
Abstract
Viral infection perturbs host cells and can be used to uncover regulatory mechanisms controlling cellular responses and susceptibility to infections. Using cell biological, biochemical, and genetic tools, we reveal that influenza A virus (IAV) infection induces global transcriptional defects at the 3′ ends of active host genes and RNA polymerase II (RNAPII) run-through into extragenic regions. Deregulated RNAPII leads to expression of aberrant RNAs (3′ extensions and host-gene fusions) that ultimately cause global transcriptional downregulation of physiological transcripts, an effect influencing antiviral response and virulence. This phenomenon occurs with multiple strains of IAV, is dependent on influenza NS1 protein, and can be modulated by SUMOylation of an intrinsically disordered region (IDR) of NS1 expressed by the 1918 pandemic IAV strain. Our data identify a strategy used by IAV to suppress host gene expression and indicate that polymorphisms in IDRs of viral proteins can affect the outcome of an infection.
Similar content being viewed by others


Main
IAV is a negative-sense single-stranded RNA virus. The IAV genome is organized into eight segments, short negative-sense noncoding ‘mini RNA chromosomes’ that are packaged inside virions after the virus completes a life cycle in infected cells. IAV, like all viruses, is an obligate parasite, because it requires cellular functions to multiply and disseminate. However, unlike most other RNA viruses, it replicates in the nucleus, a feature that places evolutionary pressure on a number of viral proteins to coopt or interfere with host chromatin-based regulatory processes in infected cells1,2.
All IAV strains encode a nonstructural protein called NS1, whose function is to antagonize host antiviral responses3. NS1-mediated host antagonism occurs through multiple mechanisms, including inhibition of sensing of the virus4 and suppression of host functions that are detrimental to the virus, such as host translation5 and inflammatory-gene expression1,6. NS1 protein sequences differ among strains. Only a few domains of NS1, such as the RNA-binding domain7 and the C-terminal domain, have been identified as pathogenic determinants in multiple strains8. Interestingly, the C terminus is among the most divergent sequences within NS1, and it is unstructured1,9,10. These features are reminiscent of IDRs, or unstructured domains often referred to as short linear modules (SLIMs), peptide motifs, or linear domains11. Pathogens often use IDRs to promote novel interactions to adapt to new hosts and to enhance transmission and virulence12.
In this work, we used influenza 1918 virus13,14, a pandemic IAV that has caused the worst pandemic known to date15, as a case study to identify novel features of IAV–host interaction and adaptation mediated by IDRs. We found that 1918 IAV encodes an NS1 that contains a unique C-terminal domain composed of a small ubiquitin-like modifier (SUMO) site embedded in a PDZ-binding domain (PDZBD) consensus sequence. This extended SUMO consensus or SUMO inside PDZBD (SUP) domain predicts site-specific SUMOylation of 1918 NS1 during infection. Using viruses bearing 1918 NS1, we discovered that IAV elicits global deregulation of RNAPII transcription termination by impairing 3′-end cleavage and termination. This effect is augmented by NS1 SUMOylation, which increases the partitioning of NS1 in nuclear granules containing 3′-end-cleavage factors. Termination defects lead to RNAPII traveling through intergenic regions and causing formation of aberrant mRNAs, thus ultimately resulting in global transcriptional downregulation. Analysis of host transcriptional responses to nonpandemic IAV indicated that IAV-induced 3′-end-termination defects are a general feature of IAV infection that is dependent on NS1 expression and is modulated by post-translational modification of the unstructured region of NS1. Our data support the idea that the study of viral polymorphic proteins can reveal important molecular events occurring during infection.
Results
A unique domain present in the NS1 of 1918 influenza virus
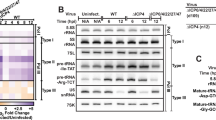
Along with its structural proteins, IAV encodes the nonstructural protein NS1, which is an antagonist of cellular antiviral responses16,17. We surveyed highly pathogenic IAV strains for the presence of unique protein domains and found a unique sequence present in the C-terminal domain of NS1 from the Brevig/mission strain (Fig. 1a and Supplementary Fig. 1a). This strain is responsible for the pandemic influenza outbreak that occurred in 1918 (ref. 14). Interestingly, different strains of IAV bear divergent NS1 C termini (NS1 tail) (Fig. 1b and Supplementary Fig. 1a,b), and NS1 tails have been linked to host tropism and virulence8,18. The C terminus of NS1 encoded by the 1918 pandemic strain (hereafter referred to as NS1) ends with the sequence 226-IKSEV-230 (Fig. 1a). This sequence is a SUMOylation consensus site (ψKxE) embedded in a PDZBD (Fig. 1a). We examined whether this domain might be a functional acceptor site for SUMO conjugation. K227 was found to be modified by SUMO, whereas the only other canonical SUMO consensus site in position 68–71 was not (Fig. 1c). To verify whether the domain might confer SUMOylation to a noncognate NS1, we generated chimeric NS1 proteins bearing residues 1–225 of H3N2 NS1 (A/New York/739/1994) and residues 226–230 of 1918 or 1918-like sequences from avian isolates. This tail swapping conferred SUMOylation in trans (Fig. 1d). We then assessed whether NS1 might be modified during infection. To do so, we generated, through reverse genetics19, a chimeric virus with the first seven segments of the A/Puerto Rico/8/1934 H1N1 strain (PR8) and the eighth segment of the 1918 strain (reassortant 7+1, hereafter referred to as NS1 virus). We used this virus to infect A549 human epithelial cells. Immunoprecipitation (IP) of NS1 followed by western blotting (WB) with anti-NS1 and anti-SUMO2/3 revealed that NS1 was SUMOylated during infection (Fig. 1e). This experiment also suggested that NS1 SUMOylation is controlled by SUMO-deconjugating enzymes, because the NS1 modification was lost after omission of the SUMO-protease inhibitor N-ethylmaleimide (NEM) from the cell extracts. Overall, our results indicated that the C terminus of NS1 from the 1918 strain was modified by SUMO within the consensus sequence embedded in a PDZBD. The modified NS1 domain is short and present in an unstructured region of the protein1,9, and it can be transferred to other substrates to confer SUMO conjugation. All these features are characteristics of SLIMs and peptide domains20. We therefore refer to this domain as the SUP SLIM or SUP peptide domain.

a, Top, Venn diagram of influenza virus NS1 proteins from human isolates and the presence of a SUMO site in the C-terminal (term) domain (tail). Bottom, amino acid sequence of the unique NS1 tail from A/Brevig Mission/1/1918 (H1N1) bearing a SUMOylation site and PDZ-domain ligand site. b, Conservation plot of amino acid sequences of NS1 among different viral isolates and hosts. The color coding represents the difference between the conservation score and the average value of the score across the protein. Protein domains of NS1 are shown at bottom. c, Ectopic expression of SUMO2 and the indicated NS1 proteins (WT NS1 and the SUMO-consensus mutants at positions K70 and K227) in A549 cells. IP with anti-FLAG and WB analysis with anti-NS1 and anti-SUMO2/3 are shown. d, IP with anti-NS1 and WB with anti-NS1 and anti-SUMO2/3 in A549 cells transfected with the indicated NS1 proteins. The tail swapping of H3N2 NS1 was performed with the sequences from the indicated viruses, all of human origin, except Hong Kong 1992 and Indonesia 2005, which are avian viruses. e, IP and anti-NS1 WB of whole cellular extract from A549 cells infected with the reassortant NS1 virus bearing segment 8 encoding 1918 NS1 (additional data in Fig. 2b). NEM, SUMO peptidase inhibitor. Uncropped blot images are shown in Supplementary Dataset 1.
Rescue and analysis of 1918 NS1-SUMO fusion virus
The observation that the PDZBD was modified by a bulky modification (SUMO, with molecular weight >10 KDa) suggests that SUMO conjugation might alter the PDZBD–PDZ protein interaction. Leveraging the PDZBD’s location at the C terminus of NS1, we engineered an NS1 covalently fused with SUMO (NS1-SUMO) (Fig. 2a). This strategy has been used to identify ubiquitin-dependent mechanisms in histone biology21. We also mutated K227 to prevent SUMO conjugation (protein denoted NS1-KR) (Fig. 2a). These mutations did not change the NS1 nuclear localization (Supplementary Fig. 2a). Using biolayer interferometry (BLI), we then determined the Kd of the interaction between a prototypical PDZ-domain-containing protein, PSD95, and the three NS1 variants (wild type (WT), R227, and SUMO). Our results showed that the Kd values of the three NS1 proteins with PSD95 were on the same order of magnitude (1.60 for NS1, 2.30 for NS1-KR, and 0.63 μM for NS1-SUMO; Fig. 2a). These results were corroborated by pulldown assays (Supplementary Fig. 2b) and by modeling of NS1 binding with three PDZ proteins (PSD95, GIPC2, and DVL1; Supplementary Fig. 2c–e and Supplementary Table 1). Overall, our results indicated that NS1 retains PDZ interactions independently of its conjugation with SUMO and suggested that SUMOylation has functions other than altering PDZ–PDZ interactions.

a, Top, schematic representations of the recombinant NS1, NS1-KR, and NS1-SUMO proteins used for BLI. Bottom, spectra showing BLI measurements of the interactions between recombinant PSD95 and WT NS1, or NS1 mutants (NS1-KR and NS1-SUMO). b, Schematic representation of WT PR8, the reassortant virus (7+1) PR8 NS1 bearing seven segments of WT PR8 and segment 8 of the 1918 influenza virus, and the reassortant virus (7+1) PR8 NS1-SUMO bearing segment 8 of the 1918 influenza virus encoding a fusion protein between NS1 and SUMO2. c, Growth curves of the indicated viruses after infection of MDCK cells at MOI = 1 (n = 6). Data are plotted as mean ± s.e.m. of n = 3 infection experiments. Plaque size from one representative experiment is shown on the right. d, Hierarchical clustering of genes with significant changes in expression (false discovery rate q <0.001) between infections with NS1 or NS1-SUMO virus at the indicated time points (hpi). Rows show the log fold change in expression (IDs not shown; color key at bottom), and columns represent different experimental conditions. Inf, infected; un, uninfected. e, Scatter plot of cellular gene expression changes in A549 cells at 6 hpi (top) or 12 hpi (bottom) with NS1 virus or NS1-SUMO virus, relative to mock-infected (mock) cells (log2 ratio). Solid and dotted lines correspond to the regression line and 95% confidence interval, respectively. Source data for a and c are available in Supplementary Dataset 2.
To probe the intrinsic role of SUMOylation of the 1918 SUP domain during infection, we engineered two viruses expressing NS1 and NS1-SUMO (hereafter referred to as NS1-SUMO virus) (Fig. 2b). We predicted that generation of the NS1-SUMO virus would be safe because fusion of SUMO to NS1 would result in decreased virulence, similarly to other NS1 fusion viruses22. For the opposite reason, the KR-mutant virus was not rescued. Indeed, as demonstrated by hemagglutination assays, the fusion of SUMO to NS1 severely impaired plaque formation (Fig. 2c). Plaque sequencing indicated that the delayed growth of the NS1-SUMO virus at low multiplicity of infection (MOI) was associated with a reversion to its WT virotype (Supplementary Fig. 2f). The decreased virulence of NS1-SUMO virus may have been driven by the formation of many defective particles, impaired assembly, inefficient cell-to-cell spreading, or indirect effects caused by SUMOylation affecting NS1 levels (Supplementary Fig. 3a).
Despite having lower pathogenicity, the NS1-SUMO virus still retained the ability to transiently infect at high MOIs. This feature allowed us to monitor early events independent of viral budding, egress, and reinfection of nearby cells. We therefore compared NS1 and NS1-SUMO viruses in experiments conducted at high MOI. A549 cells were infected with NS1 or NS1-SUMO viruses at an MOI of 3, and total RNA was extracted at 6 and 12 h postinfection (hpi). RNA-seq analysis showed that NS1 and NS1-SUMO RNA levels were comparable (Supplementary Fig. 3b). Both viruses caused altered host gene expression of approximately 1,000 shared genes (Fig. 2d,e), including upregulation of genes that are part of the host defense to infection, in agreement with the high induction of cellular-response genes after 1918 infection23. Notably, infection caused the downregulation of most transcriptionally active genes, which accounted for approximately two-thirds of genes whose expression was altered during infection (Fig. 2d). Compared with the NS1 virus, the NS1-SUMO virus displayed a higher magnitude of gene expression changes in both induced and suppressed genes at both 6 and 12 hpi (Fig. 2d). Importantly, both viruses upregulated and downregulated the same set of genes (Fig. 2e). Overall, these results suggest that NS1 affects the magnitude of induction and suppression of host genes.
SUMOylation controls NS1 oligomeric assembly and partitioning in RNA granules
NS1 binds RNA24 and forms polymers in vitro25,26. Therefore, we investigated a potential role of NS1 in interaction with RNA granules. These compartments are nucleated by RNA-binding and aggregation-prone proteins27,28 that are regulated by post-translational modifications, such as phosphorylation29, SUMOylation30, and possibly many more. SUMO enhances the valency and strength of interaction between scaffold and client proteins of RNA granules30. We therefore performed semidenaturing detergent agarose gel electrophoresis (SDD-AGE), which allows for the resolution of high-molecular-weight complexes formed as a result of protein multimerization, a feature that can influence partitioning into RNA granules. Results from this assay indicated that NS1 forms high-molecular-weight complexes during infection, and this feature is enhanced by SUMO fusion (Fig. 3a). We then took advantage of the synthetic compound biotinylated isoxazole (B-isox), which has been shown to selectively precipitate RNA-granule components31. We synthesized B-isox (Supplementary Fig. 4a,b) and performed selective precipitation from total cell extracts derived from control and infected cells. We thereby confirmed previous findings indicating that B-isox treatment induces the selective precipitation of RNA-granule proteins in a dose-dependent manner31 (Supplementary Fig. 4c,d and Supplementary Table 2). We then performed quantitative mass spectrometry analysis, which indicated that infection affected RNA-granule composition (Supplementary Table 3). SUMOylation of NS1 increased partitioning of NS1 in RNA granules (Fig. 3b) and interaction with proteins involved in mRNA processing (Fig. 3c). These findings were further supported by validation experiments, which showed NS1-SUMO-dependent enrichment with the cleavage and polyadenylation specificity factor (CPSF) and the absence of enrichment of other proteins known to be present in RNA granules (FUS and EWS) (Fig. 3c and Supplementary Table 3). Because NS1 proteins from other viral strains are known to antagonize antiviral host gene expression1,3,16,17, and recent findings indicate that phase-separated RNA granules play a key role in controlling RNAPII29,32,33,34,35, we investigated the relationship between NS1 expression and gene suppression by determining RNAPII activity and mRNA levels after infection with 1918 viruses and nonpandemic strains proficient and deficient in NS1.

a, SDD-AGE (top) and WB (bottom) analyses of NS1 and NS1-SUMO virus-infected lysates of A549. Mock, uninfected control. b, B-isox enrichment of NS1 from A549 lysate infected by NS1 and NS1-SUMO virus, as analyzed by MS. ***P = 1.98 × 10−3. c, Gene Ontology (GO) enrichment analysis for biological processes of B-isox-enriched proteins during NS1 and NS1-SUMO infection (top), WB of input and B-isox precipitates from NS1 and NS1-SUMO virus-infected A549 lysates with the indicated antibodies (bottom right). FUS and EWS are controls for resident RNA-granule proteins. d, Schematic of the approach used to detect a relative increase in transcript levels in 3′-end gene-flanking regions. For each gene, a 3′ transcript ratio (termination ratio, TR) was calculated as the average number of total RNA-seq reads per bp in 5-kb 3′ gene-flanking regions, divided by that in exonic regions. A global relative increase in 3′ transcript levels results in a horizontal transformation of the cumulative TR plot, as indicated. e,f, TR plots at termination regions of active genes (reads per kilobase per million mapped reads (RPKM) >1 in 50% of samples) in uninfected A549 cells and 6 hpi (e) or 12 hpi (f) with NS1 or NS1-SUMO virus. g, TR plots at termination regions of active genes (RPKM >1 in 50% of samples) in uninfected A549 cells and at 6 hpi with PR8 or ∆NS1 virus. h–j, TR plots at the termination region of uninfected A549 cells treated with short interfering RNA against ubiquitin conjugating enzyme E2 (siUBE2I) or a control sequence (siCtrl) (h), uninfected A549 cells transfected with either empty vector (EV), or a vector expressing GFP or SUMO (i), and A549 cells infected with PR8∆NS1 and transfected with an empty vector or a vector expressing SUMO (j). The insets show the RNA expression levels of UBE2I in siCtrl- or siUBE2I- treated cells (h) and of GFP and/or SUMO in each condition (i,j) as counts per million sequenced reads (CPM). In e–j, data are shown for n = 2 independent experiments in each condition. Uncropped blot images are shown in Supplementary Dataset 1. Source data for h–j are available in Supplementary Dataset 2.
SUMOylation of NS1 enhances suppression of the host transcriptional response via interference with RNAPII termination
Because of the known role of the CPSF complex in 3′-end cleavage and termination of RNAPII36, we first analyzed genome-wide RNAPII traveling ratios at transcriptional termination sites, namely the termination ratios (TRs), at both 6 and 12 hpi (Fig. 3d). This analysis indicated that downregulation of gene expression after infection is associated with a genome-wide deregulation of RNAPII 3′-end cleavage and termination. This deregulation was evidenced by an increase in run-through transcription, on the basis of high levels of RNA after the 3′ end at infection-regulated host genes at both 6 hpi (Fig. 3e) and 12 hpi (Fig. 3f). Notably, this effect was evident during NS1 virus infection and was magnified after NS1-SUMO virus infection (Fig. 3e,f). To determine whether run-through transcription might also be a feature of non-1918 viruses and dependent on NS1 expression, we used WT and NS1-deficient PR8 virus (hereafter referred to as dNS1 virus)6. Comparison between the termination ratios induced by these two viruses indicated that although WT PR8 virus infection caused RNAPII run-through transcription, this effect was abrogated in dNS1-virus-infected cells (Fig. 3g). Importantly, downregulation of Ube2L, an enzyme required for SUMOylation of substrate proteins, did not induce RNAPII termination defects (Fig. 3h). Additionally, overexpression of GFP or SUMO in uninfected cells did not induce RNAPII run-through (Fig. 3i). Run-through was seen in infection with WT H5N1 IAV and a modified virus encoding a GFP-fused NS1 (Supplementary Fig. 4e). Finally, overexpression of SUMO in dNS1-virus-infected cells did not affect RNAPII at the termination region (Fig. 3j). Overall, these results indicate that genome-wide deregulation of RNAPII termination (i) is a feature of IAV infection, (ii) is dependent on NS1 expression, and (iii) can be controlled by SUMOylation of the 1918 NS1 SUP.
Transcriptional shut-off during infection
RNAPII run-through during infection can be functionally relevant if it is linked to repression of factors involved in the orchestration of the antiviral response or required for the viral life cycle. To explore this possibility, we analyzed the relationship between the change in run-through transcription and expression changes in NS1- versus NS1-SUMO-infected cells by cross-comparing transcript density at the 3′ ends of genes and the percentage of gene repression. Although virtually all genes experienced increased run-through (positive signal >0 for all gene curves in Supplementary Fig. 5a in NS1-SUMO versus NS1 conditions), the effect was exacerbated at genes with decreased expression in NS1-SUMO. This result suggests that run-through transcription plays a role in the downregulation of constitutively expressed genes important for cell division (for example, CDC25A and MYC), metabolism (for example, PDK4 and RANBP6), apoptosis (for example, MCL1 and SOX4) and cell defense (for example, NFKBIA and CXCL1) (Supplementary Table 4 and representative gene-browser tracks in Supplementary Fig. 5b). Although some upregulated genes enriched in host antiviral functions (for example, IFIT1 and IFIT2) experienced run-through as well, their induction exceeded any negative effect of the increased run-through transcription seen in NS1-SUMO virus-infected cells. Notably, RNAPII run-through after infection was universally seen at all transcriptionally active genes except four (LY6E, APOL1, DEFB1, and IFI6; Supplementary Table 4), thus possibly suggesting unique features of their 3′ ends.
Overall, our data demonstrate that IAV infection can globally affect gene expression by deregulating RNAPII termination. This effect undermines the activity of many cell-defense genes and may be a general means by which viruses induce a state of transcriptional shut-off in infected cells.
Mechanism of run-through transcription
The defect in RNAPII cleavage and increased run-through into distal extragenic regions observed during viral infection may be the result of deregulated termination and/or other regulatory events that indirectly affect termination. To test whether this effect might be driven by inhibition of CPSF, we first reanalyzed published RNA-seq datasets in cells depleted of CPSF73, an essential subunit of the CPSF complex37. Our analysis indicated that loss of function of CPSF activity induced RNAPII termination defects at all active genes (Fig. 4a), recapitulating the effect caused by NS1 during infection. Because CPSF also plays a key role in alternative splicing38,39,40, we analyzed datasets from cells treated with the splicing inhibitors spliceostatin and isoginkgetin41. Splicing inhibition induced pervasive RNAPII run-through, as evidenced by the increased run-through ratio in treated cells (Fig. 4b). These data suggest that RNAPII run-through after infection may be caused by pervasive defects in splicing rather than impaired 3′-end cleavage and termination. We therefore analyzed genome-wide splicing efficiencies after infection with NS1 and NS1-SUMO viruses. Global splicing defects were detected only at 12 hpi (Fig. 4c–e), whereas deregulation of termination was already evident at 6 hpi (Fig. 3e). No increase in termination defects was evident in genes with multiple exons compared with monoexonic genes (Supplementary Fig. 6a). To gain further support for the idea that transcription termination defects precede splicing defects, we performed isoform sequencing (iso-seq), a method that allowed us to detect and define all splicing isoforms generated by a given gene on a genome-wide scale through long-read sequencing (details in Methods). Iso-seq of mRNA from A549 cells that were mock infected or infected with WT or NS1-SUMO virus indicated that infection did not have a pervasive effect on isoform expression at 6 hpi. This supports the idea that termination defects are the cause of transcriptional run-thorugh during infection. Despite this, infection induced some ‘nonphysiological’ transcripts poorly detected in mock-infected cells. These transcripts, potentially generated from interference with mRNA maturation, were increased in NS1-SUMO infection (Fig. 4f). Among the affected genes, NS1-SUMO infection had a stronger effect than NS1 infection in generating intron retention and RNAPII run-through, even for poly(A) transcripts. Furthermore, analysis of long reads mapping to viral segment 8 demonstrated that, during infection with NS1-SUMO, all mRNAs were full length (including the SUMO-encoding region) rather than truncated (Fig. 4f).

a, TR plots at the termination region in uninfected A549 cells treated with short interfering RNA against CPSF73, a subunit of the CPSF complex (siCPSF73) or a control luciferase sequence (siLuc). b, TR plots at the termination region in uninfected A549 cells treated with the indicated inhibitors and control (DMSO). c, Schematic of the approach used to detect relative accumulation of unprocessed intronic transcripts. Analogously to the TR plots, an intron/exon transcript ratio was calculated by dividing the average read coverage per bp in 5,000-bp intronic regions directly flanking an upstream exon, by the average read coverage of the upstream exon. An increase in unprocessed intronic transcripts results in horizontal transformation of the cumulative intron/exon-ratio plots, as indicated. d,e, Cumulative intron/exon ratio distributions of active genes (RPKM >1 in 50% of samples) in uninfected A549 cells and 6 hpi (d) or 12 hpi (e) with NS1 or NS1-SUMO virus (e). Data are shown for two replicates in each condition. f, Classification of all iso-seq circular consensus (CCS) reads on the basis of comparisons with GENCODE v38 transcript annotations using cuffcompare (left). Percentage of reads containing full-length RNA from segment 8 in cells infected with NS1-SUMO virus (right). ORF, open reading frame. Data underlying the analysis in f are available in Supplementary Dataset 3.
Overall, our total RNA seq and iso-seq data indicate that RNAPII run-through during IAV infection and the downregulation of most active genes is due to interference with 3′-end cleavage and termination, which in turn causes a defect in splicing at a later time as infection progresses. These results support the notion that defective RNAPII termination can delay RNAPII initiation and affect splicing fidelity42,43,44,45. Finally, downregulation of genes that experience RNAPII run-through in NS1-SUMO virus versus NS1 virus infected cells is likely to be caused by both delayed and decreased initiation and the direct effect of run-through RNAPII into neighboring genes, as suggested by the linear relationship between increased run-through and changes in gene expression (Supplementary Fig. 6b).
Finally, the finding that chemical inhibition of splicing causes RNAPII run-through, albeit to a lesser extent than during infection, indicates a connection between these two biological processes. This connection is further supported by evidence showing that last-exon splicing and RNAPII termination are mechanistically and evolutionarily linked events38,46,47. Overall, our data demonstrate that chemical- or virus-induced interference with cotranscriptional events can generate pervasive RNAPII run-through.
Discussion
By sampling sequences from highly pathogenic viruses, we identified the presence of a novel peptide domain comprising a SUMO site embedded in a PDZBD in the C-terminal region of NS1 expressed by the 1918 pandemic IAV strain. This SUP domain confers SUMOylation of NS1. During infection, SUMO enhances NS1 association with nuclear ribonucleoprotein complexes controlling RNAPII activity. In addition, infection with the NS1-SUMO virus causes pervasive RNAPII run-through into downstream extragenic DNA and nearby genes. This result prompted us to test whether infection with other IAV strains might also cause transcription termination defects. Indeed, infection with WT PR8 virus induced RNAPII run-through, an effect also recently shown by two independent studies that mapped nascent RNA-seq48 and chromatin profiling of RNAPII49 during IAV infection with multiple strains. Unlike WT PR8, NS1-deficient virus did not induce RNAPII run-through. This result provides genetic evidence for the role of NS1 in inducing aberrant RNA biogenesis. Importantly, although NS1 proteins from different strains bind transcription regulators (such as CPSF30) with different strength in vitro, recent evidence suggests that cognate interaction between viral polymerase and NS1 can increase interaction strengths and inhibitory activity during infection50. According to this finding and our results, we speculate that all IAVs might have evolved different strategies to inhibit 3′-end formation. Accordingly, pathogen-derived effector proteins tend to target the same set of host factors controlling cell functions51,52,53,54,55.
Effect of RNAPII run-through on gene induction and gene repression
Although most genes affected by RNAPII run-through were downregulated, some host antiviral genes were upregulated despite being affected by run-through. This result may possibly be because of unique regulatory sequences and events that control 3′ ends. Alternatively, an extremely high induction level and burst rate at some upregulated genes might allow them to partially bypass or escape termination inhibition. Overall, our results suggest that deregulated RNAPII termination suppresses active genes and establishes transcriptional shut-off. Many viruses cause such an effect through different strategies of host interference56,57,58,59,60. We suggest that the different mechanisms triggering host transcriptional shut-off are linked to virus-specific requirements for their life cycle. For example, IAV requires high levels of host gene transcription (or initiation events) in the first hours after infection to mediate cap-snatching and to coordinate viral-mRNA production and viral-protein expression2. As such, gene suppression must be synchronized with viral replication. Overall, our results suggest that virus-induced RNAPII run-through kinetically controls host gene expression. We show that this effect is achieved by NS1 and modulated by SUMOylation in the case of the1918 strain of IAV. Constitutive NS1 SUMOylation is associated with an increased degree of RNAPII run-through at most active loci, concomitantly with gene suppression. Interestingly, NS1-SUMO also induces increased upregulation of some host-response genes. These results suggest that SUMOylation skews NS1 activity toward the control of RNAPII termination and that constitutive SUMOylation might limit other NS1 host-antagonist function. This possibility is supported by the limited infectivity of the NS1-SUMO virus in multicycle growth, as this virus cannot dynamically control this post-translational modification. In this respect, it is important to consider that SUMOylation can also be a signal for NS1 degradation. Several transcription factors are known to function transiently on chromatin and to undergo rapid turnover61. NS1 may have adopted such a feature to provide a strategy to interfere with host transcription. Overall, our data suggest that 1918 IAV uses dynamic regulation of post-translational modifications to switch between various host-antagonizing strategies. Notably, polymorphism and mutations in SUMO-modifying enzymes exist in humans (Supplementary Fig. 6c and Supplementary Table 5), and they may potentially affect the host response to IAVs.
Evolutionary considerations
Sequence analysis of pathogen-derived polypeptides can provide valuable information about sequences governing principles of protein–protein interaction12,62. Proteins encoded by viruses, especially those that have evolved to suppress the host response or co-opt cellular signaling to increase pathogen fitness, generally display multivalent interaction surfaces that allow simultaneous binding of many host proteins and nucleic acids63,64. This functional pleiotropy can be achieved by peptide domains and IDRs65,66,67,68. Key features defining pathogen-derived linear domains and IDRs are: (i) they are located at the C or N terminus of a protein69, (ii) they are enriched in viral proteins that reside in the nuclei of infected cells62, and (iii) they contain post-translational-modification sites70. The SUP domain in NS1 of the 1918 IAV strain fits all these parameters. Most IDRs arise de novo via convergent evolution71,72, and the 1918 SUP is not conserved in other influenza viruses. These findings suggest that the 1918 SUP either evolved independently or was co-opted via viral mimicry. Previous work has identified how a circulating strain of IAV (H3N2) uses molecular mimicry to evolve histone-like sequences1. Although the 1918 IAV circulated for a short period of time, the NS1 SUP domain may have undergone high levels of mutagenesis and fixation in different strains. Intriguingly, the NS1-tail sequence of different strains is predictive of the host species from which the virus has been isolated18. Understanding the molecular mechanisms underlying this correlation can be informative in the context of surveillance and zoonosis73. Notably, the only other three sequences that contain the NS1 SUP domain have been isolated from avian species, which are key host reservoirs linked to acquisition of increased pathogenicity and pandemic potential74,75. Finally, our work shows that despite being close to the centennial of its insurgence, 1918 influenza research can still instruct us about biological principles controlling pathogens and humans.
Methods
Reagents
For chemical synthesis of B-isox, 5-(2-thienyl)-3-isoxazolecarboxylic acid (763109-71-3), N-hydroxysuccinimide (HOSu, 6066-82-6), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC-HCl, 25952-53-8), 6-amino-1-hexanol (4048-33-3), biotin (B4639), and 4-(dimethylamino)pyridine (DMAP, 1122-58-3) were purchased from Sigma-Aldrich. For B-isox-mediated precipitation of proteins, Halt Protease Inhibitor Cocktail (78438), Dynabeads MyOne Streptavidin C1 (65001), a SilverQuest Silver Staining Kit (LC6070), and GelCode Blue Stain Reagent (24590) were purchased from Thermo Fisher Scientific. For expression and purification of recombinant proteins, isopropyl β-d-1-thiogalactopyranoside (IPTG, I6758) and cOmplete EDTA-free Protease Inhibitor Cocktail Tablets (11873580001) were purchased from Sigma-Aldrich. Ni–NTA agarose (30210, Qiagen) and Nanosep Centrifugal Devices with Omega Membrane, 3K (OD003C33, Pall Corporation) were also used. For BLI, EZ-Link Sulfo-NHS-LC-biotin (21327, Thermo Fisher Scientific) and Dip and Read Streptavidin Biosensors (18-5021, ForteBio) were used. For immunofluorescence, Lipofectamine 2000 (11668027) and DAPI (D1306) were purchased from Thermo Fisher Scientific. For pulldown assays, anti-FLAG M2 Affinity Gel (A2220) was purchased from Thermo Fisher Scientific. For proteasome inhibition, MG132 (M7449) was purchased from Sigma-Aldrich. For directional RNA-seq, a Ribo-Zero Gold rRNA Removal Kit (human/mouse/rat) (MRZG12324, Illumina), Agencourt AMPure XP (A63882, Beckman Coulter Life Sciences), TruSeq Directional Library Prep Kit (RS-122-2203, Illumina), and BluePippin 2% M1 gels (BEF2010, Sage Scientific) were used. Antibodies were as follows. Anti-NS1 was from the laboratory of A.G.-S. Anti-FLAG (F7425, Sigma-Aldrich), anti-SUMO 2/3 (07-2167, Millipore), anti-FUS (A300-302A), anti-EWS (A300-418A), and anti-CPSF30 (A301-585A-M) were purchased from Bethyl Laboratories. Plasmids were as follows. pcDNA5 vectors encoding the indicated genes were used to express corresponding recombinant proteins in a mammalian system. pET28c vectors encoding the indicated genes were used to express recombinant proteins in a bacterial system. A549 cells (adenocarcinomic human alveolar basal epithelial cells), HEK293T cells (human embryonic kidney cells), and Madin–Darby canine kidney (MDCK) cells were originally obtained from the American Type Culture Collection. The cell cultures were tested and found to be mycoplasma-free.
Conservation analysis
All complete NS1 amino acid sequences were downloaded from the NCBI Influenza Virus Database for Human and Swine H1N1 and H3N2 and Avian H5N1. A multiple alignment was made of all sequences in the five strains and also for all sequences together. The multiple alignments were generated with multialign in MATLAB. For each group, the consensus sequence and conservation score were defined with seqconsensus in MATLAB. The average score was plotted across a ten-residue sliding window for each group of proteins and then normalized so that the maximum and minimum score were consistent across groups. These values were plotted in the accompanying heat map.
Modeling of PDZ–peptide–SUMO complexes
Initial models of the PDZ domains with NS1 peptides were generated in MODELLER 9.12 with the crystal structures of the PDZ domains or homology models when needed (Supplementary Table 1) and the sequences of the peptides76. Seven residues from the NS1 peptide were modeled (sequence RTIKSEV). The crystal structure of the Par-6 PDZ domain in complex with the Pals1 peptide was used as a template peptide for conformation in all NS1 complexes77. The initial PDZ–peptide complexes were then refined with the FlexPepDock server with default parameters78. The top-scoring model from FlexPepDock for each complex was selected, and the SUMO conjugated version was modeled by bonding the C-terminal glycine residue of the SUMO structure to the lysine residue in the peptide79. The possible spatial orientations of the conjugated SUMO domain were explored by varying the conformation of the lysine side chain in the peptides.
Transfection, infection, and western blotting
For transfected samples, 70%-confluent A549 cells were transfected with the vectors encoding the indicated genes with Lipofectamine for 2 d. The cells were washed with cold PBS twice, scraped and suspended in lysis buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 20 mM β-mercaptoethanol, 0.5% NP-40, 10% glycerol, 20 mM NEM, and 2× Halt protease inhibitor. The cells were then lysed at 4 °C with a BIORUPTOR 300 sonicator for ten cycles of 30 s on and 30 s off at a high level of sonication. For infected samples, 100%-confluent A549 cells were infected by the indicated viruses at the indicated MOI and collected at different time points. The cells were lysed as described above. Input and immunoprecipitated materials were analyzed by WB.
Semidenaturing detergent agarose gel electrophoresis (SDD-AGE)
The infected A549 lysates were prepared as described above and centrifuged at 2,000 r.c.f. at 4 °C for 2 min. The supernatants were removed carefully and incubated with 4× SDD-AGE loading buffer containing 2× TAE buffer, 20% glycerol, 8% SDS, and 1% bromophenol blue at room temperature for 5 min. The samples were loaded on a 1.5% agarose gel containing 0.1% SDS. The gel was run in 1× TAE buffer with 0.1% SDS at a constant voltage of 40 V at 4 °C for 3 h. Capillary transfer and WB were then performed80.
Purification of recombinant proteins
Histidine-tagged recombinant NS1, NS1-KR, NS1-SUMO, and PSD95 proteins were overexpressed in Escherichia coli BL21(DE3), OD60 nm = 0.6, with induction with IPTG at 1 mM and incubation at 30 °C overnight. Bacteria were harvested, washed with PBS twice, and suspended in lysis buffer containing 50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole, pH 8.0, with EDTA-free protease inhibitor added. The bacterial mixture was placed on ice and sonicated with a Misonix 3000 instrument for six cycles of 10 s on and 10 s off at initial output level 10. The lysate was centrifuged at 15,000 r.p.m. at 4 °C for 15 min. The soluble fraction of the lysate was mixed with Ni–NTA resin and gently rotated at 4 °C overnight. The Ni–NTA agarose was fully washed with buffer containing 50 mM NaH2PO4, 300 mM NaCl, and 20 mM imidazole, pH 8.0. The proteins were finally eluted with elution buffer containing 50 mM NaH2PO4, 300 mM NaCl, and 250 mM imidazole, pH 8.0. The purified proteins were concentrated with Nanosep (3K) centrifugal devices, and the concentrations were determined with a NanoDrop 1000 spectrophotometer. The purity of recombinant proteins was confirmed by SDS–PAGE and staining by Coomassie blue.
BLI
Recombinant PSD95 was biotinylated by reaction with Sulfo-NHS-LC-biotin with a molar coupling ratio of 1:1. Extra biotin reagent was fully removed through buffer exchange four times after the biotinylation reaction was completed. The stock of biotinylated PSD95 was diluted to 20 μg/mL in PBS, and stocks of NS1, NS1-KR, and NS1-SUMO were serially diluted to 10, 5, 2.5, and 1.25 μM in PBS. BLI assays were performed on a ForteBio OctetRED 96 instrument. Streptavidin biosensors were washed by PBS for 60 s, immobilized with biotinylated PSD95 (20 μg/mL) for 300 s, washed a second time with PBS for 60 s, associated with blank (PBS), NS1, NS1-KR, and NS1-SUMO at serially diluted concentrations (10, 5, 2.5, and 1.25 µM) for 300 s and finally dissociated by washing with PBS for 900 s. All steps were performed with agitation at 1,000 r.p.m. at 30 °C. BLI data were processed and analyzed in software Data Analysis 8.2. Steady-state analyses were used to generate Kd and R2 values81.
Pulldown assays
Purified FLAG-tagged NS1, NS1-KR, and NS1-SUMO proteins were incubated with anti-FLAG agarose in PBS under rotation at 4 °C for 1 h. The beads were fully washed with washing buffer containing 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1 mM EDTA, and 0.2% NP-40 and were then washed with the same buffer without NP-40. NS1-, NS1-KR-, and NS1-SUMO-bound agarose were incubated with purified histidine-tagged PSD95 in PBS under rotation at 4 °C for 1 h (and agarose washed by PBS was included as a control). The beads were fully washed with washing buffer. Bound proteins were then eluted by 3× FLAG peptide (0.25 mg/mL) by shaking the agarose at 1,000 r.p.m. at 4 °C for 1 h. The input and eluted proteins were run on an SDS–PAGE gel and stained by Coomassie blue.
Analysis of protein stability
A549 cells were infected with NS1 and NS1-SUMO virus for 12 h and were sequentially treated with mg132 (50 nM) or cycloheximide (50 μg/mL) for 0, 4, 8, or 24 h. Cells were lysed, and WB was performed.
Immunofluorescence
The FLAG-tagged GFP-, NS1-, NS1-KR-, and NS1-SUMO-transfected A549 lysates were washed, fixed, permeabilized, and stained with anti-GFP, anti-FLAG, and DAPI. Images were observed under a Zeiss Axio Imager Z2M microscope and processed in Zen software.
Rescue of recombinant influenza viruses
Recombinant NS1 and NS1-SUMO viruses were prepared by plasmid transfection as previously described82. Two viruses rescued contained seven segments from strain A/Puerto Rico/8/34 (PR8). For the NS1-SUMO version, a modified plasmid encoding the NS segment from strain A/Brevig Mission/1/1918, in which the two viral proteins (NS1 and NEP) are encoded in separated open reading frames was used. Plasmid pDZ_NS1 encoded the original amino acid sequence, with three silent changes to allow differentiation. The plasmid pDZ_NS1-SUMO contained the SUMO2 coding sequence as a fusion at the end of the NS1 open reading frame. Rescued viruses were amplified in embryonated chicken eggs, and the NS segment was confirmed by sequencing. All work with infectious viruses containing sequences from A/Brevig Mission/1/1918 was performed in strict accordance with CDC guidelines for biosafety level 3 (BSL3) agents at the BSL3 laboratory of the Icahn School of Medicine at Mount Sinai.
Single-cycle growth curve
Confluent monolayers of MDCK cells growing in six-well plates were infected in triplicate with the indicated viruses at an MOI of 1 PFU per cell. After 1 h adsorption, the inoculum was aspirated and replaced with 2 mL of infection medium (MEM supplemented with 1% antibiotics, 0.3% BSA, and 1 μg/mL of TPCK-treated trypsin). At the indicated times 200 μL of supernatant was collected for titration and replaced with 200 μL of fresh infection medium. Supernatants were titrated with hemagglutination assays with turkey red blood cells82.
Plaque phenotype
MDCK monolayers growing in six-well plates were infected with serial dilutions of the indicated virus. After 1 h adsorption, the inoculum was aspirated, and cells were overlaid with medium containing 0.6% agar and trypsin (1 μg/mL). After 2 d, the cells were fixed with 4% formaldehyde, and plaques were visualized with a monoclonal antibody against the viral protein NP.
Analysis of infectious viral progeny
Supernatants from a 24-h infection were analyzed with plaque assays as described above. Isolated visible plaques were suspended in PBS and inoculated onto fresh MDCK cells. When cytopathic effects were observed, viral RNA was purified from the supernatant and amplified by RT–PCR with primers specific for the NS segment. The amplified product was purified from an agarose gel and sequenced with the same primers.
RT–PCR
A549 cells were lysed with TRIzol reagent (Ambion, 15596026) and subjected to RNA extraction and DNase digestion. cDNA was synthesized with a high-capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368813) according to the manufacturer’s protocol. Primers were as follows: β-actin forward, 5′-ACCTTCTACAATGAGCTGCG-3′; β-actin reverse, 5′-CCTGGATAGCAACGTACATGG-3′; GAPDH forward, 5′-TCTGACGCTGACTGGTTAGT; GAPDH reverse, 5′-GAGGGCACAGAAAGCAATAGAG; CDC25A forward, 5′-GTGGGATGGCCTTCAGATT; and CDC25A reverse, 5′-CCATCAAGAACTAGGCAGAGAG.
Chemical synthesis of B-isox
B-isox was chemically synthesized according to a previously reported protocol with minor modifications30. A solution of 5-(2-thienyl)-3-isoxazolecarboxylic acid (a, 200 mg, 1 eq.), HOSu (239 mg, 2 eq.) and EDC-HCl (397 mg, 2 eq.) in 5 mL aqueous THF was stirred at room temperature overnight to produce chemical intermediate b. Then 6-amino-1-hexanol (243 mg, 2 eq.) was added into the mixture, and the reaction was processing by stirring at room temperature for an additional 5 h. Product c was then purified with a CombiFlash Purification System with gradient elution by MeOH and DCM. Chemical intermediate c (100 mg, 1 eq.) was next mixed with biotin (85 mg, 1 eq.), DMAP (6.7 mg, 0.2 eq.), and EDC-HCl (128 mg, 2 eq.) in 5 mL DCM. The reaction was stirred at room temperature for 3 d. The final product B-isox (d) was obtained with purity ≥98% through a CombiFlash Purification System with gradient elution with MeOH and DCM. The total yield was 11%. B-isox was characterized with HRMS. Calculations for C24H33N4O5S2, [M+H]+ and C48H65N8O10S4, [2M+H]+ were 521.1892 and 1,041.3706, respectively, and 521.2162 and 1,041.3704 were found.
B-isox-mediated precipitation
Six 15-cm dishes of 100% confluent HEK293T cells were lysed as described above. The crude lysates were centrifuged at 15,000 r.p.m. at 4 °C for 10 min to produce six supernatants (1 mL). Four supernatants (800 μL) were treated with B-isox (0, 10, 30, and 100 μM), and two supernatants were treated with B-isox (0 and 100 μM) and streptavidin–agarose as controls. The mixtures were rotated gently at 4 °C for 1 h. The mixtures were spun at 15,000 r.p.m. for 10 min, and the supernatants were removed. The pellets were washed with cold lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 20 mM β-mercaptoethanol, 0.5% NP-40, 10% glycerol, 20 mM NEM, and 2× Halt protease inhibitor) twice and dissolved in 100 μL of 2% SDS. The samples were boiled in 4× SDS loading buffer and run on an SDS–PAGE gel. The gel was silver stained. A549 cells were infected by mock, NS1, and NS1-SUMO viruses at an MOI of 3 for 12 h, and the infected lysates were precipitated by 100 μM B-isox as described above. The precipitated samples were analyzed by WB.
Preparation of peptides for mass spectrometry
B-isox-precipitated samples of A549 lysates infected by mock, NS1, and NS1-SUMO viruses were separated by SDS–PAGE. Gel bands for each sample were excised and then diced into small sections (1 mm2). Gel pieces were dehydrated in 25 mM NH4HCO3 and 50% acetonitrile (ACN) for 10 min with vortexing in two consecutive rounds and were then dried under vacuum centrifugation. Slices were rehydrated in 15 mM Tris(2-carboxyethyl)phosphine (TCEP) and 25 mM NH4HCO3, and the samples were incubated for 20 min. Freshly prepared 1 M iodoacetamide and 25 mM NH4HCO3 were then added to attain a final concentration of 50 mM iodoacetamide, and the samples were incubated in the dark for 20 min. The supernatants were removed, and 25 mM NH4HCO3 was added to cover the gel pieces. The samples were vortexed for 10 min, the supernatants were removed, and 25 mM NH4HCO3 and 50% ACN were added. After incubation with vortexing for 5 min, the supernatants were removed, and the gel slices were dried under vacuum centrifugation. A total of 0.5 μg trypsin in NH4HCO3 was added with enough volume to cover the gel slices, and the samples were incubated at 37 °C for digestion overnight. The supernatants containing extracted peptides were collected in separate low-bind tubes, and sufficient 50% ACN and 5% formic acid (FA) were added to the remaining gel slices. Samples were vortexed for 10 min, and the supernatants were combined with the previous extractions. This step was repeated with 100% CAN, and the supernatants were combined with the previously collected samples. Peptides were dried under vacuum centrifugation and suspended in 10 μL of 3.0% ACN and 0.1% FA before MS analysis.
Protein identification by liquid chromatography with tandem mass spectrometry
Digested peptides were subjected to LC–MS/MS analysis with an Easy-nLC 1000 instrument coupled to a dual-pressure linear ion trap (Velos Pro) Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Online LC separation was performed with a fused silica IntegraFrit capillary (75 mm × 25 cm) packed with 1.9 mm Reprosil-Pur C18 AQ reversed-phase resin. Peptides were eluted with a gradient of 5% to 30% ACN in 0.1% FA in 160 min, delivered at a flow rate of 300 nL/minute. For each cycle, one full MS scan (150–1,500 m/z, resolution of 120,000) in the Orbitrap was followed by 20 data-dependent MS/MS scans fragmented by normalized collision energy (setting of 35%) and acquired in the linear ion trap. Target ions already acquired in MS/MS scans were dynamically excluded for 20 s. Raw MS files were analyzed by MaxQuant version 1.3.0.3 (ref. 83), and MS/MS spectra were searched with the Andromeda search engine84 against a database containing reviewed SwissProt human and influenza protein sequences (20, 194 in total)85. All runs were analyzed simultaneously to maximize the ‘match between runs’ algorithm available in Maxquant. The multiplicity was set to 1 (as recommended for label-free experiments), and a false discovery rate of 0.01 was imposed for peptide and protein identification.
Statistical relative quantification of proteins and enrichment analysis
Normalization of raw peptide intensities, protein-level abundance inference, and differential expression analysis (log2 fold change and P values) were calculated with the open source R package MSstats version 3.3.10 (ref. 86). To ensure reproducibility, data were filtered to select for proteins identified in both biological replicas in at least one of the conditions compared. Differential expression values for proteins available in only one condition were estimated by imputation of the intensity values by random sampling from the lowest 20 normalized intensity values from the conditions in which proteins were not detected.
Gene Ontology (GO) enrichment analysis of biological processes was performed in Panther87. Data analysis and integration were carried out with the R language for statistical computing and graphics.
Directional RNA-seq
One microgram of DNase-treated RNA was depleted of rRNAs with a Ribo-Zero Gold rRNA Removal Kit (human/mouse/rat) according to the manufacturer’s instructions and purified postdepletion with a 1.6× volume of AMPure XP beads. Barcoded directional RNA-seq libraries were then prepared with a TruSeq Directional Library Prep Kit (Illumina), according to the kit instructions. PCR products were purified with a 1.8× volume of AMPure XP beads, and fragments of 300–500 bp were size-selected with BluePippin 2% M1 gels (Sage Scientific). Afterward, libraries were sequenced on the Illumina HiSeq 2500 platform in 100-bp single-end-read run format.
RNA-seq
After adaptor removal with cutadapt88 and base-quality trimming to remove 3′ read sequences if more than 20 bases with Q <20 were present, paired-end reads were mapped to the human (hg38) reference genome with STAR89, and gene-count summaries were generated with featureCounts90. Raw-fragment (that is, paired-end read) counts were then combined into a numeric matrix, with genes in rows and experiments in columns and used as input for differential gene expression analysis with the Bioconductor Limma package91 after multiple filtering steps to remove weakly expressed genes. First, gene counts were converted to fragments per kilobase per million reads (FPKM) with the RSEM package92 with default settings in strand-specific mode, and only genes with expression levels above 1 FPKM in at least 50% of samples were retained for further analysis. Additional filtering removed genes with fewer than 50 total reads across all samples or fewer than 200 nt in length. Finally, normalization factors were computed on the filtered data matrix with the weighted trimmed mean of M values (TMM) method followed by voom93 mean-variance transformation in preparation for Limma linear modeling. Data were fitted to a design matrix containing all sample groups and pairwise comparisons were performed between sample groups.
Termination ratio and intron/exon-ratio calculation
To quantify an increase in transcription beyond transcription termination sites at the 3′ ends of genes, we calculated a measure called the TR that compares the ratio between the average read coverage in 3′-gene-flanking regions and the average read coverage per base pair of exonic sequence. We defined the 3′ flanking region between +1 and +5,000 bp relative to most distal 3′ end of annotated transcripts for known genes. Analogously, to assess the accumulation of unprocessed transcripts that might be the result of a global defect in splicing, we also calculated an intron/exon transcript density ratio for all introns. This measure was defined as the average read coverage per bp in 5,000-bp intronic regions directly flanking an upstream exon, divided by the average read coverage of the upstream exon. Transcript densities in 3′-gene and exon-flanking regions were calculated after exclusion of any regions overlapping annotated genes. The termination and intron/exon transcript density ratios are analogous to the TR, as described previously94.
Transcript isoform sequencing (iso-seq)
RNA QC was performed with Qubit RNA HS Assay Kit (Thermo Fisher) and Agilent RNA Pico Kit protocols to assess the quantity and quality (RIN integrity) of the samples, respectively. Ribosomal-RNA depletion was performed according to the instructions for the Thermo Fisher Scientific RiboMinus Transcriptome Isolation Kit, human/mouse. Magnetic beads were prepared by resuspension of the RiboMinus Magnetic Beads by thorough vortexing. For each sample, 250 μl of the bead suspension was pipetted into a sterile tube. The tubes were placed on a magnetic rack for 1 min, and the supernatant was removed, discarded, and replaced with 250 μl DEPC-treated water. The tubes were placed on a magnetic rack for 1 min, and the supernatant was removed, discarded, and replaced with 250 μL hybridization buffer. The tubes were placed on a magnetic rack for 1 min; the supernatant was removed discarded, and replaced; and samples were resuspended in 100 μL hybridization buffer. The tubes were incubated at 37 °C until use. Hybridization was performed by combining 20 μL of poly(A)-tailed total RNA, 4 μL RiboMinus probe (100 pmol/μL), and 100 μL hybridization buffer. The tubes were incubated at 37 °C for 5 min. The tubes were placed on ice for 30 s and centrifuged. 124 μL of the samples was transferred to the prepared RiboMinus Magnetic beads and mixed. The tubes were incubated at 37 °C for 15 min. The tubes were placed on a magnetic rack for 1 min. The supernatant was removed and transferred to a sterile tube. RNA QC was then repeated on each sample.
Isoform generation was performed according to the iso-seq template-preparation instructions from Sequel Systems. The first-strand synthesis was performed by addition of 1–3.5 μL 3′ SMART CDS primer IIA, 1–3.5 μL RNA sample, and 0–2.5 μL nuclease-free water, for a 4.5 total μL volume. The tubes were mixed by pipetting, centrifuged briefly, and incubated in a hot-lid thermal cycler at 72 °C for 3 min. The temperature was slowly ramped to 42 °C at 0.1 °C/s for 2 min. During the incubation step, the master mix was prepared by addition of 2 μL 5× first-strand buffer, 0.25 μL 100 mM DTT, 1 μL 10 mM dNTP, 1 μL 12 μM SMARTer II A Oligonucleotide, 0.25 μL RNase inhibitor, and 1 μL SMARTScribe Reverse Transcriptase, for a total volume of 5.5 μL per reaction. The master mix was heated at 42 °C for 1 min. Aliquots of 5.5 μL master mix were added each reaction tube and mixed gently by pipetting, and the tubes were spun briefly. The tubes were incubated at 42 °C for 90 min. The reaction was terminated by heating of the tubes at 70 °C for 10 min. The first-strand reaction products were diluted by addition of 90 μL of PacBio elution buffer.
cDNA amplification was performed as follows. 24 × 50 μL PCR reactions were set up. Per sample, PCR master mix was prepared by addition of 240 μL 5× PrimeSTAR GXL Buffer, 240 μL diluted first-strand cDNA, 96 μL of 10 mM dNTP mix for each nucleotide, 16 μL 5′ PCR primer II A, 576 μL nuclease-free water, and 24 μL 1.25 U/μl PrimeSTAR GXL DNA polymerase. The samples were incubated at 98 °C for 30 s; 15 cycles of 98 °C for 10 s, 65 °C for 15 s, 68 °C for 10 min; and a final extension at 68 °C for 5 min.
For PCR-product purification, the 12 × 50 μL PCR reactions were pooled, and the samples were purified with 1× AMPure PacBio (PB), fraction 1. The remaining 12 × 50 μL PCR reactions were pooled, and the samples were purified with 0.4× AMPure PacBio, fraction 2, according to the manufacturer’s protocol. The bead purifications were repeated on fractions 1 and fraction 2, 1× and 0.4× volume, respectively. cDNA QC was performed with a Qubit dsDNA HS Assay Kit (Thermo Fisher), and the quality was assessed with an Agilent cDNA 12K Kit, according to the manufacturers’ instructions. Equal molar quantities of the two fractions per sample were pooled and subjected to SMRTbell template preparation.
SMRTbell libraries were prepared according to the manufacturer’s protocol. DNA-damage repair was performed and was followed by end repair, blunt ligation of SMRTbell adaptors, and exonuclease III and IV digestion of any unligated material. SMRTbells were purified with 1× AMPure PacBio (PB) beads and eluted in 30 μL of PacBio elution buffer. The samples were transferred to a fresh tube. The concentration was determined with a Qubit dsDNA HS Assay Kit (Thermo Fisher), and the quality was assessed with an Agilent cDNA 12K Kit or HS Kit, according to the manufacturers’ instructions.
For sequencing, SMRTbell libraries were annealed to sequencing primer v3 sequenced with 2.1 chemistry, and 10-h movies were collected on the Sequel system according to the manufacturer’s instructions. Further information on the usage of long-read sequencing can be found in ref. 95 and at https://www.pacb.com/smrt-science/smrt-sequencing/.
Statistics and reproducibility
EBayes adjusted P values of RNA-seq data were corrected for multiple testing with the Benjamini–Hochberg method and used to select genes with significant expression differences. One representative western blot from of two independent experiments is shown in the main text. t-tests in MSstats were used to calculate P values in the proteomic analysis86. Growth-curve experiments were performed two times, and the results of one representative experiment are shown in the main text.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
RNA-seq data associated with this study are available through the Gene Expression Omnibus (GEO) data repository, accession number GSE103604. Source data for Fig. 2a,c and 3h,j are available in Supplementary Dataset 2. Data underlying the analysis in Fig. 4f are available in Supplementary Dataset 3. All other data are available upon reasonable request.
References
Marazzi, I. et al. Suppression of the antiviral response by an influenza histone mimic. Nature 483, 428–433 (2012).
Rialdi, A. et al. The RNA exosome syncs IAV-RNAPII transcription to promote viral ribogenesis and infectivity. Cell 169, 679–692.e14 (2017).
Ayllon, J. & García-Sastre, A. The NS1 protein: a multitasking virulence factor. Curr. Top. Microbiol. Immunol. 386, 73–107 (2015).
Gack, M. U. et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell. Host. Microbe. 5, 439–449 (2009).
Li, S., Min, J. Y., Krug, R. M. & Sen, G. C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349, 13–21 (2006).
García-Sastre, A. et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252, 324–330 (1998).
Donelan, N. R., Basler, C. F. & García-Sastre, A. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 77, 13257–13266 (2003).
Jackson, D., Hossain, M. J., Hickman, D., Perez, D. R. & Lamb, R. A. A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl Acad. Sci. USA 105, 4381–4386 (2008).
Carrillo, B. et al. The influenza A virus protein NS1 displays structural polymorphism. J. Virol. 88, 4113–4122 (2014).
Hale, B. G. Conformational plasticity of the influenza A virus NS1 protein. J. Gen. Virol. 95, 2099–2105 (2014).
Davey, N. E. et al. Attributes of short linear motifs. Mol. Biosyst. 8, 268–281 (2012).
Gitlin, L., Hagai, T., LaBarbera, A., Solovey, M. & Andino, R. Rapid evolution of virus sequences in intrinsically disordered protein regions. PLoS. Pathog. 10, e1004529 (2014).
Taubenberger, J. K. & Morens, D. M. The pathology of influenza virus infections. Annu. Rev. Pathol. 3, 499–522 (2008).
Taubenberger, J. K. & Kash, J. C. Insights on influenza pathogenesis from the grave. Virus. Res. 162, 2–7 (2011).
Taubenberger, J. K. et al. Reconstruction of the 1918 influenza virus: unexpected rewards from the past. mBio 3, e00201–e00212 (2012).
Hale, B. G., Randall, R. E., Ortín, J. & Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89, 2359–2376 (2008).
Krug, R. M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 12, 1–6 (2015).
Obenauer, J. C. et al. Large-scale sequence analysis of avian influenza isolates. Science 311, 1576–1580 (2006).
Neumann, G., Whitt, M. A. & Kawaoka, Y. A decade after the generation of a negative-sense RNA virus from cloned cDNA: what have we learned? J. Gen. Virol. 83, 2635–2662 (2002).
Van Roey, K. et al. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 114, 6733–6778 (2014).
Zhu, Q. et al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477, 179–184 (2011).
Manicassamy, B. et al. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl Acad. Sci. USA 107, 11531–11536 (2010).
Geiss, G. K. et al. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc. Natl Acad. Sci. USA 99, 10736–10741 (2002).
Hatada, E. & Fukuda, R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J. Gen. Virol. 73, 3325–3329 (1992).
Nemeroff, M. E., Qian, X. Y. & Krug, R. M. The influenza virus NS1 protein forms multimers in vitro and in vivo. Virology 212, 422–428 (1995).
Bornholdt, Z. A. & Prasad, B. V. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature 456, 985–988 (2008).
Molliex, A. et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015).
Lin, Y., Protter, D. S., Rosen, M. K. & Parker, R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 (2015).
Kwon, I. et al. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155, 1049–1060 (2013).
Banani, S. F. et al. Compositional control of phase-separated cellular bodies. Cell 166, 651–663 (2016).
Kato, M. et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 (2012).
Harlen, K. M. & Churchman, L. S. The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 18, 263–273 (2017).
Heyn, P., Salmonowicz, H., Rodenfels, J. & Neugebauer, K. M. Activation of transcription enforces the formation of distinct nuclear bodies in zebrafish embryos. RNA. Biol. 14, 752–760 (2017).
Hnisz, D., Shrinivas, K., Young, R. A., Chakraborty, A. K. & Sharp, P. A. A phase separation model for transcriptional control. Cell 169, 13–23 (2017).
Lu, H. et al. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323 (2018).
Richard, P. & Manley, J. L. Transcription termination by nuclear RNA polymerases. Genes Dev. 23, 1247–1269 (2009).
Schlackow, M. et al. Distinctive patterns of transcription and RNA processing for human lincRNAs. Mol. Cell 65, 25–38 (2017).
Martinson, H. G. An active role for splicing in 3′-end formation. Wiley Interdiscip. Rev. RNA 2, 459–470 (2011).
Misra, A., Ou, J., Zhu, L. J. & Green, M. R. Global promotion of alternative internal exon usage by mRNA 3′ end formation factors. Mol. Cell 58, 819–831 (2015).
Misra, A. & Green, M. R. From polyadenylation to splicing: dual role for mRNA 3′ end formation factors. RNA. Biol. 13, 259–264 (2016).
Tseng, C. K. et al. Human telomerase RNA processing and quality control. Cell Rep. 13, 2232–2243 (2015).
Orphanides, G. & Reinberg, D. A unified theory of gene expression. Cell 108, 439–451 (2002).
Perales, R. & Bentley, D. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol. Cell 36, 178–191 (2009).
Mapendano, C. K., Lykke-Andersen, S., Kjems, J., Bertrand, E. & Jensen, T. H. Crosstalk between mRNA 3′ end processing and transcription initiation. Mol. Cell 40, 410–422 (2010).
Gu, B., Eick, D. & Bensaude, O. CTD serine-2 plays a critical role in splicing and termination factor recruitment to RNA polymerase II in vivo. Nucleic Acids Res. 41, 1591–1603 (2013).
Niwa, M., Rose, S. D. & Berget, S. M. In vitro polyadenylation is stimulated by the presence of an upstream intron. Genes Dev. 4, 1552–1559 (1990).
Dye, M. J. & Proudfoot, N. J. Terminal exon definition occurs cotranscriptionally and promotes termination of RNA polymerase II. Mol. Cell 3, 371–378 (1999).
Bauer, D. L. V. et al. Influenza virus mounts a two-pronged attack on host RNA polymerase II transcription. Cell Rep. 23, 2119–2129.e3 (2018).
Heinz, S. et al. Transcriptional elongation can affect 3D genome architecture. Cell (in the press).
Kuo, R. L. & Krug, R. M. Influenza a virus polymerase is an integral component of the CPSF30-NS1A protein complex in infected cells. J. Virol. 83, 1611–1616 (2009).
Pichlmair, A. et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 487, 486–490 (2012).
Rozenblatt-Rosen, O. et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495 (2012).
Marazzi, I. & Garcia-Sastre, A. Interference of viral effector proteins with chromatin, transcription, and the epigenome. Curr. Opin. Microbiol. 26, 123–129 (2015).
Heaton, N. S. et al. Targeting viral proteostasis limits influenza virus, HIV, and dengue virus infection. Immunity 44, 46–58 (2016).
Marazzi, I., Greenbaum, B. D., Low, D. H. P. & Guccione, E. Chromatin dependencies in cancer and inflammation. Nat. Rev. Mol. Cell Biol. 19, 245–261 (2018).
Maldonado, E., Cabrejos, M. E., Banks, L. & Allende, J. E. Human papillomavirus-16 E7 protein inhibits the DNA interaction of the TATA binding transcription factor. J. Cell. Biochem. 85, 663–669 (2002).
Dasgupta, A. Targeting TFIIH to inhibit host cell transcription by Rift Valley Fever Virus. Mol. Cell 13, 456–458 (2004).
Di Valentin, E. et al. Varicella-zoster virus IE63 protein represses the basal transcription machinery by disorganizing the pre-initiation complex. Biol. Chem. 386, 255–267 (2005).
Kundu, P., Raychaudhuri, S., Tsai, W. & Dasgupta, A. Shutoff of RNA polymerase II transcription by poliovirus involves 3C protease-mediated cleavage of the TATA-binding protein at an alternative site: incomplete shutoff of transcription interferes with efficient viral replication. J. Virol. 79, 9702–9713 (2005).
Fraser, K. A. & Rice, S. A. Herpes simplex virus immediate-early protein ICP22 triggers loss of serine 2-phosphorylated RNA polymerase II. J. Virol. 81, 5091–5101 (2007).
Geng, F., Wenzel, S. & Tansey, W. P. Ubiquitin and proteasomes in transcription. Annu. Rev. Biochem. 81, 177–201 (2012).
Hagai, T., Azia, A., Babu, M. M. & Andino, R. Use of host-like peptide motifs in viral proteins is a prevalent strategy in host-virus interactions. Cell Rep. 7, 1729–1739 (2014).
Daugherty, M. D. & Malik, H. S. Rules of engagement: molecular insights from host-virus arms races. Annu. Rev. Genet. 46, 677–700 (2012).
Garamszegi, S., Franzosa, E. A. & Xia, Y. Signatures of pleiotropy, economy and convergent evolution in a domain-resolved map of human-virus protein-protein interaction networks. PLoS. Pathog. 9, e1003778 (2013).
Dyson, H. J. & Wright, P. E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 (2005).
Uversky, V. N. & Dunker, A. K. Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264 (2010).
Babu, M. M., Kriwacki, R. W. & Pappu, R. V. Versatility from protein disorder. Science 337, 1460–1461 (2012).
Tompa, P., Davey, N. E., Gibson, T. J. & Babu, M. M. A million peptide motifs for the molecular biologist. Mol. Cell 55, 161–169 (2014).
Coletta, A. et al. Low-complexity regions within protein sequences have position-dependent roles. BMC Syst. Biol. 4, 43 (2010).
Beltrao, P. et al. Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 (2012).
Holt, L. J. et al. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 (2009).
Mosca, R., Pache, R. A. & Aloy, P. The role of structural disorder in the rewiring of protein interactions through evolution. Mol. Cell. Proteomics. 11, 014969 (2012).
Schrauwen, E. J. & Fouchier, R. A. Host adaptation and transmission of influenza A viruses in mammals. Emerg. Microbes Infect. 3, e9 (2014).
Miller, M. S. & Palese, P. Peering into the crystal ball: influenza pandemics and vaccine efficacy. Cell 157, 294–299 (2014).
Taft, A. S. et al. Identification of mammalian-adapting mutations in the polymerase complex of an avian H5N1 influenza virus. Nat. Commun. 6, 7491 (2015).
Eswar, N., Eramian, D., Webb, B., Shen, M. Y. & Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 (2008).
Penkert, R. R., DiVittorio, H. M. & Prehoda, K. E. Internal recognition through PDZ domain plasticity in the Par-6–Pals1 complex. Nat. Struct. Mol. Biol. 11, 1122–1127 (2004).
London, N., Raveh, B., Cohen, E., Fathi, G. & Schueler-Furman, O. Rosetta FlexPepDock web server: high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 39, W249–W253 (2011).
Reverter, D. & Lima, C. D. A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2-SUMO complex. Structure 12, 1519–1531 (2004).
Halfmann, R. & Lindquist, S. Screening for amyloid aggregation by semi-denaturing detergent-agarose gel electrophoresis. J. Vis. Exp. 17, e838 (2008).
Shah, N. B. & Duncan, T. M. Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J. Vis. Exp. 84, e51383 (2014).
Martínez-Sobrido, L. & García-Sastre, A. Generation of recombinant influenza virus from plasmid DNA. J. Vis. Exp. 42, 2057 (2010).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome. Res. 10, 1794–1805 (2011).
The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017).
Choi, M. et al. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 (2014).
Mi, H. et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189 (2017).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome. Biol. 15, R29 (2014).
Rahl, P. B. et al. c-Myc regulates transcriptional pause release. Cell 141, 432–445 (2010).
Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genomics. Proteomics. Bioinformatics. 13, 278–289 (2015).
Acknowledgements
We thank all members of the laboratories of I.M. and A.G.-S., and J. Bloom and A. Kornblihtt for valuable discussions and suggestions on the manuscript. We thank the Medicinal Chemistry Core, Integrated Screening Core, Microscopy CoRE,, and Global Health and Emerging Pathogens Institute (GHEPI) at the Icahn School of Medicine at Mount Sinai. H.v.B., I.M., and A.G.-S. are partially supported by HHSN272201400008C–Center for Research on Influenza Pathogenesis (CRIP), a NIAID-funded Center of Excellence for Influenza Research and Surveillance (CEIRS). I.M. is supported in part by the Department of Defense W911NF-14-1-0353. I.M. and H.v.B. are supported by NIH grant 1R01AN3663134. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry. This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Author information
Authors and Affiliations
Contributions
N.Z., N. Moshkina, A.R., and V.Y. performed experiments with modeling of PDZ–peptide–SUMO complexes, transfection, infection, WB, SDD-AGE, purification of recombinant proteins, BLI, pulldown assays, analysis of proteins stability, immunofluorescence, RT–PCR, chemical synthesis of B-isox, and B-isox-mediated precipitation; V.S., S.R., and B.H. performed independent validation of SUMOylation of the SUP domain; N. Mena, R.A., M.T.S.-A., J.A., and T.T. performed experiments with rescue of recombinant influenza viruses, single-cycle growth curves, plaque phenotypes, and analysis of infectious viral progeny; J.H., D.J.-M., and Y.M. performed experiments with preparation of peptides for MS, protein identification by LC–MS/MS, and statistical relative quantification of proteins and enrichment analysis; R.F., M.S., D.P., A.K., M.B., M.L.S., R.S., and H.v.B. performed directional RNA-seq, differential gene expression analysis, TR and intron/exon ratio calculation, and iso-seq analysis. S.-Y.H., D.L., and E.G. performed bioinformatics analysis on splicing and termination; B.G. performed conservation analysis; J.J., R.K.P., A.T., and N.K. provided materials and insights in experimental procedures; and A.G.-S. and I.M. supervised the project. I.M. wrote the paper, on which coauthors provided feedback.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 NS1 C-terminal diversity in different strains of influenza virus.
a, Diagram representing the C-terminus of an NS1 protein in different Influenza strains. b, Visual representation of the conservation score among NS1 proteins averaged over 10 amino acidic residues.
Supplementary Figure 2 Control experiments.
Control experiments for Fig. 2. a, IF of ectopically expressed GFP, NS1, NS1-KR and NS1-SUMO in A549 (scale bar indicates 100um). b, Pull-down assay between recombinant NS1, NS1-KR, NS1-SUMO and PSD95. PBS served as a negative control. Coomassie staining SDS-PAGE showing purified recombinant proteins (left) and eluted complexes (right). c-e, Structural modeling of SUMOylated NS1 tail and the indicated PDZ domain containing-proteins GIPC2 (c), DVL1 (d) and PSD95 (e). f, Schematic diagram of plaque selection and analysis. All plaques selected after 24 hpi with NS1-SUMO virus indicate that the virus has reverted back to NS1 virus (WT virotype) by mutagenesis of its C-term SUMO extension. This result indicates that NS1-SUMO virus can infect but it is compromised in its ability to bud and spread from cell-to-cell.
Supplementary Figure 3 Control experiments for 1918 NS1 and NS1-SUMO viruses.
a, A549 cells were mock infected (M) or infected with NS1 and NS1-SUMO virus for 12 hours and sequentially treated with mg132 (50 nM) or cycloheximide (CHX, 50 ug/mL) for 0/4/8/24hours. Lysates were immunoblotted with anti-NS1 antibody. b, Cumulative expression levels of positive- and negative-sense viral RNAs for all segments in A549 cells at 6 or 12 hours post-infection with NS1 virus (blue), or NS1-SUMO virus (red). Normalized log2 counts of viral RNAs per million sequenced reads (CPM) are shown for two biological replicate experiments.
Supplementary Figure 4 Control experiments.
Control experiments for Fig. 3. a, Synthetic route of B-isox. Reagents and conditions: (1) HOSu, EDC·HCl, THF, RT, overnight; (2) 6-amino-1-hexanol, THF, 5 hours; (3) Biotin, DMAP, EDC·HCl, DCM, 3 days. b, LC-HRMS of synthesized B-isox. c, Silver staining gel showing B-isox-mediated precipitation of proteins from cell lysate. d, Statistical Analyses of the B-isox precipitated proteins after infection. Statistical quantitation of the overlap between proteins precipitated by B-isox during infection compared to a set of 1286 B-isox-precipitated proteins from the McKnight’s lab (Cell, 2013, 155(5): 1049-1060), a set of 1786 RNA binding proteins from UniProt, a set of 126 literature-cited RNA granule proteins, and the entire human proteome. Fisher’s exact test was used to determine the significance of the overlapping between datasets. e, qPCR analysis of run through transcription downstream of the GAPDH and CDC25A genes in A549 cells infected with WT or NS1-GFP A/Vietnam/1203/2004/H5N1 influenza virus at MOI of 3 for 6 and 12 hours. Expression levels were calculated by using primer sets amplifying region downstream of termination sites and calculating fold enrichment comparing 6 hours and 12 hours time post infection (as mock infection has no detectable signal indicating absence of RNAPII run-through). Error bars correspond to mean ± s.e.m.
Supplementary Figure 5 Control experiments.
Control experiments related to Fig. 4. a, TR density at termination region plotted as a function of gene induction (red) or repression (green) during infection with NS1-SUMO virus at 6 hpi (left) and 12 hpi (right). b, Normalized RNA-Seq read coverage tracks showing increased 3’-end transcript levels for representative genes: CXCL1, MYC, NFKBIA, and CDC25A.
Supplementary Figure 6 TR region changes at specific gene sets and human variation in SUMO-modifying genes.
a, TR plots at termination region of active (RPKM > 1 in 50% of samples) multi-exonic (upper panels) and mono-exonic (lower panels) genes in uninfected A549 cells and 6 or 12 hpi with NS1 or NS1-SUMO virus. Data is shown for two replicates in each condition. b, Scatter plot showing the relationship between the log fold-change (logFC) in gene expression between NS1-SUMO virus and NS1 virus-infected A549 cells at 6 hpi, and the logFC in transcript levels in 5,000 bp 3’ gene-flanking regions. Genes with a significant increase or decrease in expression in NS1-SUMO vs NS1 infected A549 cells at 6 hpi are highlighted in red and green, respectively. All other genes are highlighted in grey. Solid and dotted lines correspond to the regression line and 95% confidence interval for genes with significant expression differences, with the R2 and significance shown at the top-left. c, Analysis of SUMO-modifying gene mutations using a compendium of protein-coding genetic variation in 60,706 humans from the ENCODE Consortium. The probability of being loss-of-function intolerant (intolerant of both heterozygous and homozygous of variants) is shown for all genes compared to SUMO-modifying genes.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–6
Supplementary Table 1
Information of PDZ domain containing proteins used in this study
Supplementary Table 2
Cross-comparison of B-isox precipitated proteins
Supplementary Table 3
Quantitative mass-spectrometry analysis of B-isox precipitated proteins
Supplementary Table 4
RNA-seq analysis of NS1 vs NS1-SUMO viral infection
Supplementary Table 5
Human polymorphism and damaging-mutations of SUMO-modifying enzymes
Supplementary Dataset 1
Uncropped WB images for Figures 1C, 1D, 1E, 3A, and 3C
Supplementary Dataset 2
Supporting data for Figures 2A, 2C, 3H, 3I, and 3J
Supplementary Dataset 3
Supporting data for Figure 4F, containing the raw circular consensus sequencing (CCS) reads, the reads after adapter trimming (cutadapt.fa), removal of Poly(A) tails (cutadapt_polA.fa), and removal of short reads <300 nt (cutadapt_polA_gt300.fa)
Rights and permissions
About this article

Cite this article
Zhao, N., Sebastiano, V., Moshkina, N. et al. Influenza virus infection causes global RNAPII termination defects. Nat Struct Mol Biol 25, 885–893 (2018). https://doi.org/10.1038/s41594-018-0124-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-018-0124-7
This article is cited by
-
The host RNA polymerase II C-terminal domain is the anchor for replication of the influenza virus genome
Nature Communications (2024)
-
mRNA 3’UTR lengthening by alternative polyadenylation attenuates inflammatory responses and correlates with virulence of Influenza A virus
Nature Communications (2023)
-
Cut site preference allows influenza A virus PA-X to discriminate between host and viral mRNAs
Nature Microbiology (2023)
-
Avian influenza viruses suppress innate immunity by inducing trans-transcriptional readthrough via SSU72
Cellular & Molecular Immunology (2022)
-
3 ′-5 ′ crosstalk contributes to transcriptional bursting
Genome Biology (2021)
