
Abstract
Ageing organisms accumulate senescent cells that are thought to contribute to body dysfunction. Telomere shortening and damage are recognized causes of cellular senescence and ageing. Several human conditions associated with normal ageing are precipitated by accelerated telomere dysfunction. Here, we systematize a large body of evidence and propose a coherent perspective to recognize the broad contribution of telomeric dysfunction to human pathologies.
Similar content being viewed by others
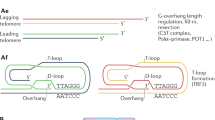

Main
Telomeres are the genomic portions at the ends of linear chromosomes. Telomeric DNA in vertebrates is made of TTAGGG repeats bound by a set of proteins that modulate their biological functions and protect them from being recognized as DNA damage that triggers a DNA damage response (DDR). As standard DNA polymerases cannot fully replicate linear DNA templates in the absence of telomerase, a DNA-template-independent DNA polymerase, and because of nucleolytic processing, DNA replication results in the generation of chromosomes with progressively shortened telomeres1. As telomeres reach a critical length, they become unable to bind enough telomere-capping proteins and are sensed as exposed DNA ends2, which activates the DDR pathways that, through the induction of the cell cycle inhibitors p21 and p16, arrest proliferation3,4. Such short telomeres, however, retain a sufficient number of telomere-binding proteins to inhibit DNA repair and avoid fusions5, and consequently fuel a persistent DNA damage signal that enforces a permanent DNA damage-induced proliferative arrest. This initiates and maintains cellular senescence, a key contributor to organismal ageing and multiple age-related diseases6,7. Activation of the DDR at telomeres (termed tDDR hereafter) results in the formation of telomere-associated DDR foci (TAFs) or telomere-induced DNA damage foci (TIFs), which are markers of cellular senescence in cultured cells and tissues (Box 1). Following telomere dysfunction, some cell types may also undergo cell death by apoptosis8,9 or autophagy10.
In addition to irreversible cell cycle arrest, cellular senescence is characterized by changes in chromatin, gene expression, organelles and cell morphology11. Importantly, senescent cells secrete a complex set of pro-inflammatory cytokines, known as the senescence-associated secretory phenotype (SASP). This alters the composition of the extracellular matrix, impairs stem cell functions, promotes cell transdifferentiation and can spread the senescence phenotype to surrounding cells, thereby causing systemic chronic inflammation12. SASP is both promoted by DDR13 and can promote DDR and TAF formation in an autocrine and paracrine fashion14,15,16.
Although conceptually appealing to explain proliferative exhaustion and cell ageing, telomere shortening is inadequate to explain ageing in non-proliferating, quiescent or terminally differentiated cells. Nevertheless, TAFs and senescence have been reported in ageing post-mitotic cells, including cardiomyocytes, adipocytes, neurons, osteocytes and osteoblasts17. These observations can be explained by an evolutionary perspective by which telomere-binding proteins inhibit DNA repair in cis18,19 to maintain the linear structure of chromosomes and to prevent fusions. As a consequence, DNA damage that occurs within telomeric repeats (tDD) resists repair, which causes persistent tDDR signalling and TAF formation also at long telomeres19,20,21. Endogenous or exogenous DNA damage is constantly generated, and the fraction that occurs at telomeres, which is less efficiently repaired, thus accumulates and induces a senescence-like phenotype (Fig. 1a).

a, Genomic DNA damage (DD) triggers a transient DNA damage response (DDR) that may not be sufficient for senescence establishment. Alternatively, an irreparable, therefore persistent, DNA damage at telomeres causes a protracted DDR and cellular senescence that are associated with SASP-mediated inflammation and consequent fibrosis. These events in a stem-cell context impair stem-cell properties and alter differentiation. Overall this contributes to organismal ageing17. b, In proliferating tissues, telomeres are shortened with cell cycle divisions and, when critically short, they trigger a DDR. In non-proliferating, post-mitotic tissues, telomere dysfunction can be driven by irreparable DNA damage within telomeres. In both cases, the persistent DDR activation sustains a senescent phenotype that is characterized by arrested proliferation and SASP activation.
Therefore, persistent tDDR activation is the shared causative event of both replicative cellular senescence caused by critically short telomeres and the senescence-like state caused by damaged telomeres in non-replicating cells (Fig. 1b). Although these events may be mechanistically distinct in origin, DNA damage at long telomeres may cause, within the time frame of organismal ageing, degradation or loss of the terminal portions of telomeres, therefore leading to telomere shortening.
In the broader context of organismal ageing, the notion that DNA is the only irreplaceable component of the cell makes a strong argument in favour of an apical role of DNA integrity in ageing. The irreparability of telomeres makes it more so.
In addition to being hard to repair, telomeric DNA is hypersensitive to oxidative DNA damage, a phenomenon recently named TelOxidation22. Oxidative stress reportedly both induces tDD without telomere shortening and accelerates telomere shortening23,24,25,26 by inhibiting telomerase27 and disrupting the recognition by telomere-binding proteins, which contributes to telomere uncapping22,28.
tDDR activation and TAF accumulation are often causally connected to other ageing-associated processes. These include mitochondrial dysfunction, altered nutrient sensing, impaired autophagy, loss of proteostasis and epigenetic dysregulation, which suggests that there is a unifying ‘telomere-centric’ mechanistic rationale for many ageing hallmarks, as also proposed in ref. 29.
Telomere dysfunction during ageing
As replicative cellular senescence is caused by telomere shortening below a critical length1, the effect of telomere shortening on organismal ageing in animal models, primarily mouse and fish species, has mostly been studied through genetic deletion of telomerase component genes, either the telomerase RNA component (Terc) or the telomerase reverse transcriptase protein (Tert), and successive inbreedings. In these models, telomere shortening progressively causes tDDR activation and cellular senescence, which recapitulates features of ageing and age-related diseases30,31. Across species, however, short telomeres do not necessarily predict short lifespan—that is, humans have shorter telomeres but longer lifespans than rodents. Instead, telomere shortening rates and the increase in short telomeres has been proposed to predict lifespan32. Consistently, one or a few critically short telomeres are sufficient to trigger a DDR and impose cellular senescence in vivo, regardless of a majority of otherwise long telomeres33. Thus, individual DDR signalling events at telomeres are key determinants of cell fate and organismal ageing. Indeed, the levels of DDR markers and TAFs increase during ageing in different mammalian tissues. Mice, despite having long telomeres and ubiquitous telomerase expression, show an age-dependent increase in dysfunctional telomeres in both proliferating and non-proliferating tissues20,34,35,36,37. Damaged telomeres also increase in the brain, liver and skin of aged baboons19,38. In humans, TAFs increase with age in the absence of telomere shortening in skin melanocytes15 and CD8+ T cells, together with other senescence markers39. Although most human data on telomeres are based on studies of leukocytes, a recent study of telomere length in tissues from 952 individuals concluded that 21 out of the 24 tissues studied show age-dependent telomere shortening40.
Several pieces of evidence support a role for tDDR as a driver of ageing and age-associated diseases. First, interventions known to increase health span such as dietary restriction41, exercise42, rapamycin43,44 and 17β-oestradiol45 can reduce the frequency of cells with TAFs. Clearance of senescent cells with senolytic strategies reduces TAF numbers in vivo36,37,41,46. Conversely, TAFs accumulate after chronic inflammation47, obesity37,41, mitochondrial dysfunction36 and impaired autophagy48, which are all known to accelerate ageing.
The most prevalent hypothesis is that it is not telomere dysfunction per se that leads to ageing and age-associated diseases but telomere dysfunction-activated tDDR that causes cellular senescence, which, also by SASP, facilitates the age-related loss of tissue functions11. Although senescence may occur independently of telomere dysfunction17 (for example, during development), the differential contribution of telomere-dependent and -independent cellular senescence to ageing and age-related disorders remains to be determined.
Here, we review the role of telomere dysfunction in the context of human ageing and age-related diseases, often termed telomeropathies, telomere biology disorders or telomere syndromes (Fig. 2 and Table 1). Although we describe diseases individually, grouped by the organs affected, individual patients with telomeropathies often show more than one clinical manifestation, which supports the notion of a “spectrum disorder”49 caused by telomere dysfunction.

Schematic representation of the age-related diseases described in this Review grouped by organs or systems. AA, aplastic anaemia; AD, Alzheimer’s disease; ALD, alcholic liver disease; AMD, age-related macular degeneration; CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis; IRI, ischaemia–reperfusion injury; MDS, myelodysplastic syndrome; NAFLD, non-alcoholic fatty liver disease; PBC, primary biliary cirrhosis; PD, Parkinson’s disease; T2D, type 2 diabetes.
Pulmonary diseases
Several lung diseases have been associated with ageing50 and causally linked to telomere dysfunction and senescent cell accumulation.
Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF), which affects approximately 3 million people worldwide, is a lung degenerative disease characterized by interstitial remodelling. Fibrosis in human ageing lungs has been associated with telomere shortening, DDR and cellular senescence51. Patients with IPF accumulate TAFs and senescence markers in lungs that increase with disease severity and activate SASP52. Moreover, circulating leukocytes and alveolar epithelial cells show shorter telomeres than age-matched individuals53. Short telomeres are an established risk factor for IPF, and 37% of familial cases and 25% of sporadic cases show telomere lengths below the tenth percentile for their age, as assessed in circulating leukocytes and alveolar epithelial cells54. In independent cohorts, a shorter telomere length was demonstrated to be a robust independent predictor of disease progression, response to therapy and death55, and to be associated with a shorter time to allograft dysfunction following lung transplant56. Causality of telomere dysfunction in familial and sporadic IPF is indicated by the detection of germline mutations in genes involved in telomere maintenance57,58.
Mouse studies indicate that telomere dysfunction can recapitulate IPF features. Mice with short telomeres induced by knockout of telomerase components show TAFs, inflammation and fibrosis in lungs59, which is aggravated by cigarette smoke60 or a low dose of bleomycin, a DNA-damaging agent61. Telomere dysfunction, specifically induced in alveolar epithelial type II cells through the deletion of the telomere proteins TRF1 or TRF2, led to impaired regeneration, inflammation and fibrosis61,62. Consistently, expression of telomerase using adeno-associated vectors (AAVs) in alveolar epithelial type II cells led to TAF reduction, decreased inflammation and improved lung function in telomerase-mutant and wild-type aged mice63,64. Additionally, genetic or pharmacological clearance of senescent cells improved lung function and health in a mouse model of bleomycin-induced IPF52.
Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease (COPD) affects around 300 million people globally and is associated with high morbidity and mortality in elderly patients65. COPD exhibits accelerated lung ageing characterized by inflammation of parenchyma and airways, chronic remodelling of the peripheral bronchi and inter-alveolar septa disruption towards emphysema. Compared with unaffected individuals, small airway epithelial cells from patients with COPD show higher levels of TAFs and senescence markers34. Cigarette smoke (a major risk factor for COPD) induced TAFs, cellular senescence and SASP in cultured primary human airway epithelial cells and fibroblasts34 and reduced telomere protection protein 1 (TPP1) levels in mouse and human lungs, which caused tDDR activation66. Short telomeres were also observed in lungs and circulating leukocytes of patients with COPD66,67. Telomerase mutations are a risk factor for emphysema among patients with COPD68.
Other lung diseases
Non-cystic fibrosis bronchiectasis is a common inflammatory lung disease characterized by irreversible dilation of the bronchi. Lung tissues from these patients show shorter telomeres and increased levels of TAFs and senescence markers69.
Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has been predicted to cause long-term lung fibrosis70. Disease severity has been correlated with patient age and leukocyte telomere length71, which is consistent with a recent finding that tDDR activation induced by telomere shortening increases the expression of ACE2, the SARS-CoV-2 receptor on human lung cells72.
Acquired bone marrow failure syndromes
Nucleated blood cells show the shortest telomeres among human tissues40. Thus, it is not surprising that impaired telomere maintenance most acutely impairs haematopoiesis.
Aplastic anaemia
Aplastic anaemia is a rare disease with a variable age of diagnosis and is characterized by pancytopaenia in the peripheral blood and markedly hypocellular bone marrow73. Around 9% of patients with acquired aplastic anaemia carry mutations in the telomerase components TERC, TERT and DKC1, which are also mutated in inherited aplastic anaemia, as in dyskeratosis congenita74,75. Additional rarer mutations in other telomere homeostasis genes have been identified76,77,78. About one-third of patients with aplastic anaemia show short telomeres in peripheral blood, which suggests that there is a causative link between aplastic anaemia pathology and telomere dysfunction75,76,79. Patients with shorter telomeres at diagnosis show a more severe disease, a poorer response to treatments, an increased risk of relapse and development of myelodysplastic syndrome (MDS; see below) and a lower survival rate75,80,81. Leukocyte telomere length in the donor, but not in the recipient, positively correlates with survival following allogenic haematopoietic cell transplantation82. The use of the androgen receptor agonist danazol in patients with aplastic anaemia with critically short telomeres improved their condition by increasing telomerase expression and lengthening telomeres. This provides support for a causative role of telomere length in this disease83.
At least two mouse models have demonstrated a causative role of telomere dysfunction in bone marrow failure. First, partial depletion of TRF1 in hematopoietic stem and progenitor cells caused TAF formation, cell depletion and a compensatory proliferation, which resulted in rapid telomere attrition and cellular senescence84. Similarly, transplantation of bone marrow from late-generation Tert-knockout mice in irradiated wild-type recipients resulted in aplastic anaemia85. In both models, danazol or AAV-mediated Tert expression improved haematopoiesis85,86.
MDS
MDS is a heterogeneous group of clonal haematopoietic disorders characterized by ineffective haematopoiesis and DNA damage accumulation in haematopoietic stem and progenitor cells87. MDS affects up to 13.2 per 100,000 people88, often in advanced age. MDS can also be secondary to chemotherapy or radiation or be associated with inherited abnormalities in DNA repair and telomere maintenance genes89. Bone marrow cells in patients with MDS have shorter telomeres than those in healthy donors90. Moreover, late-generation telomerase-deficient mice recapitulate myelodysplastic features that can be reversed by telomerase reactivation91.
Metabolic diseases
Metabolic diseases occur when the organism is incapable of efficiently converting food into energy. Here, we summarize the evidence that suggests that telomere dysfunction is a common causal factor in several of these diseases.
Metabolic syndrome
Estimated to affect one in three people in the United States, metabolic syndrome is a set of related conditions including chronic inflammation, obesity, dyslipidaemia, high blood pressure and insulin resistance co-occurring in an individual, which together increase the risk of serious cardiovascular disease. Similar to the other conditions contributing to it, obesity is associated with increased TAFs in various organs and tissues37,41,92. Short telomeres in adipose tissues are associated with metabolic disease progression, and causality was demonstrated by telomerase inactivation in mouse adipocyte precursors that led to hypertrophy and inflammation93.
Liver diseases
Telomere shortening has been implicated in hepatocyte senescence94 and in disease progression in patients with liver cirrhosis95. Patients with cirrhosis have a higher incidence of telomerase mutations and bear shorter telomeres than unaffected individuals96. Late-generation telomerase-deficient mice exposed to chronic liver injury show accelerated cirrhosis development97.
Non-alcoholic fatty liver disease (NAFLD) is characterized by an excess of hepatic fat accumulation (steatosis) and, in later stages, inflammation (non-alcoholic steatohepatitis) and fibrosis. TAF and p21 levels in hepatocytes positively correlate with NAFLD severity41, and the clearance of senescent cells in aged and obese mice reduced the fraction of TAF-positive hepatocytes and alleviated liver steatosis41. Primary biliary cirrhosis, an autoimmune disease with chronic, progressive cholestasis and liver failure, is associated with short telomeres, DDR activation and senescence marker accumulation in biliary epithelial cells98. Treatment of a mouse model of biliary liver fibrosis with a senolytic drug reduced fibrosis99. Alcoholic liver disease and chronic viral hepatitis have also been associated with telomere dysfunction in human liver biopsies100,101.
Type 2 diabetes
Type 2 diabetes (T2D) is an age-associated disease characterized by a decrease in pancreatic β-cell mass and function and insulin resistance in multiple tissues that results in hyperglycaemia. Several cross-sectional human studies of white blood cells have shown an association between T2D and short telomeres102. Mice with short telomeres display impaired insulin secretion and glucose intolerance associated with an accumulation of senescence markers, which suggests a causal role for telomere shortening in this pathology. Senescent cell clearance improved glucose homeostasis and insulin sensitivity in obese and aged mice103.
Cardiovascular diseases
Cardiovascular diseases are the leading causes of morbidity and mortality in western countries, and ageing is a major risk factor for their development104. Both telomere shortening and telomere damage have been reported as hallmarks and potential drivers of heart diseases and as indicators of therapeutic outcomes105.
Cardiac diseases
The heart exhibits cardiomyocyte hypertrophy and fibrosis during ageing that leads to increased ventricular stiffness and impaired cardiac function. During physiological ageing in humans and mice, TAFs occur independently from telomere length in post-mitotic cardiomyocytes. This is associated with the induction of p16 and p21 and a cardiac-specific form of SASP that contributes to cardiac hypertrophy and fibrosis36. The clearance of senescent cells in aged mice improves heart functions and reduces the fraction of TAF-positive cardiomyocytes without a significant effect on mean telomere length36. This observation confirms a role for cellular senescence in heart disease and hints at the negligible contribution of telomere length to cardiomyocyte senescence36. Length-independent telomere damage may result from oxidative damage, as mouse models of increased oxidative stress and mitochondrial dysfunction show early onset of age-dependent telomere dysfunction36. Consistently, cardiac ischaemia–reperfusion injury (IRI), which is associated with massive induction of oxidative stress, promotes TAF formation and senescence, and treatment with a senolytic agent improved cardiac function106.
Patients with genetic forms of hypertrophic or dilated cardiomyopathy, conditions that affect 1 in 500–2,500 people worldwide, have cardiomyocytes with shorter telomeres than age-matched individuals107,108. Short telomeres were also observed in a large independent cohort, with the severity of hypertrophic cardiomyopathy correlating with the telomere length of leukocyte telomeres109. A causal role of short and dysfunctional telomeres in cardiac diseases has been established in mice110. Late-generation telomerase-deficient mice show severe left ventricular loss of function, increased cardiomyocyte hypertrophy and decreased number of cardiomyocytes111. AAV-mediated Tert expression in adult mice improved their survival following myocardial infarction112.
Atherosclerosis
Atherosclerosis is a vascular disease that is characterized by the formation of artery plaques containing vascular smooth muscle cells (VSMCs) that potentially leads to thrombosis and myocardial infarction, and is considered a leading cause of mortality worldwide113. In a limited cohort, patients with atherosclerosis bear shorter telomeres in circulating leukocytes than healthy age-matched individuals114. VSMCs in human atherosclerotic plaques show cellular senescence markers and telomeres that are markedly shorter than those in unaffected vessels from the same individual115. Telomere dysfunction induced by VSMC-specific expression of mutant TRF2 is sufficient to increase atherosclerosis116,117. Indeed, clearance of senescent cells reduced TAF-positive cells in the medial layer of the aorta from aged and hypercholesterolaemic mice46 and alleviated plaque formation and disease progression46,118.
Skeletal disorders
Changes in bone and joint tissues that lead to osteoporosis and osteoarthritis are associated with an accumulation of senescent cells.
Osteoarthritis
Osteoarthritis is characterized by the degeneration of joint cartilage and subchondral bone, and affects more than 30 million adults in the United States (https://oaaction.unc.edu/oa-module/oa-prevalence-and-burden/). Chondrocytes, the cells that constitute the articular cartilage, show several senescence markers, including DDR activation, in osteoarthritis119,120. A causal role of senescence in this pathogenesis was established by evidence showing that removal of senescent cells reduced the development of post-traumatic and naturally occurring osteoarthritis in mouse models121. Moreover, treatment with a senolytic therapy mitigated age-dependent disc degeneration122. A link between osteoarthritis and telomere dysfunction is supported by the observations that patients with osteoarthritis have leukocytes with shorter telomeres than age-matched individuals123,124, and telomere length inversely correlates with chronic severe pain125. Moreover, cultured chondrocytes isolated from areas close to the osteoarthritis lesions from the hips of patients have shorter mean telomere length and increased levels of senescence markers than those from distal sites in the same joint126. Interestingly, the presence of ultrashort telomeres has been proposed to be a better marker than average telomere length for the extent of osteoarthritis damage127.
Osteoporosis
Osteoporosis is a chronic skeletal disorder that affects more than 200 million people worldwide. It is characterized by low bone mineral density and microarchitectural deterioration of bone tissues that can lead to an increased fracture risk128. A total of 33% of women and 20% of men over the age of 50 years are estimated to experience osteoporosis-related fractures. Increasing evidence points towards a role for telomere dysfunction and senescence in osteoporosis129. Senescent osteocytes that express p16 at high levels have been associated with age-related bone loss in mice, and their clearance increased bone strength130. Osteoporosis correlates with short telomeres in the leukocytes of patients, and long telomeres in a female cohort were associated with high bone mineral density and reduced risk of osteoporosis131. Supporting a causal role of telomere dysfunction in osteoporosis, late-generation telomerase-deficient mice recapitulate several features of osteoporosis, such as decreased bone volume, diminished osteoblast number and function, and increased porosity, with TAFs being associated with impaired osteoblast differentiation132,133.
Kidney diseases
During physiological ageing, kidneys experience detrimental structural and functional changes. Several renal pathologies such as acute kidney injury, glomerulonephritis, diabetic nephropathy, polycystic kidney disease and chronic kidney disease (CKD) have been associated with cellular senescence and telomere dysfunction134.
CKD, which is estimated to affect 15% of the adult population in the United States (https://www.kidney.org/news/newsroom/fsindex), is an independent risk factor for cardiovascular events in older people, often leading to end-stage renal disease. Dialysis and kidney transplant remain the only two major treatments. CKD shows several features of accelerated ageing, including decreased kidney weight, atrophy, sclerosis, fibrosis and a CKD-associated secretory phenotype, which is similar to SASP135. In CKD, senescence markers were observed in tubular epithelial cells, podocytes, interstitial and mesangial cells136, and their accumulation was associated with disease progression135.
DNA damage accumulates in many forms of kidney injury. Kidneys in patients with CKD exhibited an increased number of tubules positive for the DDR marker γH2AX, which was inversely correlated with the estimated glomerular filtration rate, and a greater number of phosphorylated ATR-positive cells137. Increased levels of γH2AX and phosphorylated ATM in glomeruli are associated with clinicopathological parameters in patients with IgA nephropathy, a condition that often leads to CKD138. Thus, DDR activation and senescence alone or in combination with insults such as infections, lipopolysaccharides, uraemic toxins and dialysis treatments, can contribute to CKD139,140.
In patients with uraemia, a sign of kidney damage, lymphocyte telomere length was significantly shorter than in unaffected individuals141. A study of a large cohort of patients with CKD revealed that telomere length measured in peripheral blood was a strong independent predictor of all-cause mortality142. Moreover, in a large population study, telomere shortening was associated with an increased risk for CKD progression in individuals who actively smoke and in patients with diabetes mellitus143. Telomere length predicts long-term kidney allograft function, and telomere shortening is linked to complications of kidney transplantation144.
Animal studies suggest that telomere dysfunction is causally implicated in kidney ageing, acute kidney injury and decreased recovery after insult. Mice with dysfunctional telomeres show an age-dependent decline in kidney function and morphology145 as well as reductions in renal function and regeneration after IRI146. In aged mice, clearance of senescent cells reduced glomerulosclerosis and retained blood urea nitrogen levels, which indicates that senescence contributes to these pathological alterations147. Cats with CKD also show shortened telomeres and increased numbers of senescent cells in the kidney148.
Recently, strong evidence has linked telomere shortening and dysfunction with kidney fibrosis. In two independent mouse models, short and dysfunctional telomeres were shown to sensitize kidneys to folic acid-induced toxicity that resulted in fibrosis, thereby demonstrating a key contribution of telomere dysfunction in this pathology149.
Neurodegenerative diseases
Brain ageing is characterized by a progressive decline in memory and cognition and is recognized as the greatest risk factor for neurodegenerative diseases. Senescent cells accumulate with age in the murine brain and is exacerbated in late-generation telomerase-deficient mice150, which suggests a causal role for telomere dysfunction in this process. Indeed, TAFs increase with age in hippocampal neurons in baboons19 and mice151. Age-dependent TAF increases in the brain also correlate with chronic inflammation151 and obesity37, both of which are associated with age-dependent cognitive decline. Single-cell RNA sequencing of the hippocampus of aged mice revealed an increased p16 level with age, which was stronger in microglia and oligodendrocyte progenitor cells152. Clearance of senescent cells in aged mice significantly improved cognitive function152, which indicates that senescence has an important role in age-associated cognitive impairment.
Alzheimer’s disease
Alzheimer’s disease is the most common cause of dementia and affects around 10% of people over the age of 65 years (https://www.alz.org/alzheimers-dementia/facts-figures). Cellular senescence markers have been reported in neurons and astrocytes from patients with Alzheimer’s disease and in cultured human astrocytes exposed to β-amyloid153. DNA damage and DDR markers have been observed in models of Alzheimer’s disease154,155 and in neurons in postmortem brains from patients with the disease156. Neuronal cell death, a characteristic of Alzheimer’s disease, is thought to be a consequence of microglia senescence, and telomeres in microglia were reported to be shorter in patients with the disease than in healthy individuals157.
Genetic clearance of senescent cells or senolytic treatment in tauopathy mouse models mitigated cognitive decline and neurodegeneration158,159, which suggests that cellular senescence has a causative effect in this pathology. Similarly, treatment of an Alzheimer’s disease mouse model with a senolytic agent improved memory and learning ability160.
Although blood cells from patients with Alzheimer’s disease were found to have shorter telomeres, the role of telomere length is controversial161. In a mouse model of Alzheimer’s disease, shorter telomeres were found in blood cells but not in the hippocampus compared to wild-type mice162. Across patients with amyloid pathology, leukocyte telomere length positively correlated with better cognition and memory, whereas cognitive decline over 2 years was steeper in patients with the lowest quartile of telomere length, who also have a greater chance of developing dementia163. In support of a role of short telomeres in neurodegenerative diseases, late-generation telomerase-deficient mice recapitulate Alzheimer’s disease phenotypes164. Notably, AAV-mediated Tert expression ameliorated memory impairment164. Telomerase appears to have a protective role against tau pathological hyperphosphorylation in neurons from patients165 and against amyloid-β-induced cell death in embryonic mouse hippocampal neurons in vitro166. Conversely, β-amyloid can induce telomere shortening and inhibit telomerase activity167.
Parkinson’s disease
Parkinson’s disease is a progressive disorder in which movement is impaired. The disease affects more than 10 million people worldwide (https://www.parkinson.org/Understanding-Parkinsons/Statistics), and age is the main risk factor in both sporadic and familial forms. Senescent astrocytes have been detected in postmortem brain samples from patients with Parkinson’s disease168, and senescence of dopamine neurons has been proposed to contribute to disease pathogenesis169. Although there is no clear evidence of telomere length changes in patients with Parkinson’s disease170, mice with critically short telomeres recapitulate some features of the disease, including poor performance in neuromuscular coordination tests164. Pointing to a causal role of telomere dysfunction in Parkinson’s disease, telomerase activators led to decreased levels of pathological α-synuclein protein and improved motor symptoms in a mouse model of Parkinson’s disease171.
Age-related macular degeneration
Age-related macular degeneration (AMD) is an eye disease that affects the macula region in the retina and is the most common cause of irreversible blindness in older people worldwide, affecting about 67 million people in Europe alone172. Multiple senescence markers were detected in retinal tissues of AMD animal models and in patients with AMD173.
Different conclusions have been reached regarding the association between leukocyte telomere length and AMD174,175, but a double-blinded study of a small number of patients with early AMD showed that treatment with a telomerase activator significantly improved macular function176.
Reduced fertility
Currently, up to 25% of couples are affected by infertility (https://www.who.int/reproductivehealth/topics/infertility/burden/en/), and advancing age, especially in females, is associated with reproductive decline. Telomere shortening has been associated with reduced fertility in several ways177,178,179,180. Women undergoing in vitro fertilization tend to have shorter leukocyte telomere length than healthy individuals177, and among them, patients with polycystic ovary syndrome with low telomerase activity and short telomeres in granulosa cells, which support oocyte maturation, show an earlier onset of infertility178. Similarly, patients with premature ovarian insufficiency have shorter telomeres and reduced telomerase activity in leukocytes and granulosa cells than healthy individuals179. Short telomeres in human oocytes and polar bodies, which are extruded after meiotic divisions, are associated with aneuploidy in oocytes and early-stage embryos, which is probably due to aberrant chromosome segregation during meiosis180. According to a case report, a woman with dyskeratosis congenita, a telomeropathy associated with a reduced ovarian reserve, responded poorly to hormonal treatment before in vitro fertilization and her oocytes contained critically short telomeres181.
Among males, those with infertility tend to have shorter telomeres in sperm than unaffected men182. Short telomeres have been associated with poor sperm quality in males with normozoosperm183 and with sperm aneuploidy in men with idiopathic infertility184.
Causality of the associations mentioned above is supported by the observation that both sperm and oocytes from mice with critically short telomeres show decreased potential of fertilization and development185. Similar observations have been made in zebrafish and killifish186,187.
Conclusions and therapeutic opportunities
Here, we summarized age-related conditions that bear an often-unappreciated underlying cause in telomere dysfunction, either in the form of telomere shortening or telomere DNA damage. The identification of a cause can suggest therapeutic options. Preclinical activities in this direction include attempts to counteract telomere shortening or to counteract tDDR activation and cellular senescence. As senotherapies have been reviewed elsewhere188, we will not discuss them here.
To counteract telomere shortening, induction of telomerase activity, either after reactivation of endogenous TERT expression83,86 or by its exogenous delivery85,112,164, has been proposed. Because the TERT promoter responds to sex hormones, androgen therapy, based on the clinical use of danazol, a synthetic testosterone, has been part of clinical practice for many years in the treatment of aplastic anaemia with some efficacy83. A natural plant compound named TA-65 has been reported to boost telomerase activity and lengthen telomeres in mice189. Small-molecule inhibitors of PAPD5, a non-canonical poly(A) polymerase that destabilizes TERC RNA, have been shown to rescue TERC levels in induced pluripotent stem cells from patients with dyskeratosis congenita190. The clinical values of these approaches189,190 remain unclear, and they are unlikely to effectively treat patients who carry inactivating genetic mutations in telomerase genes or in genes necessary for its function.
Therapeutic benefits have been observed for AAV-mediated delivery of TERT in animal models of aplastic anaemia, IPF and Alzheimer’s disease164,191. So far, this is the most advanced preclinical therapeutic programme192, although the potential impact of the reported extra-telomeric activities of TERT193 remains to be addressed. Also, this approach cannot benefit patients carrying mutations in genes supporting telomerase activity. Although efficacy has been observed in cardiomyocytes194,195, it remains unclear whether inducing telomerase activity can be beneficial in non-proliferating cells, as in vitro studies indicate that telomerase expression is not able to prevent the accumulation of persistent double-strand breaks at telomeres after exogenous genotoxic insults19,20. Finally, in a clinical context, the use of immunogenic viral vectors such as AAV may reduce the possibility of repeated treatments.
As it is not telomere dysfunction per se that causes cellular senescence or apoptosis but the pathways that it engages, namely tDDR activation, another approach is to blunt the consequences of telomere dysfunction, that is, DNA damage signalling (Box 2). Although it may be dangerous to inhibit DNA damage signalling and repair throughout the genome, an opportunity may lie in the selective DDR inhibition at dysfunctional telomeres. The discovery that DDR activation depends on noncoding RNAs, such as damage-induced long non-coding RNAs (dilncRNA) and DNA damage-response RNAs (DDRNA), generated at exposed DNA ends, including dysfunctional telomeres, make such RNAs attractive targets for potential therapeutic interventions196. Antisense oligonucleotides (ASOs) are an emerging class of drugs targeting RNAs, with eight products presently on the market and many more in advanced clinical trials197. The reported ability of telomeric ASOs (tASOs) to blunt DDR activation specifically at telomeres in vivo in mice72,198,199 and to improve health span and lifespan in a progeria animal model199 provides preliminary but promising bases for their clinical application. An advantage of this approach is its broad activity independent from the genetic defects underlying telomere dysfunction, its ability to act at both critically short and damaged telomeres and in non-proliferating cells, and its nonviral delivery. Limitations of their use may lie in their tissue distribution, the need for safety evaluations and their relatively less-studied mechanism of action.
Of course, the different telomere biology of mice, the most commonly used animal model, with longer telomeres and widespread telomerase expression, compared with humans has to be considered when translating any potential therapy to the clinic.
However, for the benefit of patients, both TERT expression and tASO approaches, by acting upstream in the pathological cascade, are expected, and some have demonstrated, to make a promising impact on the many consequences of telomere dysfunction, such as impaired proliferation, inflammation and fibrosis.
Finally, geared with these tools, it is probable that researchers will be able to ascertain and broaden the impact of telomere dysfunction in more diseases.
References
Harley, C. B., Futcher, A. B. & Greider, C. W. Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 (1990).
de Lange, T. How telomeres solve the end-protection problem. Science 326, 948–952 (2009).
d’Adda di Fagagna, F. et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198 (2003).
Herbig, U., Jobling, W. A., Chen, B. P., Chen, D. J. & Sedivy, J. M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell 14, 501–513 (2004).
Cesare, A. J. & Karlseder, J. A three-state model of telomere control over human proliferative boundaries. Curr. Opin. Cell Biol. 24, 731–738 (2012).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011 (2017).
Schumacher, B., Pothof, J., Vijg, J. & Hoeijmakers, J. H. J. The central role of DNA damage in the ageing process. Nature 592, 695–703 (2021).
d’Adda di Fagagna, F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer 8, 512–522 (2008).
Karlseder, J., Broccoli, D., Dai, Y., Hardy, S. & de Lange, T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 283, 1321–1325 (1999).
Nassour, J. et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659–663 (2019).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J. & Kirkland, J. L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972 (2013).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Acosta, J. C. et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 (2008).
Victorelli, S. et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 38, e101982 (2019).
Razdan, N., Vasilopoulos, T. & Herbig, U. Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Aging Cell 17, e12838 (2018).
Di Micco, R., Krizhanovsky, V., Baker, D. & d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 22, 75–95 (2021).
Bae, N. S. & Baumann, P. A RAP1/TRF2 complex inhibits nonhomologous end-joining at human telomeric DNA ends. Mol. Cell 26, 323–334 (2007).
Fumagalli, M. et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 (2012).
Hewitt, G. et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 708–708 (2012).
Fumagalli, M., Rossiello, F., Mondello, C. & d’Adda di Fagagna, F. Stable cellular senescence is associated with persistent DDR activation. PLoS ONE 9, e110969 (2014).
Jacome Burbano, M. S., Cherfils-Vicini, J. & Gilson, E. Neutrophils: mediating TelOxidation and senescence. EMBO J. 40, e108164 (2021).
Fouquerel, E. et al. Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol. Cell 75, 117–130.e6 (2019).
von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344 (2002).
Lagnado, A. et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 40, e106048 (2021).
Barnes, R. P. et al. Telomeric 8-oxoguanine drives rapid premature senescence in the absence of telomere shortening. Preprint at bioRxiv https://doi.org/10.1101/2021.05.05.442662 (2021).
Fouquerel, E. et al. Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 23, 1092–1100 (2016).
Opresko, P. L., Fan, J., Danzy, S., Wilson, D. M. 3rd & Bohr, V. A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 33, 1230–1239 (2005).
Chakravarti, D., LaBella, K. A. & DePinho, R. A. Telomeres: history, health, and hallmarks of aging. Cell 184, 306–322 (2021).
Henriques, C. M., Carneiro, M. C., Tenente, I. M., Jacinto, A. & Ferreira, M. G. Telomerase is required for zebrafish lifespan. PLoS Genet. 9, e1003214 (2013).
Blasco, M. A. et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25–34 (1997).
Whittemore, K., Vera, E., Martínez-Nevado, E., Sanpera, C. & Blasco, M. A. Telomere shortening rate predicts species life span. Proc. Natl Acad. Sci. USA 116, 15122–15127 (2019).
Hemann, M. T., Strong, M. A., Hao, L.-Y. & Greider, C. W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 107, 67–77 (2001).
Birch, J. et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L1124–L1137 (2015).
Farr, J. N. et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 31, 1920–1929 (2016).
Anderson, R. et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 38, e100492 (2019).
Ogrodnik, M. et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 29, 1233 (2019).
Herbig, U., Ferreira, M., Condel, L., Carey, D. & Sedivy, J. M. Cellular senescence in aging primates. Science 311, 1257 (2006).
Martinez-Zamudio, R. I. et al. Senescence-associated β-galactosidase reveals the abundance of senescent CD8+ T cells in aging humans. Aging Cell 20, e13344 (2021).
Demanelis, K. et al. Determinants of telomere length across human tissues. Science 369, eaaz6876 (2020).
Ogrodnik, M. et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 8, 15691 (2017).
Bianchi, A. et al. Moderate exercise inhibits age-related inflammation, liver steatosis, senescence, and tumorigenesis. J. Immunol. 206, 904–916 (2021).
Correia-Melo, C. et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742 (2016).
Correia-Melo, C. et al. Rapamycin improves healthspan but not inflammaging in nfκb1–/– mice. Aging Cell 18, e12882 (2019).
Stout, M. B. et al. 17α-Estradiol alleviates age-related metabolic and inflammatory dysfunction in male mice without inducing feminization. J. Gerontol. A Biol. Sci. Med. Sci. 72, 3–15 (2017).
Roos, C. M. et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977 (2016).
Jurk, D. et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2, 4172 (2014).
Cassidy, L. D. et al. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat. Commun. 11, 307 (2020).
Holohan, B., Wright, W. E. & Shay, J. W. Cell biology of disease: telomeropathies: an emerging spectrum disorder. J. Cell Biol. 205, 289–299 (2014).
Schneider, J. L. et al. The aging lung: physiology, disease, and immunity. Cell 184, 1990–2019 (2021).
Lee, S. et al. Molecular programs of fibrotic change in aging human lung. Nat. Commun. 12, 6309 (2021).
Schafer, M. J. et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 8, 14532 (2017).
Alder, J. K. et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl Acad. Sci. USA 105, 13051–13056 (2008).
Cronkhite, J. T. et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 729–737 (2008).
Stuart, B. D. et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir. Med. 2, 557–565 (2014).
Newton, C. A. et al. Telomere length in patients with pulmonary fibrosis associated with chronic lung allograft dysfunction and post-lung transplantation survival. J. Heart Lung Transplant. 36, 845–853 (2017).
Armanios, M. Y. et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 356, 1317–1326 (2007).
Stuart, B. D. et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 47, 512–517 (2015).
Chen, R. et al. Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J. Biol. Chem. 290, 30813–30829 (2015).
Alder, J. K. et al. Telomere length is a determinant of emphysema susceptibility. Am. J. Respir. Crit. Care Med. 184, 904–912 (2011).
Povedano, J. M., Martinez, P., Flores, J. M., Mulero, F. & Blasco, M. A. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 12, 286–299 (2015).
Alder, J. K. et al. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl Acad. Sci. USA 112, 5099–5104 (2015).
Povedano, J. M. et al. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. eLife 7, e31299 (2018).
Pineiro-Hermida, S. et al. Telomerase treatment prevents lung profibrotic pathologies associated with physiological aging. J. Cell Biol. https://doi.org/10.1083/jcb.202002120 (2020).
James, S. L. et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1789–1858 (2018).
Ahmad, T. et al. Shelterin telomere protection protein 1 reduction causes telomere attrition and cellular senescence via sirtuin 1 deacetylase in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 56, 38–49 (2017).
Houben, J. M. et al. Telomere shortening in chronic obstructive pulmonary disease. Respir. Med. 103, 230–236 (2009).
Stanley, S. E. et al. Telomerase mutations in smokers with severe emphysema. J. Clin. Invest. 125, 563–570 (2015).
Birch, J. et al. Telomere dysfunction and senescence-associated pathways in bronchiectasis. Am. J. Respir. Crit. Care Med. 193, 929–932 (2016).
Michalski, J. E., Kurche, J. S. & Schwartz, D. A. From ARDS to pulmonary fibrosis: the next phase of the COVID-19 pandemic? Transl. Res. https://doi.org/10.1016/j.trsl.2021.09.001 (2021).
Aviv, A. Telomeres and COVID-19. FASEB J. 34, 7247–7252 (2020).
Sepe, S. et al. DNA damage response at telomeres boosts the transcription of SARS-CoV-2 receptor ACE2 during aging. EMBO Rep. https://doi.org/10.15252/embr.202153658 (2021).
Vaht, K. et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica 102, 1683–1690 (2017).
Yamaguchi, H. et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood 102, 916–918 (2003).
Yamaguchi, H. et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med. 352, 1413–1424 (2005).
Savage, S. A. et al. Genetic variation in telomeric repeat binding factors 1 and 2 in aplastic anemia. Exp. Hematol. 34, 664–671 (2006).
Guo, Y. et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood 124, 2767–2774 (2014).
Savage, S. A. et al. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 82, 501–509 (2008).
Brümmendorf, T. H., Maciejewski, J. P., Mak, J., Young, N. S. & Lansdorp, P. M. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood 97, 895–900 (2001).
Scheinberg, P. et al. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. JAMA 304, 1358–1364 (2010).
Sakaguchi, H. et al. Peripheral blood lymphocyte telomere length as a predictor of response to immunosuppressive therapy in childhood aplastic anemia. Haematologica 99, 1312–1316 (2014).
Gadalla, S. M. et al. Association between donor leukocyte telomere length and survival after unrelated allogeneic hematopoietic cell transplantation for severe aplastic anemia. JAMA 313, 594–602 (2015).
Townsley, D. M. et al. Danazol treatment for telomere diseases. N. Engl. J. Med. 374, 1922–1931 (2016).
Beier, F., Foronda, M., Martinez, P. & Blasco, M. A. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood 120, 2990–3000 (2012).
Bär, C. et al. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood 127, 1770–1779 (2016).
Bär, C., Huber, N., Beier, F. & Blasco, M. A. Therapeutic effect of androgen therapy in a mouse model of aplastic anemia produced by short telomeres. Haematologica 100, 1267–1274 (2015).
Zhou, T. et al. Myelodysplastic syndrome: an inability to appropriately respond to damaged DNA? Exp. Hematol. 41, 665–674 (2013).
Lubeck, D. P. et al. Systematic literature review of the global incidence and prevalence of myelodysplastic syndrome and acute myeloid leukemia. Blood 128, 5930–5930 (2016).
Savage, S. Beginning at the ends: telomeres and human disease [version 1; peer review: 4 approved]. F1000Res. https://doi.org/10.12688/f1000research.14068.1 (2018).
Sashida, G. et al. Telomere dynamics in myelodysplastic syndrome determined by telomere measurement of marrow metaphases. Clin. Cancer Res. 9, 1489–1496 (2003).
Colla, S. et al. Telomere dysfunction drives aberrant hematopoietic differentiation and myelodysplastic syndrome. Cancer Cell 27, 644–657 (2015).
Eckhardt, B. A. et al. Accelerated osteocyte senescence and skeletal fragility in mice with type 2 diabetes. JCI Insight 5, e135236 (2020).
Gao, Z. et al. Age-associated telomere attrition in adipocyte progenitors predisposes to metabolic disease. Nat. Metab. 2, 1482–1497 (2020).
Wiemann, S. U. et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 16, 935–942 (2002).
Kitada, T., Seki, S., Kawakita, N., Kuroki, T. & Monna, T. Telomere shortening in chronic liver diseases. Biochem. Biophys. Res. Commun. 211, 33–39 (1995).
Hartmann, D. et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology 53, 1608–1617 (2011).
Rudolph, K. L., Chang, S., Millard, M., Schreiber-Agus, N. & DePinho, R. A. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 287, 1253–1258 (2000).
Sasaki, M., Ikeda, H., Yamaguchi, J., Nakada, S. & Nakanuma, Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatology 48, 186–195 (2008).
Moncsek, A. et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/−) mice. Hepatology 67, 247–259 (2018).
Ikeda, H. et al. Large cell change of hepatocytes in chronic viral hepatitis represents a senescent-related lesion. Hum. Pathol. 40, 1774–1782 (2009).
Wilson, C. L. et al. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 6, 6818 (2015).
Jeanclos, E. et al. Shortened telomere length in white blood cells of patients with IDDM. Diabetes 47, 482–486 (1998).
Narasimhan, A., Flores, R. R., Robbins, P. D. & Niedernhofer, L. J. Role of cellular senescence in type II diabetes. Endocrinology https://doi.org/10.1210/endocr/bqab136 (2021).
Foreman, K. J. et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet 392, 2052–2090 (2018).
Boniewska-Bernacka, E., Pańczyszyn, A. & Klinger, M. Telomeres and telomerase in risk assessment of cardiovascular diseases. Exp. Cell. Res. 397, 112361 (2020).
Dookun, E. et al. Clearance of senescent cells during cardiac ischemia–reperfusion injury improves recovery. Aging Cell 19, e13249 (2020).
Chimenti, C. et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ. Res. 93, 604–613 (2003).
Chang, A. C. Y. et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl Acad. Sci. USA 115, 9276–9281 (2018).
Chatterjee, S. et al. Leukocyte telomere length correlates with hypertrophic cardiomyopathy severity. Sci. Rep. 8, 11227 (2018).
Wong, L. S. et al. Telomere biology in cardiovascular disease: the TERC–/– mouse as a model for heart failure and ageing. Cardiovasc. Res. 81, 244–252 (2009).
Leri, A. et al. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 22, 131–139 (2003).
Bär, C. et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat. Commun. 5, 5863 (2014).
Barquera, S. et al. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch. Med. Res. 46, 328–338 (2015).
Samani, N. J., Boultby, R., Butler, R., Thompson, J. R. & Goodall, A. H. Telomere shortening in atherosclerosis. Lancet 358, 472–473 (2001).
Matthews, C. et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ. Res. 99, 156–164 (2006).
Wang, J. et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 132, 1909–1919 (2015).
Uryga, A. K. et al. Telomere damage promotes vascular smooth muscle cell senescence and immune cell recruitment after vessel injury. Commun. Biol. https://doi.org/10.1038/s42003-021-02123-z (2021).
Childs, B. G. et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477 (2016).
McCulloch, K., Litherland, G. J. & Rai, T. S. Cellular senescence in osteoarthritis pathology. Aging Cell 16, 210–218 (2017).
Coryell, P. R., Diekman, B. O. & Loeser, R. F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 17, 47–57 (2021).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Novais, E. J. et al. Long-term treatment with senolytic drugs dasatinib and quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 12, 5213 (2021).
Poonpet, T. et al. Association between leukocyte telomere length and angiogenic cytokines in knee osteoarthritis. Int. J. Rheum. Dis. 21, 118–125 (2018).
Zhai, G. et al. Reduction of leucocyte telomere length in radiographic hand osteoarthritis: a population-based study. Ann. Rheum. Dis. 65, 1444–1448 (2006).
Sibille, K. T. et al. Accelerated aging in adults with knee osteoarthritis pain: consideration for frequency, intensity, time, and total pain sites. Pain Rep. 2, e591 (2017).
Price, J. S. et al. The role of chondrocyte senescence in osteoarthritis. Aging Cell 1, 57–65 (2002).
Harbo, M. et al. The relationship between ultra-short telomeres, aging of articular cartilage and the development of human hip osteoarthritis. Mech. Ageing Dev. 134, 367–372 (2013).
Sozen, T., Ozisik, L. & Basaran, N. C. An overview and management of osteoporosis. Eur. J. Rheumatol. 4, 46–56 (2017).
Pignolo, R. J., Law, S. F. & Chandra, A. Bone aging, cellular senescence, and osteoporosis. JBMR Plus 5, e10488 (2021).
Farr, J. N. & Khosla, S. Cellular senescence in bone. Bone 121, 121–133 (2019).
Valdes, A. M. et al. Telomere length in leukocytes correlates with bone mineral density and is shorter in women with osteoporosis. Osteoporos. Int. 18, 1203–1210 (2007).
Wang, H. et al. Impairment of osteoblast differentiation due to proliferation-independent telomere dysfunction in mouse models of accelerated aging. Aging Cell 11, 704–713 (2012).
Brennan, T. A. et al. Mouse models of telomere dysfunction phenocopy skeletal changes found in human age-related osteoporosis. Dis. Model. Mech. 7, 583–592 (2014).
Sturmlechner, I., Durik, M., Sieben, C. J., Baker, D. J. & van Deursen, J. M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 13, 77–89 (2017).
Wang, W. J., Cai, G. Y. & Chen, X. M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget 8, 64520–64533 (2017).
Schroth, J., Thiemermann, C. & Henson, S. M. Senescence and the aging immune system as major drivers of chronic kidney disease. Front. Cell Dev. Biol. 8, 564461 (2020).
Kishi, S. et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Invest. 129, 4797–4816 (2019).
Hayashi, K. et al. Association of glomerular DNA damage and DNA methylation with one-year eGFR decline in IgA nephropathy. Sci. Rep. 10, 237 (2020).
Ramírez, R. et al. Stress-induced premature senescence in mononuclear cells from patients on long-term hemodialysis. Am. J. Kidney Dis. 45, 353–359 (2005).
Tsirpanlis, G. et al. Telomerase activity is decreased in peripheral blood mononuclear cells of hemodialysis patients. Am. J. Nephrol. 26, 91–96 (2006).
Famulski, K. S. & Halloran, P. F. Molecular events in kidney ageing. Curr. Opin. Nephrol. Hypertens. 14, 243–248 (2005).
Fazzini, F. et al. Results from the German Chronic Kidney Disease (GCKD) study support association of relative telomere length with mortality in a large cohort of patients with moderate chronic kidney disease. Kidney Int. 98, 488–497 (2020).
Raschenberger, J. et al. Association of relative telomere length with progression of chronic kidney disease in two cohorts: effect modification by smoking and diabetes. Sci. Rep. 5, 11887 (2015).
Domański, L. et al. Effect of delayed graft function, acute rejection and chronic allograft dysfunction on kidney allograft telomere length in patients after transplantation: a prospective cohort study. BMC Nephrol. 16, 23 (2015).
Schildhorn, C. et al. Renal phenotype of young and old telomerase-deficient mice. Mech. Ageing Dev. 150, 65–73 (2015).
Westhoff, J. H. et al. Telomere shortening reduces regenerative capacity after acute kidney injury. J. Am. Soc. Nephrol. 21, 327–336 (2010).
Baker, D. J. et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Quimby, J. M. & Lunn, K. F. Mirtazapine as an appetite stimulant and anti-emetic in cats with chronic kidney disease: a masked placebo-controlled crossover clinical trial. Vet. J. 197, 651–655 (2013).
Saraswati, S., Martínez, P., Graña-Castro, O. & Blasco, M. A. Short and dysfunctional telomeres sensitize the kidneys to develop fibrosis. Nat. Aging 1, 269–283 (2021).
Jurk, D. et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004 (2012).
Fielder, E. et al. Anti-inflammatory treatment rescues memory deficits during aging in nfkb1−/− mice. Aging Cell 19, e13188 (2020).
Ogrodnik, M. et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 20, e13296 (2021).
Saez-Atienzar, S. & Masliah, E. Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat. Rev. Neurosci. 21, 433–444 (2020).
Fielder, E., von Zglinicki, T. & Jurk, D. The DNA damage response in neurons: die by apoptosis or survive in a senescence-like state? J. Alzheimers Dis. 60, S107–S131 (2017).
Thadathil, N. et al. DNA double-strand break accumulation in Alzheimer’s disease: evidence from experimental models and postmortem human brains. Mol. Neurobiol. 58, 118–131 (2021).
Shanbhag, N. M. et al. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 7, 77 (2019).
Flanary, B. E., Sammons, N. W., Nguyen, C., Walker, D. & Streit, W. J. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 10, 61–74 (2007).
Bussian, T. J. et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 (2018).
Musi, N. et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17, e12840 (2018).
Zhang, P. et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728 (2019).
Boccardi, V., Pelini, L., Ercolani, S., Ruggiero, C. & Mecocci, P. From cellular senescence to Alzheimer’s disease: the role of telomere shortening. Ageing Res. Rev. 22, 1–8 (2015).
Martínez-González, K., Islas-Hernández, A., Martínez-Ezquerro, J. D., Bermúdez-Rattoni, F. & Garcia-delaTorre, P. Telomere length and oxidative stress variations in a murine model of Alzheimer’s disease progression. Eur. J. Neurosci. 52, 4863–4874 (2020).
Koh, S. H. et al. Telomere shortening reflecting physical aging is associated with cognitive decline and dementia conversion in mild cognitive impairment due to Alzheimer’s disease. Aging (Albany NY) 12, 4407–4423 (2020).
Whittemore, K. et al. Telomerase gene therapy ameliorates the effects of neurodegeneration associated to short telomeres in mice. Aging (Albany NY) 11, 2916–2948 (2019).
Spilsbury, A., Miwa, S., Attems, J. & Saretzki, G. The role of telomerase protein TERT in Alzheimer’s disease and in tau-related pathology in vitro. J. Neurosci. 35, 1659–1674 (2015).
Zhu, H., Fu, W. & Mattson, M. P. The catalytic subunit of telomerase protects neurons against amyloid β-peptide-induced apoptosis. J. Neurochem. 75, 117–124 (2000).
Wang, J. et al. New insights in amyloid beta interactions with human telomerase. J. Am. Chem. Soc. 137, 1213–1219 (2015).
Chinta, S. J. et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 22, 930–940 (2018).
Riessland, M. et al. Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons. Cell Stem Cell 25, 514–530.e8 (2019).
Forero, D. A. et al. Telomere length in Parkinson’s disease: a meta-analysis. Exp. Gerontol. 75, 53–55 (2016).
Wan, T., Weir, E. J., Johnson, M., Korolchuk, V. I. & Saretzki, G. C. Increased telomerase improves motor function and alpha-synuclein pathology in a transgenic mouse model of Parkinson’s disease associated with enhanced autophagy. Prog. Neurobiol. 199, 101953 (2021).
Li, J. Q. et al. Prevalence and incidence of age-related macular degeneration in Europe: a systematic review and meta-analysis. Br. J. Ophthalmol. 104, 1077–1084 (2020).
Lee, K. S., Lin, S., Copland, D. A., Dick, A. D. & Liu, J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflammation 18, 32 (2021).
Immonen, I., Seitsonen, S., Saionmaa, O. & Fyhrquist, F. Leucocyte telomere length in age-related macular degeneration. Acta Ophthalmol. 91, 453–456 (2013).
Weng, X. et al. Leukocyte telomere length is associated with advanced age-related macular degeneration in the Han Chinese population. Exp. Gerontol. 69, 36–40 (2015).
Dow, C. T. & Harley, C. B. Evaluation of an oral telomerase activator for early age-related macular degeneration—a pilot study. Clin. Ophthalmol. 10, 243–249 (2016).
Czamanski-Cohen, J. et al. Cell-free DNA and telomere length among women undergoing in vitro fertilization treatment. J. Assist. Reprod. Genet. 32, 1697–1703 (2015).
Li, Y. et al. Telomere length is short in PCOS and oral contraceptive does not affect the telomerase activity in granulosa cells of patients with PCOS. J. Assist. Reprod. Genet. 34, 849–859 (2017).
Xu, X. et al. Impaired telomere length and telomerase activity in peripheral blood leukocytes and granulosa cells in patients with biochemical primary ovarian insufficiency. Hum. Reprod. 32, 201–207 (2017).
Treff, N. R., Su, J., Taylor, D. & Scott, R. T. Jr. Telomere DNA deficiency is associated with development of human embryonic aneuploidy. PLoS Genet. 7, e1002161 (2011).
Robinson, L. G. Jr et al. Impaired reproductive function and fertility preservation in a woman with a dyskeratosis congenita. J. Assist. Reprod. Genet. 37, 1221–1225 (2020).
Thilagavathi, J. et al. Analysis of sperm telomere length in men with idiopathic infertility. Arch. Gynecol. Obstet. 287, 803–807 (2013).
Rocca, M. S. et al. Sperm telomere length as a parameter of sperm quality in normozoospermic men. Hum. Reprod. 31, 1158–1163 (2016).
Cariati, F. et al. Investigation of sperm telomere length as a potential marker of paternal genome integrity and semen quality. Reprod. Biomed. Online 33, 404–411 (2016).
Liu, L., Blasco, M., Trimarchi, J. & Keefe, D. An essential role for functional telomeres in mouse germ cells during fertilization and early development. Dev. Biol. 249, 74–84 (2002).
Anchelin, M. et al. Premature aging in telomerase-deficient zebrafish. Dis. Model. Mech. 6, 1101–1112 (2013).
Harel, I. et al. A platform for rapid exploration of aging and diseases in a naturally short-lived vertebrate. Cell 160, 1013–1026 (2015).
Robbins, P. D. et al. Senolytic drugs: reducing senescent cell viability to extend health span. Annu. Rev. Pharmacol. Toxicol. 61, 779–803 (2021).
de Jesus, B. B. et al. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell 10, 604–621 (2011).
Nagpal, N. et al. Small-molecule PAPD5 inhibitors restore telomerase activity in patient stem cells. Cell Stem Cell 26, 896–909.e8 (2020).
Martinez, P. & Blasco, M. A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 216, 875–887 (2017).
Bernardes de Jesus, B. et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 4, 691–704 (2012).
Martinez, P. & Blasco, M. A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 11, 161–176 (2011).
Yeh, J. K., Lin, M. H. & Wang, C. Y. Telomeres as therapeutic targets in heart disease. JACC Basic Transl. Sci. 4, 855–865 (2019).
Chatterjee, S. et al. Telomerase therapy attenuates cardiotoxic effects of doxorubicin. Mol. Ther. 29, 1395–1410 (2021).
Michelini, F. et al. From “cellular” RNA to “smart” RNA: multiple roles of RNA in genome stability and beyond. Chem. Rev. 118, 4365–4403 (2018).
Crooke, S. T., Baker, B. F., Crooke, R. M. & Liang, X. H. Antisense technology: an overview and prospectus. Nat. Rev. Drug Discov. https://doi.org/10.1038/s41573-021-00162-z (2021).
Rossiello, F. et al. DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat. Commun. 8, 13980 (2017).
Aguado, J. et al. Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson–Gilford progeria syndrome. Nat. Commun. 10, 4990 (2019).
Lai, T. P., Wright, W. E. & Shay, J. W. Comparison of telomere length measurement methods. Philos. Trans. R Soc. Lond. B Biol. Sci. https://doi.org/10.1098/rstb.2016.0451 (2018).
Takai, H., Smogorzewska, A. & de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556 (2003).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009).
Michelini, F. et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 19, 1400–1411 (2017).
Sharma, S. et al. MRE11–RAD50–NBS1 complex is sufficient to promote transcription by RNA polymerase II at double-strand breaks by melting DNA ends. Cell Rep. 34, 108565 (2021).
Francia, S. et al. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 488, 231–235 (2012).
Pessina, F. et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 21, 1286–1299 (2019).
Acknowledgements
We thank S. Sepe, C. Tripodo and P. Kordowitzki for critically reading the manuscript. Funding in D.J.’s laboratory was provided by grants R01AG 68182-1, the Ted Nash Long Life Foundation and P30DK084567. Funding in J.F.P.’s laboratory was provided by grants 1R01AG 68048-1, 1UG3CA 268103-1, UL1TR 02377-1, the Ted Nash Long Life Foundation and P01 AG 62413-1. F.d’A.d.F.’s laboratory is supported by an ERC advanced grant (TELORNAGING–835103), AIRC-IG (21762), Telethon (GGP17111), AIRC 5×1000 (21091), an ERC PoC grant (FIREQUENCER–875139), Progetti di Ricerca di Interesse Nazionale (PRIN) 2015 “ATR and ATM-mediated control of chromosome integrity and cell plasticity”, Progetti di Ricerca di Interesse Nazionale (PRIN) 2017 “RNA and genome Instability”, Progetto AriSLA 2021 “DDR & ALS”, POR FESR 2014-2020 Regione Lombardia (InterSLA project), FRRB–Fondazione Regionale per la Ricerca Biomedica–under the frame of EJP RD, the European Joint Programme on Rare Diseases with funding from the European Union’s Horizon 2020 research and innovation programme under the EJP RD COFUND-EJP number 825575. Figure 1 was originally created with BioRender.com.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
F.R. and F.d’A.d.F. are inventors on the patent applications PCT/EP2013/059753 and PCT/EP2016/068162. J.F.P. and D.J. declare no competing interests. I.F.O.M. has applied for European and US patent applications (EP 13721970.5, US 14/400,131 and 15/476,800), covering the use of antisense oligonucleotides targeting RNA species generated at the site of DNA damage, and for AU, BR, CA, CN, EA, EP, JP, KR, MX and US patent applications (AU2016300141, BR1120180017825, CA 2993128, CN 2016800566045, EA 201890379, EP 16750690.6, JP 2018504223, KR 20187005777, MX/A/2018001126, US 15/748,133 and US 17/065,409), covering the use of antisense oligonucleotides for the treatment of cancer characterized by alternative lengthening of telomeres and non-cancer conditions associated with telomere dysfunction, that list F.d’A.d.F. and F.R.
Peer review
Peer review information
Nature Cell Biology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rossiello, F., Jurk, D., Passos, J.F. et al. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol 24, 135–147 (2022). https://doi.org/10.1038/s41556-022-00842-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-022-00842-x
This article is cited by
-
Silencing PinX1 enhances radiosensitivity and antitumor-immunity of radiotherapy in non-small cell lung cancer
Journal of Translational Medicine (2024)
-
Insight into telomere regulation: road to discovery and intervention in plasma drug-protein targets
BMC Genomics (2024)
-
A single-cell atlas of lung homeostasis reveals dynamic changes during development and aging
Communications Biology (2024)
-
Cell senescence in liver diseases: pathological mechanism and theranostic opportunity
Nature Reviews Gastroenterology & Hepatology (2024)
-
mTert induction in p21-positive cells counteracts capillary rarefaction and pulmonary emphysema
EMBO Reports (2024)
