
Abstract
To discover effective drugs for COVID-19 treatment amongst already clinically approved drugs, we developed a high throughput screening assay for SARS-CoV-2 virus entry inhibitors using SARS2-S pseudotyped virus. An approved drug library of 1800 small molecular drugs was screened for SARS2 entry inhibitors and 15 active drugs were identified as specific SARS2-S pseudovirus entry inhibitors. Antiviral tests using native SARS-CoV-2 virus in Vero E6 cells confirmed that 7 of these drugs (clemastine, amiodarone, trimeprazine, bosutinib, toremifene, flupenthixol, and azelastine) significantly inhibited SARS2 replication, reducing supernatant viral RNA load with a promising level of activity. Three of the drugs were classified as histamine receptor antagonists with clemastine showing the strongest anti-SARS2 activity (EC50 = 0.95 ± 0.83 µM). Our work suggests that these 7 drugs could enter into further in vivo studies and clinical investigations for COVID-19 treatment.
Similar content being viewed by others


Introduction
Since the outbreak of COVID-19, there have been ~15 million confirmed cases worldwide, and this number continues to grow [1]. Whole-genome sequencing of samples from patients with COVID-19 pneumonia resulted in the identification of a beta-coronavirus that had never been seen before that was different from but related to SARS-CoV and MERS-CoV [2]. There are multiple sequence segments in the receptor-binding domain of the S protein of SARS-CoV-2 that have a high degree of homology with SARS-CoV [2, 3]. Moreover, some studies have confirmed that SARS-CoV-2 can bind to the ACE2 receptor on the cell surface, as does SARS-CoV, which then mediates virus infection [4,5,6]. Therefore, inhibitors targeting the S protein of SARS-CoV-2 may become potential drugs for the treatment of COVID-19 by preventing viral entry.
Given the urgent need for specific antiviral drugs, drug repurposing is an effective strategy for identifying available drugs and promising molecular leads for novel drug development, which can shorten the time and reduce the costs compared with de novo drug discovery [7]. Through this method, some FDA-approved drugs have already been tested in clinical trials, including arbidol, favipiravir, and hydroxychloroquie [8,9,10,11], both domestically and internationally. However, most of these drugs have made a limited contribution to the control of the pandemic [12]. It is widely accepted that SARS-CoV-2 is likely to persist as a pandemic for a longer period than anticipated 3 months ago. Therefore, there will remain a need to develop novel antiviral drugs with high activity and specificity that will be of critical benefit for our long-term control of this emerging disease. Several viral proteins have been suggested as drug targets and are currently being investigated, such as the 3CL protease, which had been selected as a target for both SARS-CoV and SARS-CoV-2 virus [13, 14].
In the work described in this article, we adopted a method for screening a unique set of approved drugs with a clear known targeted cellular pathway that could possibly be effective against SARS-CoV-2 infection in vitro. We found that seven of the drugs screened in vitro were able to effectively inhibit SARS-CoV-2 infection via blockade of the viral entry step. Of these, antihistamine drugs might be of benefit to COVID-19 patients because of their anti-SARS2 activity and additional anti-inflammatory effects. We suggest that further in vivo studies and clinical investigations should be undertaken for the seven anti-SARS2 drugs we report here.
Materials and methods
Cell lines
Huh-7 cells were obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Vero E6 and 293 T cell lines were obtained from ATCC (Manassas, VA, USA). All cell lines were grown in Dulbecco’s modified Eagle medium (DMEM, Thermo Fisher, Shanghai, China) supplemented with 10% fetal bovine serum (FBS) as described previously [15, 16].
Plasmid and pseudovirus production
To generate expression plasmids for the SARS-CoV and SARS-CoV-2 spike proteins (SARS-S, SARS2-S), the coding sequence of each protein was obtained from the published viral genome (GenBank: AY278491 for SARS-CoV, GenBank: MN908947 for SARS-CoV-2) and optimized for mammalian expression. An optimized coding sequence was chemically synthesized (Inovogen, Chengdu,China) and cloned into the pcDNA3.1 vector (Thermo Fisher, Shanghai, China), which was verified by sequencing.
To generate a MERS spike protein (MERS-S) expression plasmid, the codon-optimized gene of the full-length S protein of MERS-CoV (GenBank: AFS88936) with replacement of the N-terminal signal peptide (aa 1–17) with the CD5 signal sequence [17] was synthesized (Inovogen, Chengdu,China) and inserted into the pcDNA3.1 vector, and it was also verified by sequencing.
To produce viral spike protein-pseudotyped HIV virions, the HIV backbone vector pNL4-3. Luc. R-E- was used for pseudovirus packaging. 293 T cells were cotransfected with the respective envelope protein expression plasmid plus the pNL4.3LucR-E- plasmid using X-tremeGENE DNA HP Transfection Reagent (Roche) according to the manufacturer’s instructions. At 72 h post-transfection, supernatants were harvested and filtered through 0.45 μm filters, after which the filtered supernatant was further concentrated 10-fold by ultracentrifugation with a filter cartridge with a 100 kDa cutoff, aliquoted, and finally stored at −80 °C as a stock. The p24 level of the pseudovirus stock solution was determined using an ELISA kit according to the manufacturer´s manual (ab218268, Abcam).
Approved drug library and compound plate preparation
A library of 1800 approved drugs was obtained from Target Molecule Corp. The compounds collected in this library are drugs approved by the Food and Drug Administration (FDA), the European Medicines Agency, or the China Food and Drug Administration or are included in the US Pharmacopeia Dictionary, the British Pharmacopoeia, the European Pharmacopoeia, the Japanese Pharmacopoeia, or the Chinese Pharmacopoeia Dictionary. All compounds were predissolved in DMSO at 10 mM in a 96-well plate format with a 2D barcode.
For pseudovirus-based assay screening, compound plates were prepared using an automated liquid handling robotics platform (Tecan Freedom Evo). Briefly, to prepare the assay plates, 5 µL of stock DMSO solution from the compound library was added into 96-well round bottom plates containing 245 µL DMEM and thoroughly mixed. Each plate contained 8 wells without any compound in the first column as a control. These compound plates were freshly prepared on the day that the assay was performed.
Pseudovirus-based SARS-CoV-2 antiviral assay
For high throughput screening, Huh-7 cells were preseeded in 96-well white and transparent plates one day before the assay was performed and incubated overnight. The approved drugs in 96-well assay plate format were freshly prepared as described above. To set up the antiviral assay, 50 µL of compound working solution from each well of the compound plate was transferred into a white and a transparent cell culture plate. A 50 µL volume of culture medium per well was added to the clear plates (for the MTT assay) and cultured for 2 days for cell viability assessment. The white plates were first subjected to 30 min of preincubation; then, 50 µL pseudovirus (100 ng p24/mL) was added to each well, and the plates were further incubated for 2 days. The final concentration of each of the compounds in the HTS screening was 20 µM. Cell viability was assayed by the MTT method. For luciferase reporter assays, cells were lysed in 1× Glo Lysis Buffer (Promega). The luciferase activity was assessed by a bioluminometer using the Bright-Glo luciferase assay system (Promega) according to the manufacturer’s instructions. Viability and infectivity were calculated as percentages of the control value without any added compound.
To determine the CC50 and EC50, the compounds were serially diluted and assayed as described for the HTS screening, and the CC50 and EC50 were calculated using 4-parameter regression.
Antiviral activity assay using native SARS-CoV-2 virus
SARS-CoV-2 (WIV04) [18] was passaged in Vero E6 cells and titered by a plaque assay. Vero E6 cells were treated with drugs at the indicated concentrations and exposed to SARS-CoV-2 virus at an MOI of 0.01. Cells cocultured with virus were maintained in DMEM with 2% FBS, and the supernatants were collected at 24 h post infection. Magnetic Bead Virus RNA Extraction Kits (Shanghai Fine Gene Biotech, FG438) were used for viral RNA extraction. Viral RNA was quantified by real-time RT-PCR with TaqMan probes targeting the RBD2 gene.
Data analysis
For the primary SARS2-S-HIV HTS assays, the data for the 1800 approved drugs were pooled and analyzed. Cell viability and pseudovirus infectivity were calculated as percentages versus those of the control wells without drug.
Results
Discovery of anti-SARS2 entry inhibitors among candidates in the approved drug library
A collection of 1800 drugs (approved for their respective indications in or before 2018) with defined molecular targets was used as the starting point for assessing anti-SARS2 activity targeting S protein function for drug repurposing to combat the 2019-nCoV pandemic. The workflow chart of HTS screening is shown in Fig. 1.

A library of 1800 approved drugs was screened to determine the effects on blocking SARS2-S pseudovirus infection activity (@20 µM). A set of 154 drugs was selected as the primary hits and further screened for confirmation and to exclude nonspecific entry inhibition activity. Fifteen drugs were selected as specific SARS-CoV-2 entry inhibitor candidates. Experiments using a native clinical SARS-CoV-2 strain confirmed that these seven drugs were active viral entry inhibitors against SARS-CoV-2 infection in vitro.
Cell viability and pseudovirus infection were calculated as percentages of the values of the control without any drug, and the results of the primary HTS screening are shown in Fig. 2. Most drugs plotted in the upper right area were nontoxic and not active. We set different criteria for defining a primary hit. First, drugs that allowed a retention of viability ≥50% and a viability/infectivity ratio ≥5 were selected as having high activity but low toxicity (71 compounds). A second group was then chosen that resulted in a viability between 20% and 50%, while the viability/infectivity ratio was greater than 10 (40 compounds). These drugs were categorized as showing good activity with moderate toxicity. Finally, drugs that caused cell viability to decrease to less than 20% but with a viability/infectivity ratio greater than 20 were also selected because they showed very high activity as well as high toxicity (43 compounds). According to these criteria, primary screening identified 154 drugs that actively inhibited SARS2-S protein-mediated pseudovirus infection, and the selected primary hit compounds are shown in different colors in Fig. 1. These 154 drugs were all confirmed to not have any anti-HIV activity (used as a reporter to exclude drugs with off-target effects due to interfering with postentry activity).

Data from the primary screening experiments using the library of 1800 approved drugs were plotted as the cell viability (%) vs SARS2-S pseudovirus infectivity (%) in Huh-7 cells. Cell viability was calculated as a percentage of the OD@570 readout by the MTT method for each well against that of the control well with no drug. Infectivity was calculated in the same way using a luciferase activity readout. Compounds that met different primary criteria were plotted in color (Red: 20% ≥viability, viability/infectivity ≥20; Blue: 50% ≥viability ≥20%, viability/infectivity ≥10; Green: viability ≥50%, viability/infectivity ≥5).
In the second screening, the activity of the active drugs was verified at 20 µM, and the VSV-G-pseudotyped virus infection assay was carried out in parallel to identify false positive drugs, the activity of which was due to their influence on post internalization and fusion processes rather than viral entry. Drugs with a ratio of VSV-G infectivity to SARS2-S infectivity greater than 2 were selected and subjected to a third round of testing.
In the third round assay, the drugs were serially diluted at 7 doses to determine the CC50 values in Huh-7 cells, and the EC50 values for both pseudoviruses (SARS2-S and VSV-G) were also determined. After the third round assay, 15 drugs remained that were shown to have selective activity against SARS2-S infection with little interference in VSV-G-mediated infection (Table 1). Information on these 15 drugs, including structures, currently approved indications and clinical dosages, is listed in Supplementary Table S1. The results showed that all 15 drugs had significantly higher EC50 values in the VSV-G pseudovirus assays, indicating that these approved drugs may be able to specifically interfere with SARS-CoV-2 viral spike protein function. The selective index (SI, CC50/EC50) of each of these drugs was above 10, but all showed considerable toxicity relative to the activity window.
Approved drugs repurposed for SARS-CoV-2 treatment as entry inhibitors
Thus, we identified a collection of 15 approved drugs that could interfere with SARS-CoV-2 viral spike protein function. Therefore, we tested these drugs using native clinical SARS-CoV-2 virus (strain WIV04) in Vero E6 cells.
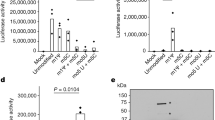
At first, the drugs were given at 10 µM and found to show no cytotoxic effects that could be observed in Vero E6 cells at this level by the MTT method. The results revealed that 7 of these 15 drugs were able to significantly inhibit SARS-CoV-2 virus replication, reducing the supernatant viral RNA load by 10-fold at 24 h post infection (Fig. 3). Next, repeat experiments on these 7 drugs using serial dilutions were conducted in Vero E6 cells using a clinical isolate of the SARS-CoV-2 virus. The CC50 values and EC50 values of these drugs against SARS-CoV-2 virus were determined and are shown in Fig. 4. The EC50 values of these drugs against native SARS-CoV-2 virus replication in Vero E6 cells were all below 5 µM, with a corresponding CC50 greater than 15 µM. We observed similar values for the EC50 values in the Vero E6 experiment with the native virus compared with the results from the Huh-7 pseudovirus assay. All seven drugs had EC90 values far below the CC50 values, which implies that these active drugs almost completely inhibited SARS-CoV-2 multiplication in Vero E6 cells with no noticeable toxicity.

Fifteen drugs selected by the pseudovirus assay were tested for inhibitory activity against native SARS-CoV-2 virus replication in Vero E6 cells at 24 h post infection at a final concentration of 10 µM. Culture supernatant was collected, and the viral genome RNA load was quantified by qRT-PCR methodology as described in “Methods”. Data are shown as the viral RNA load decrease (in log10 scale) vs the DMSO control. Remdesivir was used as a positive control at 10 µM as well. Data are shown as the means ± SEM of two independent experiments.

Antiviral activity of the seven selected drugs against native SARS-CoV-2 replication. Vero E6 cells were treated with drugs at the indicated concentrations and infected with SARS-CoV-2 virus at an MOI = 0.01. Culture supernatants were collected at 24 h post infection, and viral RNA was quantified by qRT-PCR with a TaqMan probe targeting the RBD region of the S gene (circle, solid line). Data are shown as the viral genome load vs the DMSO control (mean ± SEM, n = 6). Cell viability after drug treatment was assayed by the MTT method (square, dashed line); data are shown as percentages of the DMSO control (mean ± SEM, n = 2).
We found that 3 of these active drugs (clemastine, trimeprazine and azelastine) are histamine receptor antagonists, among which clemastine had the best EC90 (2.01 ± 0.94 µM) and EC50 (0.95 ± 0.83 µM) values. Clemastine caused no cytotoxic effects when given at concentrations below 20 µM, with an SI > 20. Trimeprazine and azelastine showed less activity, which implies the shared structural and activity relationships among these histamine antagonists. Amiodarone, which is used to control arrhythmia via inhibition of ATP-sensitive potassium channels, also showed activity against SARS2 infection (EC50 = 1.69 µM) with a good SI index (SI > 11.83). Bosutinib is used in cancer therapy and showed anti-SARS2 entry activity with an EC50 = 2.45 µM and an SI = 7.08. Toremifene is a classic estrogen receptor antagonist used against breast cancer and is also reported to possess anti-EBOV activity. We found that toremifene is also an active inhibitor of SARS2 infection with an EC50 = 2.5 µM (SI > 8). Flupenthixol is widely used for psychotic conditions and acts via a mechanism that blocks postsynaptic dopamine receptors. Our work revealed that it also had anti-SARS2 activity with an EC50 = 2.95 µM (SI = 6.25).
Moreover, we hypothesized that these drugs could also inhibit SARS-CoV viral spike protein function and possibly MERS-CoV viral spike protein function. Hence, we assayed their ability to inhibit SARS-CoV and MERS-CoV S pseudovirus infection in Huh-7 cells. The results showed that these drugs could indeed inhibit the entry of SARS-CoV- and MERS-CoV-S-pseudotyped virus (Table 2). All these drugs showed EC50 values for both the SARS-CoV and MERS-CoV S pseudovirus similar to those observed in the SARS-CoV-2 pseudovirus assay, suggesting that they might have paninhibitory activity against coronavirus infection. Nonetheless, 3 drugs, azelastine, clemastine, and toremifene, still showed higher EC90 values.
In conclusion, we report for the first time that it may be possible to identify novel anti-SARS2 drugs by repurposing approved drugs targeting the virus entry process via a pseudovirus assay method. We identified 7 drugs that are active against native virus infection in vitro, validating the pseudovirus method for identifying novel anti-SARS2 drugs and facilitating compound discovery. We anticipate that these drugs might be beneficial for COVID-19 treatment, but we remain very cautious. Nonetheless, these drugs could be very good lead compounds for development of novel drugs against SARS-CoV-2 as well as SARS-CoV.
Discussion
According to the results presented here, we found that seven already approved drugs had significant anti-SARS-CoV-2 activity. All of them have been used in the clinic for other specific indications for many years, and their clinical properties and safety profiles have been well addressed. We are currently moving forward to test the anti-SARS-CoV-2 activity of these seven drugs in animal models, such as hACE2 transgenic mice and nonhuman primates, by employing a challenge with the native virus. We believe these drugs could be efficacious for the clinical treatment of the COVID-19 pandemic as well as provide novel targets and lead compounds for anti-SARS-CoV-2 drug development.
The SARS-CoV-2 virus emerged as a novel human pathogen that resembles SARS-CoV in many respects. Research on this novel pathogen is restricted to high-level biosafety environments, which usually do not have the instruments necessary for the large-scale screening of antiviral compounds. Pseudovirus technology efficiently overcomes this difficulty and enables the screening of active compounds against the viral entry process in a biosafety level 2 laboratory. In our primary assay, we used the ratio of pseudovirus infectivity to cell viability as the main parameter to quantify the activity of compounds and set different criteria for high-, moderate- and low-toxicity compounds. For most reporter gene-based HTS assays, reporter gene expression requires a suitable cellular inner environment, and compounds that affect cellular metabolic processes not involving our desired target would produce false positives. The activity of compounds could be influenced by their toxicity, and luciferase activity would be inhibited further if the compound was even more toxic. We would identify many more false positive compounds due to toxicity effects if we used infectivity alone as an evaluation parameter. In the pandemic period three months ago, we had limited resources available, and the criteria we employed in our work were derived from our previous experience in cell-based HTS assays used to discover suitable drugs for repurposing. However, approved drugs with high inhibitory activity regardless of toxicity would be further evaluated in the future, and some of them might be promising leading compounds for novel drug development.
As a powerful tool, pseudotyped virus technology has some limitations and drawbacks. Pseudotyped viruses can mimic the entry process mediated by exogenous viral envelope proteins that belong to the class I fusion type, while viral genome release and reporter protein expression in post fusion steps proceed much like those for the HIV virus. Therefore, any compound that interferes with the intracellular steps of the HIV virus life cycle would be active in our primary HTS assay. A VSV-G-pseudotyped virus assay was employed in our secondary screening in an attempt to exclude any compound that was active in this manner. We used this strategy to ensure that our hit compounds specifically targeted the SARS-CoV-2 S protein and to document that such compounds probably acted via specific binding sites on the S protein. However, this introduced the possibility that we would miss compounds that are active as nonspecific entry inhibitors, such as chloroquine, at the cost of target specificity. We hope that the seven active drugs will be appropriate lead compounds for developing more active drugs for SARS-CoV-2 and related viruses.
An interesting finding of this screen was that 3 of the 7 identified drugs were categorized as histamine receptor antagonists, also known as antihistamines. Trimeprazine tartrate is used to treat convulsions, neurasthenia, pruritus, eczema, allergic dermatitis, and other conditions [19, 20]. Azelastine hydrochloride is used to treat allergic rhinitis. Clemastine is used to relieve hay fever and allergy symptoms. Antihistamine drugs have been reported in the past to have certain antiviral effects [21,22,23]. In a previous report that used the SARS-CoV-2 viral protein interactome to identify antiviral drugs, Gordon et al. reported that clemastine is an active antiviral agent targeting the Sigma 1 and Sigma 2 receptors [24]. They reported that clemastine had an EC50 of ~2.3 µM using the viral NP staining method, which is very close to our result (0.95 ± 0.83 µM). We also found other histamine receptor antagonists that showed activity against SARS2 pseudovirus infection, including nortriptyline, perphenazine, promethazine, and chlorprothixene. However, they failed to show activity in the native SARS-CoV-2 infection assay, which implies that histamine receptor antagonist activity per se may not be related to anti-SARS-CoV-2 activity in vitro. Trimeprazine has not been reported to interact with sigma receptors, but the similar compound chlorpromazine has been shown to have a high affinity for the sigma receptor [25] (and chlorpromazine was selected here among the first 154 primary drug hits but showed a limited toxicity/activity window in the secondary screen). This implies that trimeprazine could share a similar mechanism with chlorpromazine.
Taken together, these results highlight the importance of the sigma receptor in the SARS-CoV-2 virus life cycle. The relationship of the sigma receptor affinity and anti-SARS-CoV-2 (as well as anti-SARS-CoV) activity of these histamine receptor antagonists is intriguing, but more thorough investigation is needed in the future.
SARS-CoV-2 virus symptoms primarily present in the respiratory system [26]. Mast cells located in the submucosa of the respiratory tract are known to be activated by the SARS-CoV-2 virus [27]. Once activated, degranulation occurs, releasing histamine and proteases, and abnormal production and release of these mediators from mast cells can further exacerbate SARS-CoV-2-induced inflammation in the respiratory system. Antihistamines block airway inflammation and bronchoconstriction provoked by histamine release from mast cells and are widely used to treat allergic conjunctivitis, rhinitis, and sinusitis. Therefore, anti-allergic medications may also ease the symptoms of COVID-19. However, we have found no reports in the literature concerning the use of antihistamines or other anti-allergic therapies in COVID-19 treatment. We propose that the antihistamine drug clemastine could be a promising new agent for COVID-19 treatment by virtue of its inhibition of SARS-CoV-2 virus infection and its anti-inflammatory effect.
Apart from H1 receptor antagonists, other active agents identified in our screen included flupenthixol, a dopamine receptor antagonist used in antipsychotic treatment. Flupenthixol has an extensive receptor spectrum and interacts with a variety of dopamine and serotonin binding sites to exert antipsychotic and antidepressant effects [28]. We found no reports on the antiviral activity of flupenthixol, but the related drug chlorprothixene may have anti-herpes simplex virus 2 activity [29]. However, this drug had no anti-SARS-CoV-2 activity in our assay.
Bosutinib, an inhibitor of the Abelson (Abl) kinase signaling pathway, was found to strongly inhibit SARS-CoV-2 in vitro with an EC50 = 2.45 ± 0.12 μM and low toxicity. Excessive activation of the ABL pathway can lead to chronic myeloid leukemia (CML), and bosutinib was originally developed and approved as an inhibitor of this pathway for the treatment of CML [30]. We found that the related kinase inhibitor imatinib showed weak activity in pseudovirus screening as well. Some studies have found that Abelson kinase inhibitors such as imatinib and dasatinib are effective inhibitors of SARS-CoV and MERS-CoV virus fusion [31, 32]. They all have an inhibitory effect on coronavirus in vitro, which further confirms the importance of the ABL kinase signaling pathway in the coronavirus life cycle. Moreover, imatinib mesylate has been shown to prevent the entry of Ebola virus, poxvirus, and coxsackievirus [33,34,35]. All of the above findings indicate that the ABL kinase signaling pathway may be important for the entry of multiple viruses.
Amiodarone hydrochloride is an ion channel inhibitor used to treat arrhythmias. Previous studies have found that amiodarone can inhibit SARS-CoV infection by altering the late endosome [36]. Because SARS-CoV-2 and SARS-CoV have a high degree of similarity [3], amiodarone may use a similar mechanism to suppress the replication of SARS-CoV-2. Amiodarone has shown antiviral activity in more than one study, and other works have shown that it can also effectively inhibit the entry of Ebola virus in vitro by interfering with the endocytosis pathway and inhibiting proteolysis [37,38,39].
Selective estrogen receptor modulators have shown potential antiviral effects in the clinic, and toremifene is a typical representative of this class. Some research has found that toremifene can also block entry of MERS-CoV, SARS-CoV [32, 40, 41], and Ebola virus [42] in some cell lines. The antiviral activity of toremifene has nothing to do with its effect on the classic estrogen receptor-related pathway [43]. Toremifene can interact with viral membrane glycoproteins to disrupt their stability, thereby preventing the fusion of viral membranes and endosomal membranes and thus exerting antiviral effects [44, 45].
Remdesivir was initially believed to be effective for the treatment of COVID-19, and many clinical trials are ongoing. On 1st May, the US FDA issued an emergency use authorization for the investigational antiviral drug remdesivir for the treatment of suspected or laboratory-confirmed COVID-19 in adults and children hospitalized with severe disease. One study showed that remdesivir improves oxygen support by 68% in critically ill patients [46]. However, due to the inability to recruit enough patients, the two clinical trials conducted by remdesivir in China, one for severe patients and the other for mild and normal patients, were terminated early [47]. In vitro studies have shown that remdesivir has an inhibitory effect on SARS-CoV-2, with an EC50 of 0.77 μM [48], which is close to the EC50 of clemastine and better than that of the other 6 active drugs identified in our assay. Remdesivir acts as a polymerase inhibitor to affect the intracellular viral RNA replication process, which differs from the mechanism of the seven active drugs we discovered here that target the viral entry process. Therefore, these active drugs could be used along with remdesivir, as they may act in a synergistic manner for COVID-19 treatment.
Further investigation of these seven active repurposed drugs is urgently required, especially studies of them in vivo activities using suitable animal models, which are quite limited in the current situation. These drugs have been used for decades and are safe, but it is unclear as to whether these approved drugs have pharmacokinetics that are suitable for COVID-19 antiviral treatment, and we suggest that their use merits caution and further study in the context of the treatment of COVID-19.
Our approach of combining pseudovirus screening and wild-type virus assays is an efficient and resource-saving strategy for rapid drug repurposing and active compound discovery in an effort to control emerging viruses and may also identify panactive agents. Furthermore, the possibilities for the relief of syndromes by multitargeted therapies are extended in this way; for example, drugs that directly target the virus and alleviate inflammatory conditions can be revealed, as we demonstrated in this study. More positively, these results and conclusions offer a promising starting point for specific SARS-CoV-2 antiviral agent development.
References
https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/.
Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63:457–60.
Chan JFW, Kok KH, Zhu Z, Chu H, To KKW, Yuan S, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221–36.
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–20.
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–4.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–3.
Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Disco. 2004;3:673–83.
Wang Z, Chen X, Lu Y, Chen F, Zhang W. Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Biosci Trends. 2020;14:64–8.
Chen C, Huang J, Cheng Z, Wu J, Chen S, Zhang Y, et al. Favipiravir versus Arbidol for COVID-19: a randomized clinical trial. medRxiv. 2020. https://doi.org/10.1101/2020.03.17.20037432.
Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrobial Agents. 2020;56:105949. https://doi.org/10.1016/j.ijantimicag.2020.105949.
Chen Z, Hu J, Zhang Z, Jiang S, Han S, Yan D, et al. Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. medRxiv. 2020. https://doi.org/10.1101/2020.03.22.20040758.
Li H, Yang L, Liu FF, Ma XN, He PL, Tang W, et al. Overview of therapeutic drug research for COVID-19 in China. Acta Pharmacol Sin. 2020;41:1133–40.
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–93.
Chen S, Chen LL, Luo HB, Sun T, Chen J, Ye F, et al. Enzymatic activity characterization of SARS coronavirus 3C-like protease by fluorescence resonance energy transfer technique. Acta Pharmacol Sin. 2005;26:99–106.
Yang L, Shi LP, Chen HJ, Tong XK, Wang GF, Zhang YM, et al. Isothiafludine, a novel non-nucleoside compound, inhibits hepatitis B virus replication through blocking pregenomic RNA encapsidation. Acta Pharmacol Sin. 2014;35:410–8.
Tong XK, Qiu H, Zhang X, Shi LP, Wang GF, Ji FH, et al. WSS45, a sulfated alpha-D-glucan, strongly interferes with Dengue 2 virus infection in vitro. Acta Pharmacol Sin. 2010;31:585–92.
Zhao G, Du L, Ma C, Li Y, Li L, Poon VK, et al. A safe and convenient pseudovirus-based inhibition assay to detect neutralizing antibodies and screen for viral entry inhibitors against the novel human coronavirus MERS-CoV. Virol J. 2013;10:266.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3.
Harling DW. Trimeprazine tartrate and convulsions. Anaesthesia. 1995;50:97–8.
France KG, Blampied NM, Wilkinson P. A multiple-baseline, double-blind evaluation of the effects of trimeprazine tartrate on infant sleep disturbance. Exp Clin Psychopharmacol. 1999;7:502–13.
Schafer A, Cheng H, Xiong R, Soloveva V, Retterer C, Mo F, et al. Repurposing potential of 1st generation H1-specific antihistamines as anti-filovirus therapeutics. Antivir Res. 2018;157:47–56.
Xu W, Xia S, Pu J, Wang Q, Li P, Lu L, et al. The antihistamine drugs carbinoxamine maleate and chlorpheniramine maleate exhibit potent antiviral activity against a broad spectrum of influenza viruses. Front Microbiol. 2018;9:2643.
Chen J, Dai L, Goldstein A, Zhang H, Tang W, Forrest JC, et al. Identification of new antiviral agents against Kaposi’s sarcoma-associated herpesvirus (KSHV) by high-throughput drug screening reveals the role of histamine-related signaling in promoting viral lytic reactivation. PLoS Pathog. 2019;15:e1008156.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–68.
Largent BL, Wikstrom H, Gundlach AL, Snyder SH. Structural determinants of sigma receptor affinity. Mol Pharmacol. 1987;32:772–84.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Kritas SK, Ronconi G, Caraffa A, Gallenga CE, Ross R, Conti P. Mast cells contribute to coronavirus-induced inflammation: new anti-inflammatory strategy. J Biol Regul Homeost Agents. 2020;34:9–14.
Wenzel JM, Su ZI, Shelton K, Dominguez HM, von Furstenberg VA, Ettenberg A. The dopamine antagonist cis-flupenthixol blocks the expression of the conditioned positive but not the negative effects of cocaine in rats. Pharmacol Biochem Behav. 2013;114–115:90–6.
Kristiansen JE, Andersen LP, Vestergaard BF, Hvidberg EF. Effect of selected neuroleptic agents and stereo-isomeric analogues on virus and eukaryotic cells. Pharmacol Toxicol. 1991;68:399–403.
Cuellar S, Vozniak M, Rhodes J, Forcello N, Olszta D. BCR-ABL1 tyrosine kinase inhibitors for the treatment of chronic myeloid leukemia. J Oncol Pharm Pract. 2018;24:433–52.
Coleman CM, Sisk JM, Mingo RM, Nelson EA, White JM, Frieman MB. Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and middle east respiratory syndrome coronavirus fusion. J Virol. 2016;90:8924–33.
Dyall J, Coleman CM, Hart BJ, Venkataraman T, Holbrook MR, Kindrachuk J, et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother. 2014;58:4885–93.
García M, Cooper A, Shi W, Bornmann W, Carrion R, Kalman D, et al. Productive replication of Ebola virus is regulated by the c-Abl1 tyrosine kinase. Sci Transl Med. 2012;4:123ra24.
Reeves PM, Smith SK, Olson VA, Thorne SH, Bornmann W, Damon IK, et al. Variola and monkeypox viruses utilize conserved mechanisms of virion motility and release that depend on abl and SRC family tyrosine kinases. J Virol. 2011;85:21–31.
Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124:119–31.
Stadler K, Ha HR, Ciminale V, Spirli C, Saletti G, Schiavon M, et al. Amiodarone alters late endosomes and inhibits SARS coronavirus infection at a post-endosomal level. Am J Respir Cell Mol Biol. 2008;39:142–9.
Cristiano Salata DM, Francesco M, Cristina P, Arianna C, Aldo B, Giorgio P. Amiodarone affects Ebola virus binding and entry into target cells. New Microbiol. 2018;41:162–4.
Salata C, Baritussio A, Munegato D, Calistri A, Ha HR, Bigler L, et al. Amiodarone and metabolite MDEA inhibit Ebola virus infection by interfering with the viral entry process. Pathog Dis. 2015;73:ftv032. https://doi.org/10.1093/femspd/ftv032.
Madrid PB, Panchal RG, Warren TK, Shurtleff AC, Endsley AN, Green CE, et al. Evaluation of ebola virus inhibitors for drug repurposing. ACS Infect Dis. 2015;1:317–26.
Cong Y, Hart BJ, Gross R, Zhou H, Frieman M, Bollinger L, et al. MERS-CoV pathogenesis and antiviral efficacy of licensed drugs in human monocyte-derived antigen-presenting cells. PLoS One. 2018;13:e0194868.
de Wilde AH, Jochmans D, Posthuma CC, Zevenhoven-Dobbe JC, van Nieuwkoop S, Bestebroer TM, et al. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother. 2014;58:4875–84.
Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, et al. FDA-approved selective estrogen receptor modulators inhibit ebola virus infection. Sci Transl Med. 2013;5:190ra79.
Janakiram NB, Rao CV. Molecular markers and targets for colorectal cancer prevention. Acta Pharmacol Sin. 2016;29:1–20.
Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, et al. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature. 2016;535:169–72.
Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, et al. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One. 2013;8:e56265.
Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, et al. Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med. 2020;382:2327–36.
https://clinicaltrials.gov/ct2/results?cond=&term=remdesivir&cntry=&state=&city=&dist=.
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–71.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFC1200604). Experiments related to SARS-CoV-2 were carried out at the National Biosafety Laboratory, Wuhan, Chinese Academy of Sciences. We are particularly grateful to Tao Du and Lun Wang from the Zhengdian Biosafety Level 3 Laboratory and the team running the laboratory for their work. The pNL4.3.Luc.R-.E- plasmid was kindly gifted by Dr Shi-bo Jiang and Dr Lu Lu from Fudan University.
Author information
Authors and Affiliations
Contributions
RJP, YCZ, JX, and SQX worked as a team to perform validation assay involving live SARS2 virus in biosafety level 3 laboratory in Wuhan Institute of Virology; LY performed HTS screening assay; HL conducted pseudovirus package and validation assay; XNM helped in data collection and analysis in HTS assay; YZ, FHZ, PLH, and WT participated in the experiments; XKT designed project, constructed plasmid and wrote the manuscript; BZ and JPZ supervised the whole project.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
About this article

Cite this article
Yang, L., Pei, Rj., Li, H. et al. Identification of SARS-CoV-2 entry inhibitors among already approved drugs. Acta Pharmacol Sin 42, 1347–1353 (2021). https://doi.org/10.1038/s41401-020-00556-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00556-6
Keywords
This article is cited by
-
Molecular docking as a tool for the discovery of novel insight about the role of acid sphingomyelinase inhibitors in SARS- CoV-2 infectivity
BMC Public Health (2024)
-
A high-throughput screening system for SARS-CoV-2 entry inhibition, syncytia formation and cell toxicity
Biological Procedures Online (2023)
-
Early intervention with azelastine nasal spray may reduce viral load in SARS-CoV-2 infected patients
Scientific Reports (2023)
-
A model for network-based identification and pharmacological targeting of aberrant, replication-permissive transcriptional programs induced by viral infection
Communications Biology (2022)
-
Discovery of potential small molecular SARS-CoV-2 entry blockers targeting the spike protein
Acta Pharmacologica Sinica (2022)
