and unpleasant emotion. Neuropsychologia. 1997;35:1437–44.
McManis MH, Bardley MM, Berg WK, Cuthbert BN, Lang PJ. Emotional reactivity in children: Verbal, physiological, and behavioral responses to affective pictures. Psychophysiology. 2001;38:222–31.
Bradley MM, Cogispoti M, Sabatinelli D, Lang PJ. Emotion and motivation II: sex differences in picture processing. Emotion. 2001;1:300–19.
Wrase J, Klein S, Gruesser SM, Hermann D, Flor H, Mann K, et al. Gender differences in the processing of standardized emotional visual stimuli in humans: a functional magnetic resonance imaging study. Neurosci Lett. 2003;348:41–5.
Swaab DF, Chung WC, Kruijver FP, Hofman MA, Hestiantoro A. Sex differences in the hypothalamus in the different stages of human life. Neurobiol Aging. 2003;24 (Suppl 1):S1–19.
Bao AM, Hestiantoro A, Van Someren EJ, Swaab DF, Zhou JN. Colocalization of corticotropin-releasing hormone and oestrogen receptor-alpha in the paraventricular nucleus of the hypothalamus in mood disorders. Brain. 2005;128:1301–13.
van den Pol AN. The magnocellular and parvocellular paraventricular nucleus of rat: intrinsic organization. J Comp Neurol. 1982;206:317–45.
Finley KH. The capillary bed of the paraventricular and supraoptic nuclei of the hypothalamus. Res Publ Assoc Res Nerv Ment Dis. 1937;18:94–109.
Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci. 1983;6:269–324.
Herman JP, Ostrander MM, Mueller NK, Figueiredo H. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1201–13.
McClellan KM, Stratton MS, Tobet SA. Roles for gamma-aminobutyric acid in the development of the paraventricular nucleus of the hypothalamus. J Comp Neurol. 2010;518:2710–28.
Stratton MS, Budefeld T, Majdic G, Tobet S. Embryonic GABA-B receptor blockade alters adult hypothalamic structure and anxiety- and depression-like behaviors in mice [abstract]. In: Society of Neuroscience 41st Annual Meeting. Washington, D.C.; 2011.
Frahm KA, Schow MJ, Tobet SA. The vasculature within the paraventricular nucleus of the hypothalamus in mice varies as a function of development, subnuclear location, and GABA signaling. Horm Metab Res. 2012;44:619–24.
Goncharuk VD, Van Heerikhuize J, Swaab DF, Buijs RM. Paraventricular nucleus of the human hypothalamus in primary hypertension: activation of corticotropin-releasing hormone neurons. J Comp Neurol. 2002;443:321–31.
Mesulam MM. Patterns in behavioral neuroanatomy: association areas, the limbic system, and hemispheric specialization. In: Mesulam MM, editor. Principles of behavioral neurology. Philadelphia: F.A. Davis Company; 1985. pp. 1–58.
Goldstein JM, Jerram M, Poldrack R, Ahern T, Kennedy DN, Seidman LJ, et al. Hormonal cycle modulates arousal circuitry in women using functional magnetic resonance imaging. J Neurosci. 2005;25:9309–16.
Jacobs EG, Holsen LM, Lancaster K, Makris N, Whitfield-Gabrieli S, Remington A, et al. 17β-Estradiol differentially regulates stress circuitry activity in healthy and depressed women. Neuropsychopharmacology. 2015;40:566–76.
Zuloaga DG, Carbone DL, Hiroi R, Chong DL, Handa RJ. Dexamethasone induces apoptosis in the developing rat amygdala in an age-, region-, and sex-specific manner. Neuroscience. 2011;199:535–47.
Schow MJ, Knoll JG, Frahm KA, Zhang Q, Tobet S. Vascular development and sex differences in the region of the paraventricular nucleus of the hypothalamus [abstract]. In: Society of Neuroscience 40th Annual Meeting. San Diego, CA; 2010.
Frahm KA, Schow MJ, Tobet SA. Differences in vasculature branching in GABABR1 knockout mice in the paraventricular nucleus of the hypothalamus [abstract]. In: Society for Neuroscience 40th Annual Meeting. San Diego, CA; 2010.
Kermani P, Hempstead B. Brain-derived neurotrophic factor: a newly described mediator of angiogenesis. Trends Cardiovasc Med. 2007;17:140–3.
Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–47.
Warner-Schmidt JL, Duman RS. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci USA. 2007;104:4647–52.
Cao L, Jiao X, Zuzga DS, Liu Y, Fong DM, Young D, et al. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet. 2004;36:827–35.
Duman RS. Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med. 2004;5:11–25.
Simerly RB, Chang C, Muramatsu M, Swanson LW. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. 1990;294:76–95.
Suzuki S, Handa RJ. Estrogen receptor-beta, but not estrogen receptor-alpha, is expressed in prolactin neurons of the female rat paraventricular and supraoptic nuclei: comparison with other neuropeptides. J Comp Neurol. 2005;484:28–42.
Carbone DL, Handa RJ. Sex and stress hormone influences on the expression and activity of brain-derived neurotrophic factor. Neuroscience. 2013;239:295–303.
Carbone DL, Zuloaga DG, Lacagnina AF, McGivern RF, Handa R. Exposure to dexamethasone during late gestation causes female-specific decreases in core body temperature and prepro-thyrotopin releasing hormone expression in the paraventricular nucleus of the hypothalamus in rats. Physiol Behav. 2012;108:6–12.
Hossain A, Hajman K, Charitidi K, Erhardt S, Zimmermann U, Knipper M, et al. Prenatal dexamethasone impairs behavior and the activation of the BDNF exon IV promoter in the paraventricular nucleus in adult offspring. Endocrinology. 2008;149:6356–65.
Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–54.
Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. 2006;147:2909–15.
Ambach G, Palkovits M. Blood supply of the rat hypothalamus. II. Nucleus paraventricularis. Acta Morphol Acad Sci Hung. 1974;22:311.
Menendez A, Alvarez-Uria M. The development of vascularization in the postnatal rat paraventricular nucleus: a morphometric analysis. J Hirnforsch. 1987;28:325–9.
Zacchigna S, Lambrechts D, Carmeliet P. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9:169–81.
Frahm KA, Handa RJ, Tobet SA. Embryonic exposure to dexamethasone affects nonneuronal cells in the adult paraventricular nucleus of the hypothalamus. J Endocr Soc. 2018;2:140–53.
Petropoulos S, Gibb W, Matthews SG. Developmental expression of multidrug resistance phosphoglycoprotein (P-gp) in the mouse fetal brain and glucocorticoid regulation. Brain Res. 2010;1357:9–18.
Wilson AC, Clemente L, Liu T, Bowen RL, Meethal SV, Atwood CS. Reproductive hormones regulate the selective permeability of the blood-brain barrier. Biochim Biophys Acta. 2008;1782:401–7.
Schulz KM, Sisk CL. The organizing actions of adolescent gonadal steroid hormones on brain and behavioral development. Neurosci Biobehav Rev. 2016;70:148–58.
Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. 2013;339:1095–9.
Bilbo SD, Block CL, Bolton JL, Hanamsagar R, Tran PK. Beyond infection - Maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp Neurol. 2018;299:241–51.
Bittle J, Stevens HE. The role of glucocorticoid, interleukin-1β, and antioxidants in prenatal stress effects on embryonic microglia. J Neuroinflammation. 2018;15:44.
Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating angiotensin II gains access to the hypothalamus and brainstem during hypertension via breakdown of the blood brain barrier. Hypertension. 2014;63:572–9.
Oquendo MA, Echavarria G, Galfalvy HC, Grunebaum MF, Burke A, Barrera A, et al. Lower cortisol levels in depressed patients with comorbid post-traumatic stress disorder. Neuropsychopharmacology. 2003;28:591–8.
Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160:1554–65.
Rubin RT, Poland RE, Lesser IM, Winston RA, Blodgett AL. Neuroendocrine aspects of primary endogenous depression. I. Cortisol secretory dynamics in patients and matched controls. Arch Gen Psychiatry. 1987;44:328–36.
Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. 2001;49:1023–39.
Barden N. Implication of the hypothalamic-pituitary-adrenal axis in the physiopathology of depression. J Psychiatry Neurosci. 2004;29:185–93.
Heim C, Newport DJ, Wagner D, Wilcox MM, Miller AH, Nemeroff CB. The role of early adverse experience and adulthood stress in the prediction of neuroendocrine stress reactivity in women: a multiple regression analysis. Depress Anxiety. 2002;15:117–25.
Meynen G, Unmehopa UA, Heerikhuize JJ, Hofman MA, Swaab DF, Hoogendijk WJ. Increased arginine vasopressin mrna expression in the human hypothalamus in depression: a preliminary report. Biol Psychiatry. 2006;60:892–5.
van Amelsvoort TAMJ, Abel KM, Robertson DMR, Daly E, Critchley H, Whitehead M, et al. Prolactin response to d-fenfluramine in postmenopausal women on and off ERT: comparison with young women. Psychoneuroendocrinology. 2001;26:493–502.
Webster MJ, Knable MB, O’Grady J, Orthmann J, Weickert CS. Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol Psychiatry. 2002;7:985–94. 924
Cottrell EC, Seckl JR, Holmes MC, Wyrwoll CS. Foetal and placental 11beta-HSD2: a hub for developmental programming. Acta Physiol (Oxf). 2014;210:288–95.
Holmes MC, Wyrwoll C, Seckl J. Fetal programming of adult behaviour by stress and glucocorticoids. Psychoneuroendocrinology. 2015;61:9.
Maccari S, Darnaudery M, Morley-Fletcher S, Zuena AR, Cinque C, Van Reeth O. Prenatal stress and long-term consequences: implications of glucocorticoid hormones. Neurosci Biobehav Rev. 2003;27:119–27.
Takahashi LK, Kalin NH. Early developmental and temporal characteristics of stress-induced secretion of pituitary-adrenal hormones in prenatally stressed rat pups. Brain Res. 1991;558:75–8.
Henry C, Kabbaj M, Simon H, Le Moal M, Maccari S. Prenatal stress increases the hypothalamo-pituitary-adrenal axis response in young and adult rats. J Neuroendocrinol. 1994;6:341–5.
Shoener JA, Baig R, Page KC. Prenatal exposure to dexamethasone alters hippocampal drive on hypothalamic-pituitary-adrenal axis activity in adult male rats. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1366–1373.
Bhatnagar S, Lee TM, Vining C. Prenatal stress differentially affects habituation of corticosterone responses to repeated stress in adult male and female rats. Horm Behav. 2005;47:430–8.
McCormick CM, Smythe JW, Sharma S, Meaney MJ. Sex-specific effects of prenatal stress on hypothalamic-pituitary-adrenal responses to stress and brain glucocorticoid receptor density in adult rats. Brain Res Dev Brain Res. 1995;84:55–61.
Richardson HN, Zorrilla EP, Mandyam CD, Rivier CL. Exposure to repetitive versus varied stress during prenatal development generates two distinct anxiogenic and neuroendocrine profiles in adulthood. Endocrinology. 2006;147:2506–17.
Tobe I, Ishida Y, Tanaka M, Endoh H, Fujioka T, Nakamura S. Effects of repeated maternal stress on FOS expression in the hypothalamic paraventricular nucleus of fetal rats. Neuroscience. 2005;134:387–95.
Hiroi R, Carbone DL, Zuloaga DG, Bimonte-Nelson HA, Handa RJ. Sex-dependent programming effects of prenatal glucocorticoid treatment on the developing serotonin system and stress-related behaviors in adulthood. Neuroscience. 2016;320:43–56.
Kamphuis PJ, Bakker JM, Broekhoven MH, Kunne C, Croiset G, Lentjes EG, et al. Enhanced glucocorticoid feedback inhibition of hypothalamo-pituitary-adrenal responses to stress in adult rats neonatally treated with dexamethasone. Neuroendocrinology. 2002;76:158–69.
Brunton PJ, Donadio MV, Yao ST, Greenwood M, Seckl JR, Murphy D, et al. 5alpha-Reduced neurosteroids sex-dependently reverse central prenatal programming of neuroendocrine stress responses in rats. J Neurosci. 2015;35:666–77.
Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34.
Kohler CA, Freitas TH, Stubbs B, Maes M, Solmi M, Veronese N, et al. Peripheral alterations in cytokine and chemokine levels after antidepressant drug treatment for major depressive disorder: systematic review and meta-analysis. Mol Neurobiol. 2017;55:4195–206.
Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204:313–21.
Lin YL, Wang S. Prenatal lipopolysaccharide exposure increases depression-like behaviors and reduces hippocampal neurogenesis in adult rats. Behav Brain Res. 2014;259:24–34.
Enayati M, Solati J, Hosseini MH, Shahi HR, Saki G, Salari AA. Maternal infection during late pregnancy increases anxiety- and depression-like behaviors with increasing age in male offspring. Brain Res Bull. 2012;87:295–302.
Giovanoli S, Notter T, Richetto J, Labouesse MA, Vuillermot S, Riva MA, et al. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J Neuroinflamm. 2015;12:221.
Hanamsagar R, Bilbo SD. Sex differences in neurodevelopmental and neurodegenerative disorders: Focus on microglial function and neuroinflammation during development. J Steroid Biochem Mol Biol. 2016;160:127–33.
Gilman SE, Hornig M, Ghassabian A, Hahn J, Cherkerzian S, Albert PS, et al. Socioeconomic disadvantage, gestational immune activity, and neurodevelopment in early childhood. Proc Natl Acad Sci USA. 2017;114:6728–33.
Majidi-Zolbanin J, Doosti MH, Kosari-Nasab M, Salari AA. Prenatal maternal immune activation increases anxiety- and depressive-like behaviors in offspring with experimental autoimmune encephalomyelitis. Neuroscience. 2015;294:69–81.
Zuloaga DG, Carbone DL, Quihuis A, Hiroi R, Chong DL, Handa RJ. Perinatal dexamethasone-induced alterations in apoptosis within the hippocampus and paraventricular nucleus of the hypothalamus are influenced by age and sex. J Neurosci Res. 2012;90:1403–12.
Handa RJ, Burgess LH, Kerr JE, O’Keefe JA. Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Horm Behav. 1994;28:464–76.
Berk M, Williams LJ, Jacka FN, O’Neil A, Pasco JA, Moylan S, et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013;11:200.
Messay B, Lim A, Marsland AL. Current understanding of the bi-directional relationship of major depression with inflammation. Biol Mood Anxiety Disord. 2012;2:4.
Müller N. Immunology of major depression. Neuroimmunomodulation. 2013;21:123–30.
Rosenblat JD, Cha DS, Mansur RB, McIntyre RS. Inflamed moods: a review of the interactions between inflammation and mood disorders. Prog Neuro-Psychopharmacol Biol Psychiatry. 2014;53:23–34.
Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71:171–86.
Valkanova V, Ebmeier KP, Allan CL. CRP, IL-6 and depression: a systematic review and meta-analysis of longitudinal studies. J Affect Disord. 2013;150:736–44.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57.
Wright JL, Merchant RE. Blood-brain barrier changes following intracerebral injection of human recombinant tumor necrosis factor-alpha in the rat. J Neurooncol. 1994;20:17–25.
Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8.
Pinto VLM, Brunini T, Ferraz MR, Okinga A, Mendes-Ribeiro AC. Depression and cardiovascular disease: role of nitric oxide. Cardiovasc Hematol Agents Med Chem. 2008;6:142–9.
Pizzi C, Santarella L, Costa M, Manfrini O, Flacco M, Capasso L, et al. Pathophysiological mechanisms linking depression and atherosclerosis: an overview. J Biol Regul Homeost Agents. 2011;26:775–82.
Rawdin B, Mellon S, Dhabhar F, Epel E, Puterman E, Su Y, et al. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav Immun. 2013;31:143–52.
VanRyzin JW, Pickett LA, McCarthy MM. Microglia: driving critical periods and sexual differentiation of the brain. Dev Neurobiol. 2017;78:580–92.
Hanamsagar R, Alter MD, Block CS, Sullivan H, Bolton JL, Bilbo SD. Generation of a microglial developmental index in mice and in humans reveals a sex difference in maturation and immune reactivity. Glia. 2017;65:1504–20.
Sorge RE, Mapplebeck JCS, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18:1081–3.
Bale TL. The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialogues Clin Neurosci. 2016;18:459–64.
Jasarevic E, Howard CD, Misic AM, Beiting DP, Bale TL. Stress during pregnancy alters temporal and spatial dynamics of the maternal and offspring microbiome in a sex-specific manner. Sci Rep. 2017;7:44182.
Gur TL, Shay L, Palkar AV, Fisher S, Varaljay VA, Dowd S, et al. Prenatal stress affects placental cytokines and neurotrophins, commensal microbes, and anxiety-like behavior in adult female offspring. Brain Behav Immun. 2017;64:50–8.
Rose DR, Careaga M, Van de Water J, McAllister K, Bauman MD, Ashwood P. Long-term altered immune responses following fetal priming in a non-human primate model of maternal immune activation. Brain Behav Immun. 2017;63:60–70.
Veru F, Laplante DP, Luheshi G, King S. Prenatal maternal stress exposure and immune function in the offspring. Stress. 2014;17:133–48.
Douros K, Moustaki M, Tsabouri S, Papadopoulou A, Papadopoulos M, Priftis KN. Prenatal maternal stress and the risk of asthma in children. Front Pediatr. 2017;5:202.
O’Regan D, Kenyon CJ, Seckl JR, Holmes MC. Prenatal dexamethasone ‘programmes’ hypotension, but stress-induced hypertension in adult offspring. J Endocrinol. 2008;196:343–52.
Hadoke PW, Lindsay RS, Seckl JR, Walker BR, Kenyon CJ. Altered vascular contractility in adult female rats with hypertension programmed by prenatal glucocorticoid exposure. J Endocrinol. 2006;188:435–42.
Igosheva N, Taylor PD, Poston L, Glover V. Prenatal stress in the rat results in increased blood pressure responsiveness to stress and enhanced arterial reactivity to neuropeptide Y in adulthood. J Physiol. 2007;582:665–74.
Moritz KM, Dodic M, Jefferies AJ, Wintour EM, DeMatteo R, Singh RR, et al. Haemodynamic characteristics of hypertension induced by prenatal cortisol exposure in sheep. Clin Exp Pharmacol Physiol. 2009;36:981–7.
Rogers JM, Ellis-Hutchings RG, Grey BE, Zucker RM, Norwood J Jr., Grace CE, et al. Elevated blood pressure in offspring of rats exposed to diverse chemicals during pregnancy. Toxicol Sci. 2014;137:436–46.
Gwathmey TM, Shaltout HA, Rose JC, Diz DI, Chappell MC. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension. 2011;57:620–6.
O’Sullivan L, Cuffe JS, Koning A, Singh RR, Paravicini TM, Moritz KM. Excess prenatal corticosterone exposure results in albuminuria, sex-specific hypotension, and altered heart rate responses to restraint stress in aged adult mice. Am J Physiol Renal Physiol. 2015;308:F1065–1073.
Tang JI, Kenyon CJ, Seckl JR, Nyirenda MJ. Prenatal overexposure to glucocorticoids programs renal 11beta-hydroxysteroid dehydrogenase type 2 expression and salt-sensitive hypertension in the rat. J Hypertens. 2011;29:282–9.
Igosheva N, Klimova O, Anishchenko T, Glover V. Prenatal stress alters cardiovascular responses in adult rats. J Physiol. 2004;557:273–85.
Kiguti LRA, Borges CS, Mueller A, Silva KP, Polo CM, Rosa JL, et al. Gender-specific impairment of in vitro sinoatrial node chronotropic responses and of myocardial ischemia tolerance in rats exposed prenatally to betamethasone. Toxicol Appl Pharmacol. 2017;334:66–74.
Lee JH, Zhang J, Flores L, Rose JC, Massmann GA, Figueroa JP. Antenatal betamethasone has a sex-dependent effect on the in vivo response to endothelin in adult sheep. Am J Physiol Regul Integr Comp Physiol. 2013;304:R581–587.
Shaltout HA, Rose JC, Chappell MC, Diz DI. Angiotensin-(1-7) deficiency and baroreflex impairment precede the antenatal Betamethasone exposure-induced elevation in blood pressure. Hypertension. 2012;59:453–8.
Arya V, Demarco VG, Issar M, Hochhaus G. Contrary to adult, neonatal rats show pronounced brain uptake of corticosteroids. Drug Metab Dispos. 2006;34:939–42.
O’Regan D, Kenyon CJ, Seckl JR, Holmes MC. Glucocorticoid exposure in late gestation in the rat permanently programs gender-specific differences in adult cardiovascular and metabolic physiology. Am J Physiol Endocrinol Metab. 2004;287:E863–870.
Carbone DL, Zuloaga DG, Hiroi R, Foradori CD, Legare ME, Handa RJ. Prenatal dexamethasone exposure potentiates diet-induced hepatosteatosis and decreases plasma IGF-I in a sex-specific fashion. Endocrinology. 2012;153:295–306.
Samuelsson AM, Ohrn I, Dahlgren J, Eriksson E, Angelin B, Folkow B, et al. Prenatal exposure to interleukin-6 results in hypertension and increased hypothalamic-pituitary-adrenal axis activity in adult rats. Endocrinology. 2004;145:4897–911.
Wei Y, Du W, Xiong X, He X, Ping Y, Deng Y, et al. Prenatal exposure to lipopolysaccharide results in myocardial remodelling in adult murine offspring. J Inflamm (Lond). 2013;10:35.
Chen X, Tang Y, Gao M, Qin S, Zhou J, Li X. Prenatal exposure to lipopolysaccharide results in myocardial fibrosis in rat offspring. Int J Mol Sci. 2015;16:10986–96.
Zhao S, Zhang H, Cao D, Liu Y, Li X. Lipopolysaccharide exposure during pregnancy leads to aortic dysfunction in offspring rats. PLoS ONE. 2014;9:e102273.
Loria AS, Pollock DM, Pollock JS. Early life stress sensitizes rats to angiotensin II-induced hypertension and vascular inflammation in adult life. Hypertension. 2010;55:494–9.
Cuffe JS, Burgess DJ, O’Sullivan L, Singh RR, Moritz KM. Maternal corticosterone exposure in the mouse programs sex-specific renal adaptations in the renin-angiotensin-aldosterone system in 6-month offspring. Physiol Rep. 2016;4(8):e12754. https://doi.org/10.14814/phy2.12754.
Dagan A, Gattineni J, Habib S, Baum M. Effect of prenatal dexamethasone on postnatal serum and urinary angiotensin II levels. Am J Hypertens. 2010;23:420–4.
Shaltout HA, Rose JC, Figueroa JP, Chappell MC, Diz DI, Averill DB. Acute AT(1)-receptor blockade reverses the hemodynamic and baroreflex impairment in adult sheep exposed to antenatal betamethasone. Am J Physiol Heart Circ Physiol. 2010;299:H541–7.
Bogdarina I, Haase A, Langley-Evans S, Clark AJ. Glucocorticoid effects on the programming of AT1b angiotensin receptor gene methylation and expression in the rat. PLoS ONE. 2010;5:e9237.
Dodic M, McAlinden AT, Jefferies AJ, Wintour EM, Cock ML, May CN, et al. Differential effects of prenatal exposure to dexamethasone or cortisol on circulatory control mechanisms mediated by angiotensin II in the central nervous system of adult sheep. J Physiol. 2006;571:651–60.
Marshall AC, Shaltout HA, Nautiyal M, Rose JC, Chappell MC, Diz DI. Fetal betamethasone exposure attenuates angiotensin-(1-7)-Mas receptor expression in the dorsal medulla of adult sheep. Peptides. 2013;44:25–31.
Guo DF, Uno S, Ishihata A, Nakamura N, Inagami T. Identification of a cis-acting glucocorticoid responsive element in the rat angiotensin II type 1A promoter. Circ Res. 1995;77:249–57.
Cottrell EC, Seckl JR. Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci. 2009;3:19.
Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Horm Behav. 2011;59:279–89.
Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest. 1998;101:2174–81.
Dumortier O, Theys N, Ahn MT, Remacle C, Reusens B. Impairment of rat fetal beta-cell development by maternal exposure to dexamethasone during different time-windows. PLoS ONE. 2011;6:e25576.
Drake AJ, Raubenheimer PJ, Kerrigan D, McInnes KJ, Seckl JR, Walker BR. Prenatal dexamethasone programs expression of genes in liver and adipose tissue and increased hepatic lipid accumulation but not obesity on a high-fat diet. Endocrinology. 2010;151:1581–7.
Cleasby ME, Kelly PA, Walker BR, Seckl JR. Programming of rat muscle and fat metabolism by in utero overexposure to glucocorticoids. Endocrinology. 2003;144:999–1007.
Jorns A, Sennholz C, Naujok O, Lenzen S. Beta cell mass regulation in the rat pancreas through glucocorticoids and thyroid hormones. Pancreas. 2010;39:1167–72.
Blondeau B, Lesage J, Czernichow P, Dupouy JP, Breant B. Glucocorticoids impair fetal beta-cell development in rats. Am J Physiol Endocrinol Metab. 2001;281:E592–9.
Garofano A, Czernichow P, Breant B. Beta-cell mass and proliferation following late fetal and early postnatal malnutrition in the rat. Diabetologia. 1998;41:1114–20.
Gesina E, Tronche F, Herrera P, Duchene B, Tales W, Czernichow P, et al. Dissecting the role of glucocorticoids on pancreas development. Diabetes. 2004;53:2322–9.
Rojas J, Chavez-Castillo M, Chavez-Castillo M, Cabrera M, Cabrera M, Bermudez V, et al. Glucococorticoid-induced death of pancreatic Beta cells: an organized chaos. JOP. 2015;16:11–9.
Matthews LC, Hanley NA. The stress of starvation: glucocorticoid restraint of beta cell development. Diabetologia. 2011;54:223–6.
Shen CN, Seckl JR, Slack JM, Tosh D. Glucocorticoids suppress beta-cell development and induce hepatic metaplasia in embryonic pancreas. Biochem J. 2003;375:41–50.
Miranda RA, Torrezan R, de Oliveira JC, Barella LF, da Silva Franco CC, Lisboa PC, et al. HPA axis and vagus nervous function are involved in impaired insulin secretion of MSG-obese rats. J Endocrinol. 2016;230:27–38.
Prates KV, de Oliveira JC, Malta A, Matiusso CCI, Miranda RA, Ribeiro TA, et al. Sympathetic innervation is essential for metabolic homeostasis and pancreatic beta cell function in adult rats. Mol Cell Endocrinol. 2018;462:119–26.
Croizier S, Prevot V, Bouret SG. Leptin controls parasympathetic wiring of the pancreas during embryonic life. Cell Rep. 2016;15:36–44.
Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, et al. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J Clin Invest. 2004;113:96–105.
Aguirre GA, De Ita JR, de la Garza RG, Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J Transl Med. 2016;14:3.
Boucher J, Tseng YH, Kahn CR. Insulin and insulin-like growth factor-1 receptors act as ligand-specific amplitude modulators of a common pathway regulating gene transcription. J Biol Chem. 2010;285:17235–45.
Dunger D, Yuen K, Ong K. Insulin-like growth factor I and impaired glucose tolerance. Horm Res. 2004;62 (Suppl 1):101–7.
van Haeften TW, Twickler TB. Insulin-like growth factors and pancreas beta cells. Eur J Clin Invest. 2004;34:249–55.
Laron Z. Insulin--a growth hormone. Arch Physiol Biochem. 2008;114:11–6.
Di Cola G, Cool MH, Accili D. Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J Clin Invest. 1997;99:2538–44.
Nugent BM, Bale TL. The omniscient placenta: metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol. 2015;39:28–37.
Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci USA. 2013;110:5169–74.
Howerton CL, Bale TL. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc Natl Acad Sci USA. 2014;111:9639–44.
Bouret SG. Early life origins of obesity: role of hypothalamic programming. J Pediatr Gastroenterol Nutr. 2009;48 (Suppl 1):S31–8.
Smith JT, Waddell BJ. Developmental changes in plasma leptin and hypothalamic leptin receptor expression in the rat: peripubertal changes and the emergence of sex differences. J Endocrinol. 2003;176:313–9.
Yura S, Itoh H, Sagawa N, Yamamoto H, Masuzaki H, Nakao K, et al. Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab. 2005;1:371–8.
Attig L, Solomon G, Ferezou J, Abdennebi-Najar L, Taouis M, Gertler A, et al. Early postnatal leptin blockage leads to a long-term leptin resistance and susceptibility to diet-induced obesity in rats. Int J Obes (Lond). 2008;32:1153–60.
Bouret SG, Draper SJ, Simerly RB. Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J Neurosci. 2004;24:2797–805.
Shin BC, Dai Y, Thamotharan M, Gibson LC, Devaskar SU. Pre- and postnatal calorie restriction perturbs early hypothalamic neuropeptide and energy balance. J Neurosci Res. 2012;90:1169–82.
Mela V, Diaz F, Lopez-Rodriguez AB, Vazquez MJ, Gertler A, Argente J, et al. Blockage of the neonatal leptin surge affects the gene expression of growth factors, glial proteins, and neuropeptides involved in the control of metabolism and reproduction in peripubertal male and female rats. Endocrinology. 2015;156:2571–81.
Lok A, Mocking RJ, Ruhe HG, Visser I, Koeter MW, Assies J, et al. Longitudinal hypothalamic-pituitary-adrenal axis trait and state effects in recurrent depression. Psychoneuroendocrinology. 2012;37:892–902.
Goldstein JM, Holsen L, Huang G, Hammond BD, James-Todd T, Cherkerzian S, et al. Prenatal stress-immune programming of sex differences in comorbidity of depression and obesity/metabolic syndrome. Dialogues Clin Neurosci. 2016;18:425–36.
Acknowledgements
The authors would like to thank Michelle Shafer, Jessica Stowell, and Sara Cherkerzian, Sc.D. for their help in preparing this review for publication. The work for this manuscript was, in part, supported by translational program projects from the Arizona Biomedical Research Commission ADHS14-00003606 (2014–2017; Handa, PI) and OWRH-NIMH P50 MH082679 (2007–2013; Goldstein, Tobet, PIs).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Goldstein, J.M., Hale, T., Foster, S.L. et al. Sex differences in major depression and comorbidity of cardiometabolic disorders: impact of prenatal stress and immune exposures. Neuropsychopharmacol 44, 59–70 (2019). https://doi.org/10.1038/s41386-018-0146-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0146-1
This article is cited by
-
Sex- and suicide-specific alterations in the kynurenine pathway in the anterior cingulate cortex in major depression
Neuropsychopharmacology (2024)
-
A sex-specific genome-wide association study of depression phenotypes in UK Biobank
Molecular Psychiatry (2023)
-
Impacts of gender and lifestyle on the association between depressive symptoms and cardiovascular disease risk in the UK Biobank
Scientific Reports (2023)
-
Depression, aging, and immunity: implications for COVID-19 vaccine immunogenicity
Immunity & Ageing (2022)
-
Evaluation of inflammatory and metabolic impairments regarding depression dimensions: a case control study
Middle East Current Psychiatry (2022)
