
Abstract
Herpes simplex virus type-1 (HSV-1) is a human pathogenic member of the Alphaherpesvirinae subfamily of herpesviruses. The HSV-1 genome is a large double-stranded DNA specifying about 85 protein coding genes. The latest surveys have demonstrated that the HSV-1 transcriptome is much more complex than it had been thought before. Here, we provide a long-read sequencing dataset, which was generated by using the RSII and Sequel systems from Pacific Biosciences (PacBio), as well as MinION sequencing system from Oxford Nanopore Technologies (ONT). This dataset contains 39,096 reads of inserts (ROIs) mapped to the HSV-1 genome (X14112) in RSII sequencing, while Sequel sequencing yielded 77,851 ROIs. The MinION cDNA sequencing altogether resulted in 158,653 reads, while the direct RNA-seq produced 16,516 reads. This dataset can be utilized for the identification of novel HSV RNAs and transcripts isoforms, as well as for the comparison of the quality and length of the sequencing reads derived from the currently available long-read sequencing platforms. The various library preparation approaches can also be compared with each other.
Design Type(s) | transcription profiling by high throughput sequencing design • parallel group design |
Measurement Type(s) | total RNA |
Technology Type(s) | RNA sequencing assay |
Factor Type(s) | temporal_interval • assay material selection • cap analysis of gene expression assay • enzymatic amplification • size fractionation • nucleic acid library construction protocol • Technology Platform |
Sample Characteristic(s) | Human alphaherpesvirus 1 |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others
Background & Summary
Herpes simplex virus type-1 (HSV-1) is a human pathogenic herpesvirus belonging to the family of Herpesviridae. HSV-1 is associated with orofacial infections1. This is a highly contagious and lifelong infection that is widespread in the world2. HSV-1 is a prototype herpesvirus, which is used as a model organism in the study of molecular pathogenesis of the lytic and latent viral infection, and it is also used as a gene delivery vector3,4 for basic research and gene therapy5, as well as in oncolytic virotherapy6. HSV-1 has a large (~152 kbp) double-stranded DNA genome and a very complex transcriptome7.
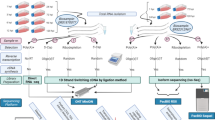
Here, we provide a large RNA-seq dataset derived from third- and fourth-generation8 long-read sequencing (LRS) methods (Fig. 1). The previously applied short-read sequencing approaches are inefficient in distinguishing between embedded gene products, RNA isoforms and overlapping transcripts. This problem can be circumvented by LRS methods that are capable of identifying full-length transcripts9–13. Our major aim with this study was to provide a dataset that can be suitable for deeply characterizing the complexity of the HSV-1 transcriptome profile and for obtaining a detailed picture on the transcript-level variations. In order to achieve this goal, the currently available LRS techniques, the Pacific Biosciences (PacBio) and the Oxford Nanopore Technologies (ONT) platforms were used for the characterisation of the HSV-1 lytic transcriptome. We used the RSII7 and the Sequel isoform sequencing (Iso-Seq) approaches for the analyses of both the polyadenylated fraction and the total transcriptome of the HSV-1 transcripts. The amplified Iso-Seq method was used for cDNA sequencing. We used the ONT MinION technique for both cDNA and direct (d)RNA sequencing for the investigations of the poly(A) RNA molecules.

Data flow diagram shows the detailed overview of this research.
The utilized sequencing methods along with the various library preparation approaches were as follows, PacBio: oligo(d)T-primed and random-hexamer primed Iso-Seq protocol with or without size-selection, as well as ONT: cDNA-sequencing, cDNA-sequencing using 5′ Cap-selected transcripts, and dRNA sequencing. These techniques allow for the detection of novel transcript isoforms (splice and length variants), as well as several novel protein-coding RNAs, non-coding (nc)RNAs, and polycistronic transcripts. This LRS dataset is also useful for the investigation of the transcriptional overlaps. Additionally, these data can be used to compare the “sequencing by synthesis” method with the nanopore-based approach. The various library preparation approaches can also be compared with each other.
We used 17 SMRT Cells for the sequencing of our samples on RSII system, using the P6-C4 chemistry. The movie time was 240 or 360 per SMRT Cell. Samples were also run on a single Sequel SMRT Cell using P6-C4 chemistry with 600 min movie time. Altogether 7 MinION flow cells were applied for sequencing the different ONT libraries. The raw reads were aligned to the HSV-1 reference genome in each case (X14112).
The PacBio RSII sequencing generated 38,601 reads of inserts (ROIs) using oligo(d)T-primed reverse transcription (RT), while, using the Sequel platform, we obtained 77,851 HSV-1 specific ROIs. Overall, the different ONT methods generated 175,169 viral reads (Table 1). The average read lengths are as follows: 1,536 and 1,000 bp for the no size-selected oligo(d)T-primed and random-primed RSII, respectively; 2,104 for the Sequel, while the obtained read lengths are 1,466, 1,067 and 1,111 bp for the MinION cDNA, Cap-selected cDNA, and direct RNA-seq methods (Table 2, Fig. 2).

Error bars represents the standard deviance (SD).
The dataset generated here is expected to provide valuable information for the herpesvirus studies and will open new possibilities for the investigation of HSV-1 at the molecular level. Our data provides a wide-ranging resource for understanding, comparing, and analysing the utilized library preparation and sequencing methods, as well as this valuable data can be used to design novel bioinformatics pipelines and also to test the currently available ones.
Here, we provide a comprehensive overview about preparation methods of sequencing libraries, as well as a description of the data (Figure 1, Table 3). Detailed statistics about the read quality, such as insertions, deletions, and mismatches, as well as the coverages of the pre-processed data (binary alignment (BAM)) can be found in Table 4 and Fig. 3. The data show that the ONT MinION sequencing resulted in relatively high error rates for insertions, deletions and mismatches. The best read quality (i.e. less insertions, deletions and mismatch) – as expected – was obtained from the PacBio runs. The composition of the errors of the three sequencers (RSII, Sequel, MinION) are different. Mismatches are the most common errors in ONT, while the insertions are the least frequent errors in ONT (consistent with the previously published data14). Intriguingly, contrary to others’ datasets14, deletions are the major errors in our RSII data; the Sequel shows the same pattern as was expected14. The read length distribution of the samples is visualized in Fig. 4 and Fig. 5 Porechop tool (https://github.com/rrwick/Porechop) was used to carry out an analysis on the 5′ and 3′ adapters, with which we were able to determine the orientation of the sequencing reads (Table 5 (available online only), Table 6, Fig. 6). About 90% of the PacBio reads could be categorized as forward or reverse oriented read, but only 66%, 58% and 49% from the 1D-seq, Cap-seq and random libraries could be categorized, respectively.

(a) Percentage of deletions; error bar represents the SD values. (b) Percentage of insertions with SD values. (c) Percentage of mismatches given together with SD values. (d) Average read coverages across the HSV-1 genome.

(a) The average distribution of read lengths which align to the HSV-1 genome; binned to 500 bp intervals. *Percentage values are in Figure 3. (b) The divided bar chart shows the number of reads at log10 scale, binned to 100 bp intervals. The samples can be seen broken down according to the library preparation and/or sequencing approaches in C–E. (c) Read lengths distributions at log10 scale, binned to 1,000 bp ranges. This figure shows the RSII size-selected samples. It can be seen that the majority of the reads mapped to the HSV-1 genome are shorter than 3,000 bps, most of the reads are between 1,001–2,000 bps, independently from the focus of the size selection. (d) This bar chart shows that the same library preparation method (Isoform sequencing with no size selection using Clontech SMARTer PCR cDNA Synthesis Kit) result in different read length distribution using the two PacBio platforms: 40% of the Sequel reads are longer than 2,000 bp, while only 10% of the RSII reads belong to this size range. (e) The average read length using the MinION nanopore sequencing is below 2,000 bp. There is no random primed read above 9,000 bp. Intriguingly, the longest reads are derived from the direct RNA sequencing approach (15,000–19,000 bp).

This heat map-like illustration shows the distribution of read counts within 500 bp intervals. This dataset shows that the Sequel sequencer resulted in longer average read lengths (~79% of the reads are longer than 1,500 bp). The size selection of the RSII samples had a significant effect on the average read lengths: even if most of the reads are within 1,000–2,000 bp, the distribution of the read counts is different: 0.8–2 kb: 87,560%; 2–3 kb: 73,493%; 3–5 kb: 73,543%; >5 kb: 69,487% of the reads are within this range. The MinION sequencing on the poly(A)-selected samples shows about the same read length distribution as the non-size-selected RSII (the majority of the reads is within the 1,000–2,000 bp interval, however, the distribution of the MinION reads is smoother. The Cap-selected samples and the direct RNA samples have a peak at the lower range: about 30–35% of the reads are between 501–1,000 and 1,001–1,500 bp in both samples. The random primed samples (RSII and MinION) generated the shortest average read lengths.

(a) This figure shows the percentages of read counts derived from different PacBio RSII poly(A) sequencing. (b) This bar chart compares the data derived from the two PacBio approaches. We could detect the adapters on more than 90% Sequel reads, while we obtained in a poorer result from RSII. (c) This diagram compares the ONT’s own library preparation approach (1D) with the Lexogen Teloprime Cap-selection protocol, which used for a library, run on a MinION flow cell. (d) We were not able to identify the orientation of about 50% of reads using the random-primed approach, while dRNA-seq could detect adapters on about 20% of the reads. (e) Segmented pie chart illustrates the distribution of reads with (labelled as: Fw and Rev) or without orientation (labelled as: No orientation). Fifty-six % of this dataset derived from the different nanopore sequencing approaches, 26.5% from Sequel and the remaining less than 20% is from the RSII. A high ratio (~90%) of reads from PacBio RSII and Sequel sequencing could be categorized as Fw or Rev, however Porechop were not able to identify the adapters and thus the orientation of at least about 30% of reads.
The ratio of the complete (full-length) reads in the different samples were also calculated (Fig. 7). The data shows that the random-primed cDNA sequencing as well as the dRNA sequencing resulted in very few complete reads, which has a technical reason: random primed sequencing could gain complete reads when the random primer binds to the 3′-end of the RNA. The small number of full-length reads, from the dRNA sequencing is consistent with our previous findings: we observed that the dRNA sequencing resulted in poor 5′ and 3′ read ends15,16. It can also be seen that nanopore sequencing produces a large number of incomplete reads; these short fragments are probably removed from PacBio sequencing by the MagBead loading.

Bar chart represents the full-length and partial read distributions in the different libraries.
Methods
A part of the methods section (PacBio RSII sequencing) is an expanded version of descriptions in our related work7, however, the largest part of the data including the entire PacBio Sequel and ONT sequencing data has not yet been published elsewhere.
The various library preparation and sequencing methods utilized in this study are shown in Figure 1.
Viruses, cells and infection
The Vero immortalized kidney epithelial cell line, derived from the kidney of an African green monkey (Cercopithecus aethiops) was used for maintaining and propagating HSV-1. The cell culture was grown in Dulbecco’s modified Eagle medium (DMEM, Gibco/Thermo Fisher Scientific) with 10% Foetal Bovine Serum (Gibco/Thermo Fisher Scientific) and 100 μl/ml Penicillin-Streptomycin 10 K/10 K Mixture (Lonza), and in a 37 °C incubator with a humidified atmosphere of 5% CO2 in air. The viral stocks were prepared by infecting rapidly-growing semi-confluent cells with viruses at a multiplicity of infection (MOI) of 1 plaque-forming unit (pfu)/cell. The infected cells were incubated until complete cytopathic effect was observed. Three times freeze-thaw cycles were applied and it was followed by a centrifugation step at 10,000×g for 15 min. Cells were infected in a suspension with HSV-1 at an MOI of 1, then they were incubated for 1 h. The virus suspension was removed and cells were washed with phosphate-buffered saline (PBS). This step was followed by the addition of fresh medium to the cells and they were incubated for 1, 2, 4, 6, 8, or 12 h for the RSII sequencing and for 1, 2, 3, 4, 5, 6, 8, 12, 18, or 24 h for the Sequel and MinION runs.
RNA purification
Total RNA extraction
Total RNA isolation was carried out by using the NucleoSpin® RNA kit (Macherey-Nagel) following the kit’s recommendations and our previously published methods12. In sum, the viral infected cells were lysed in a lysis puffer (containing chaotropic ions which inhibit ribonucleases, supplied by the kit). DNase I (provided by the kit) treatment was carried out during the purification. The RNA samples, which were bound to a silica membrane, were eluted in RNase-free water. The potential residual DNA contamination was eliminated by applying additional DNase treatment; the Ambion® TURBO DNA-freeTM Kit was used (Thermo Fisher Scientific). The final concentrations of the RNA samples were measured by Qubit® 2.0 Fluorometer using Qubit RNA BR Assay Kit (Life Technologies) and then they were stored at −80 °C until further use. The RNA samples taken from each time points were mixed for library preparation and sequencing.
Ribosomal RNA depletion
For the random hexamer-primed cDNA sequencing, the RNA samples were handled with the ribodepletion kit (Epicentre Ribo-ZeroTM Magnetic Kit H/M/R, Epicentre/Illumina), to remove the ribosomal (r)RNAs.
Isolation of polyadenylated RNA
For the sequencing of the polyadenylated [polyA(+)] fraction of the samples, RNAs were purified by using the Qiagen Oligotex mRNA Mini Kit, following to the “Spin Columns” protocol of the kit.
The final concentrations of the rRNA depleted and the PolyA(+) RNA samples were determined through use of the Qubit RNA HS Assay Kit (Life Technologies).
Preparation of cDNAs and sequencing libraries
PacBio SMRTbell library preparation
Full-length cDNAs were prepared according to the PacBio Isoform Sequencing (Iso-Seq) protocol by using the Clontech SMARTer PCR cDNA Synthesis Kit. We applied the no Size Selection protocol for the study of short RNAs, while we carried out size selection by using SageELFTM and BluePippinTM Size-Selection Systems (Sage Science) for the isolation of long transcripts. Reverse transcription (RT) reactions were primed with oligo(d)T (supplied by the Clontech Kit) or random hexamer primer (custom designed, ordered from IDT DNA). Samples were amplified by PCR using KAPA HiFi Enzyme (Kapa Biosystems), according to the above mentioned PacBio protocol. In short, primary denaturation was done at 95 °C for 2 min. This step was followed by 16 cycles for PA-sequencing, as well as 20 or 30 cycles for random hexamer-primed samples (the optimal cycle was determined in the optimization step) at 98 °C for 20 s (denaturation), 65 °C for 15 s (annealing) 72 °C for 4 min (extension). 72 °C was used for the final extension, it was set to 5 min. (n: 18 cycles was set to the No size-selection protocol, and the same PCR setting was applied for size-selected samples.
PCR products were mixed together and then size selected with the SageELFTM System following the PacBio’s protocol. Size-selected cDNAs were amplified with KAPA enzyme using the above mentioned conditions. The cDNAs with a size over 5 kb was size-selected again, with the BluePippinTM System to remove the short samples. Five-hundred ng of each non-size-selected sample was used for the generation of the SMRTbell templates, with the PacBio DNA Template Prep Kit 1.0. The quantity of the size-selected cDNAs used in the SMRTbell template preparation reaction was based on the following protocols: Procedure & Checklist – Isoform Sequencing (Iso-SeqTM) using the Clontech SMARTer PCR cDNA Synthesis Kit and SageELFTM Size Selection System & BluePippinTM Size-Selection Systems. The DNA/Polymerase Binding Kit P6 and v2 primers were used to generate SMRTbell library-DNA polymerase complexes. The ready complexes were bound to MagBeads using the PacBio MagBead Binding Kit.
The volume of the sequencing primer for the annealing, and the polymerase (P5 or P6) for the binding was determined using the PacBio Calculator version 2.3.1.1. by adding the concentrations and the average insert sizes of SMRTbell templates.
The polymerase-template complexes were bound to MagBeads, loaded onto SMRT Cells and sequenced on the RSII instrument.
The Binding Calculator (PacBio) was used to calculate the amount of samples to be used for sequencing, following MagBead one-cell per well (OCPW) method. The P6v2 binding kit was used, the on-plate concentration was set to 0.05 nM. The insert sizes were set based on the applied size-selections: 1000, 2500 and 6000 bp sizes were chosen.
Briefly, the sequencing primer was diluted in PacBio Elution Buffer (EB) to 150 nM. The annealing step was carried out using 1 μl template (cc: ~20 ng/μl), the diluted sequencing primer and primer buffer (10×). The concentration of this reaction mixture was 0.8333 nM. Annealing step was set to 20 °C for 30 min. The DNA polymerase enzyme was diluted to a final concentration of 50 nM in PacBio Binding Buffer (BB) v2, and then it was bound to the annealed sample followed by the addition of DTT, dNTP and BB. The sample complex (final concentration was set to 0.5 nM) was incubated at 30 °C for 4 h. The complex (0.5 μl) was added to 18.5 μl MagBead BB (final concentration was set to 0.0125 nM). The detailed protocol of the MagBeads preparation is as follows: 73.9 μl MagBeads were washed with MagBead Wash Buffer, then it was replaced by MagBead BB (73.9 μl). The washed MagBead was used to bound to the sample-complex. Nineteen μl from the complex was added to the washed MagBeads and then they were incubated in a HulaMixer (Life Technologies) at 4 °C for 30 min. After this binding step, the sample was purified with BB (19 μl), then with 19 μl WB. The final elution was in 19 μl BB. The MagBead-bound sample complexes were loaded for sequencing.
The MagBead One Cell Per Well protocol was applied for the sequencing. Sequencing runs were performed by using the PacBio RS II sequencer or Sequel platform. Size-selected and no size-selected samples were run on RSII, while the Sequel platform was applied for the no size-selected samples. The DNA Sequencing Reagent 4.0 was used for the reactions. Two hundred and forty min or 360 min movie lengths were set for the RSII runs, while 600 min was applied for the Sequel sequencing (one movie was recorded for each SMRT Cell).
Direct RNA sequencing
To avoid the potential false-priming effect of PCR reactions, an amplification free method was used for the analysis of HSV-1 transcripts. For this, the ONT’s Direct RNA sequencing (DRS) protocol (Version: DRS_9026_v1_revM_15Dec2016) was utilized. A total RNA sample containing an equal amount from the 10 time points (1, 2, 3, 4, 5, 6, 8, 12, 18, 24 h pi) was used to isolate the polyadenylated RNA fraction. This sample (115 ng) was then used as template for the RT, it was added to the RT adapter (supplied by the DRS kit) (Table 7). T4 DNA ligase (2 M U/ml; New England BioLabs) was also added to the reaction. The reaction mixture was incubated at room temperature for 10 min. SuperScript (SS)III RT enzyme (Life Technologies) was added to the RNA-cDNA hybrid, and the generation of the first-strand cDNA was carried out according to the ONT DRS kit’s recommendations: the reaction was done at 50 °C for 50 min, then the SSIII was inactivated at 70 °C (10 min). The RNase OUT (40 U/μl; Life Technologies) recombinant RNase inhibitor–treated Agencourt AMPure XP Magnetic Beads (Beckman Coulter) was used to purify the sample (2U RNase OUT per 1 μl bead), then it was eluted in 20 μl Nuclease-Free Water (Ambion/Thermo Fisher Scientific). T4 DNA ligase and NEBNext Quick Ligation Reaction Buffer (New England BiceoLabs) were used to ligate the purified sample to the RMX adapter. The ligation was done at room temperature. The incubation time was 10 min. The adapter-ligated sample was purified with the RNase OUT-handled XP beads using Wash Buffer (DRS Kit), and then eluted in 21 μl Elution Buffer (DRS Kit). The quantification of the ready libraries was performed using the Qubit 2.0 Fluorometer as well as Qubit dsDNA HS Assay Kit (both from Life Technologies). The R9.4 SpotON Flow Cell was used for MinION sequencing.
Oxford Nanopore 1D cDNA sequencing
Samples were sequenced on a MinION device from the ONT according to the ONT 1D Strand switching cDNA by ligation protocol (Version: SSE_9011_v108_revS_18Oct2016). The sequencing libraries were generated by using the ONT Ligation Sequencing Kit 1D (SQK-LSK108). The PolyA(+)-selected RNA fraction or the rRNA-depleted sample was used for cDNA production. Identical quantity of RNA samples from each of the applied time points were mixed together.
Thirty-one ng from the Poly(A+)-selected, or 50 ng from the rRNA-depleted sample was used as template for the RT reactions. An anchored oligo(d)T primer [(VN)T20; ordered from Bio Basic, Canada, (Table 7)] or a modified random-primer (Table 7) was used for priming the reactions. The RNA-primer mixture was incubated at 65 °C for 5 min. After this short step, a strand-switching oligo [containing three O-methyl-guanine RNA bases (PCR_Sw_mod_3G; Bio Basic, Canada)], buffer and DTT [both are derived from the SuperScript IV Reverse Transcriptase kit (Life Technologies)] were added to the reactions. The samples were handled with a recombinant RNase inhibitor (RNase OUTTM, Life Technologies); the incubation was carried out at 42 °C 2 min.
The SuperScript IV Reverse Transcriptase enzyme (200 unit) was added to the sample. The RT reaction was carried out in a Veriti Cycler (Applied Biosystems) at 50 °C for 10 min. It was followed by the strand-switching step at 42 °C for 10 min. Samples were heated to 80 °C (10 min) to inactivate the enzymes. Samples were amplified by PCR with KAPA HiFi DNA Polymerase (Kapa Biosystems) and Ligation Sequencing Kit Primer Mix (1D Kit). Five μl from the cDNAs were used as template in each of the PCR reactions. The Veriti PCR machine was set as follows: initial denaturation 95 °C, 30 sec (1 cycle); denaturation 95 °C, 15 sec (15 cycles); annealing 62 °C 15 sec (15 cycles); extension 65 °C 4 min (15 cycles); final extension step 65 °C, 10 min. NEBNext End repair/dA-tailing Module (New England Biolabs) was utilized for repairing the DNA ends, while the NEB Blunt/TA Ligase Master Mix (New England Biolabs) was used for ligating the adapters (1D kit). The Beckman Coulter Agencourt AMPure XP beads were used to purify the DNA after each of the enzymatic steps. The concentration of the samples was measured by using the Qubit Fluorometer 2.0 (Life Technologies) and the Qubit (ds)DNA HS Assay Kit (Life Technologies). The libraries were sequenced on the ONT R9.4 SpotON Flow Cells.
MinION cDNA sequencing on Cap-selected samples
To generate full-length cDNA from capped and polyadenylated RNAs, the “all-in-one” protocol from the Lexogen was applied using the TeloPrime Full-Length cDNA Amplification Kit. A 2.2 μg mixture from the different total RNAs (1, 2, 3, 4, 5, 6, 8, 12, 18 and 24 h pi) was used for the RT. For this, the sample was mixed with RT buffer and a specific primer (both are the part of the kit, Table 7). The reaction started at a 30 sec incubation at 70 °C, then it was followed by a 1 min step at 37 °C. At this point, the reaction was kept at 37 °C. The reverse transcriptase and the additional reagents (derived from the kit) were mixed with the sample and then the incubation was containing at 37 °C for 2 min. The next step of the RT reaction was carried out at 46 °C for 50 min. Sample was purified by using the kit’s Silica columns. The double-strand (ds) specific ligase enzyme (Lexogen kit) was used to join the adapter to the cDNA, the reaction was performed at 25 °C, overnight, then the sample was purified using the silica membranes of the Lexogen kit. The dscDNAs were generated by using the Enzyme Mix and the Second-Strand Mix (Lexogen kit). The cDNA production was performed in a Veriti cycler. The following protocol was applied: 98 °C for 90 sec, 62 °C for 60 sec, 72 °C for 5 min (16 cycles), hold at 25 °C. The sample concentration was detected by using Qubit 2.0 and Qubit dsDNA HS quantitation assay (Life Technologies). The specificity of the obtained product was checked by using real-time PCR. The Rotor-Gene Q qPCR machine (Qiagen), a gene specific primer (us9, 10 μM each, ordered from IDT DNA, Table 8), and the ABsolute qPCR SYBR Green Mix (Thermo Fisher Scientific) was applied. The preliminary denaturation was carried out at 94 °C for 15 min, then 35 cycles of 94 °C for 25 sec, 60 °C 25 sec and 72 °C 6 sec was applied.
The cDNAs from polyadenylated-capped RNA samples were used for library preparation for ONT sequencing. The 1D Strand switching cDNA by ligation method was used. After the end-repair, the samples were ligated to the 1D adapters. Finally, they were measured on the ONT R9.4 SpotON Flow Cells.
Read processing
The SMRT Analysis v2.3.0 (PacBio RSII), the SMRT Link (PacBio Sequel) and the Albacore v2.0.1 (ONT MinION) software packages were used for base calling (Figure 1). The reads were mapped by using GMAP and the following setting was applied: Minimum Full Passes = 1, Minimum Predicted Accuracy = 90, Minimum Length of Reads of Insert = 1, Maximum Length of Reads of Insert = No Limit. These consensus reads were mapped using GMAP17, with the following settings: gmap -d Genome.fa --nofails -f samse File.fastq > Mapped_file.sam. The quality information was acquired by using custom made routines18.
The Porechop v.0.2.3 software was used to determine the orientation of the sequencing reads. For this, a modified adapter.py file was used, where we added the various, library-specific adapter sequences (Table 6). The first mapped nucleotide downstream the “END” adapter was labelled the 5′ end of a read, while the last mapping nucleotide upstream of the “START” (polyA tail: A20 or 3′ adapters) was designated the 3′ end of the read. Reads lacking an adapter on both ends, or with 5′ or 3′ adapters on both ends, can be discarded from further in silico analysis.
Code Availability
1.SMRT Analysis v2.3.0: https://s3.amazonaws.com/files.pacb.com/software/smrtanalysis/2.3.0/smrtanalysis-patch_2.3.0.140936.p5.run
2.SMRT Link v5.1.0: https://downloads.pacbcloud.com/public/software/installers/smrtlink_5.1.0.26412.zip
3.Albacore v2.0.1: https://github.com/Albacore/albacore
4.GMAP: http://research-pub.gene.com/gmap/ (version 2015-12-31)
5.Custom routines have been archived on Github (https://doi.org/10.5281/zenodo.1034511).
6.Porechop v.0.2.3: https://github.com/rrwick/Porechop
Data Records
Data from MinION and Sequel sequencing have been uploaded to the European Nucleotide Archive (Data Citation 1) - contains BAM files. The RSII raw sequencing files, processed data files as well as metadata have been submitted to the Gene Expression Omnibus repository (Data Citation 2). All reads were mapped to the X14112 genome build. The provided sequencing data can be used without restrictions.
Technical Validation
The quantity of the purified total RNAs, the polyA-selected RNAs, the ribodepleted RNA samples, as well as the cDNA samples and the final sequencing libraries were detected by Qubit 2.0 (Life Technologies) fluorometer using the Qubit RNA Broad-Range, High Sensitivity RNA and High Sensitivity dsDNA Assay Kits.
Usage Notes
Our provided dataset was primarily generated to analyse the potential splice variants, as well as transcriptional start and stop site variations, the isoforms and the complexity of HSV-1 transcriptome. The provided raw RSII data files can be utilized to develop novel base calling algorithms or read processing tools, as well as to improve the currently existing bioinformatics software. The uploaded BAM files contain reads already aligned to the X14112 HSV-1 reference genome using GMAP v2017-04-2417.
These mapped reads can be further analysed by using different long-read aligners (e.g. NGMLR: https://github.com/philres/ngmlr; GraphMap: https://github.com/isovic/graphmap, etc.), or bioinformatics tools (e.g. samtools19 and bedtools20). These data can be visualized by utilizing different programs such as the Geneious21, IGV22, or Artemis23. The files contain terminal poly(A) sequences as well as the 5′and 3′ adapter sequences, which can be utilized to determine the orientations of the reads. Novel long-read sequencing pipelines e.g.: SQANTI24 can be tested using this dataset.
Additional information
How to cite this article: Boldogkői, Z. et al. Transcriptomic study of Herpes simplex virus type-1 using full-length sequencing techniques. Sci. Data. 5:180266 doi: 10.1038/sdata.2018.266 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Miller, C. S. & Redding, S. W. Diagnosis and management of orofacial herpes simplex virus infections. Dent. Clin. North. Am. 36, 879–895 (1992).
Marchi, S., Trombetta, C. M., Gasparini, R., Temperton, N. & Montomoli, E. Epidemiology of herpes simplex virus type 1 and 2 in Italy: a seroprevalence study from 2000 to 2014. J. Prev. Med. Hyg. 58, E27–E33 (2017).
Lim, F., Khalique, H., Ventosa, M. & Baldo, A. Biosafety of gene therapy vectors derived from herpes simplex virus type 1. Curr. Gene. Ther. 13, 478–491 (2013).
Filip, L. HSV-1 as a Model for Emerging Gene Delivery Vehicles. ISRN Virology 2013, 397243 (2013).
Grant, K. G., Krisky, D. M., Ataai, M. M. & Glorioso, J. C. Engineering cell lines for production of replication defective HSV-1 gene therapy vectors. Biotechnol. Bioeng. 102, 1087–1097 (2009).
Kaur, B., Chiocca, E. A. & Cripe, T. P. Oncolytic HSV-1 Virotherapy: Clinical Experience and Opportunities for Progress. Curr. Pharm. Biotechnol. 13, 1842–1851 (2012).
Tombácz, D. et al. Long-Read Isoform Sequencing Reveals a Hidden Complexity of the Transcriptional Landscape of Herpes Simplex Virus Type 1. Front. Microbiol 8, 1079 (2017).
Niedringhaus, T. P., Milanova, D., Kerby, M. B., Snyder, M. P. & Barron, A. E. Landscape of Next-Generation Sequencing Technologies. Anal Chem. 83, 4327–4341 (2011).
Sharon, D., Tilgner, H., Grubert, F. & Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 31, 1009–1014 (2013).
Tilgner, H., Grubert, F., Sharon, D. & Snyder, M. P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. USA 111, 9869–9874 (2014).
Rhoads, A. & Au, K. F. PacBio Sequencing and Its Applications. Genomics, Proteomics Bioinformatics 13, 278–289 (2015).
Tombácz, D. et al. Full-Length Isoform Sequencing Reveals Novel Transcripts and Substantial Transcriptional Overlaps in a Herpesvirus. PLoS One. 11, e0162868 (2016).
Chen, S. Y., Deng, F., Jia, X., Li, C. & Lai, S. J. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Sci. Rep 7, 7648 (2017).
Weirather, J. L. et al. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 6, 100 (2017).
Moldován, N. et al. Third-generation Sequencing Reveals Extensive Polycistronism and Transcriptional Overlapping in a Baculovirus. Sci Rep 8, 8604 (2018).
Moldován, N., Tombácz, D., Szűcs, A., Csabai, Z., Snyder, M. & Boldogkői, Z. Multi-Platform Sequencing Approach Reveals a Novel Transcriptome Profile in Pseudorabies Virus. Front. Microbiol. 8, 2708 (2018).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Szunyike. Source code for: Long-read sequencing of the HCMV transcriptome with the Pacific Biosciences RSII platform: Long-read sequencing data statistics. Zenodohttps://zenodo.org/record/1034511 (2017).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatic 16, 944–945 (2000).
Tardaguilla, M. et al. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 28, 396–411 (2018).
Data Citations
European Nucleotide Archive PRJEB25433 (2018)
Gene Expression Omnibus GSE97785 (2017)
Acknowledgements
This work was supported by the Swiss-Hungarian Cooperation Programme: SH/7/2/8 to Z.B., by the NIH Centers of Excellence in Genomic Science (CEGS) Center for Personal Dynamic Regulomes: 5P50HG00773502 to M.S., and by the Bolyai János Scholarship of the Hungarian Academy of Sciences to D.T.
Author information
Authors and Affiliations
Contributions
Z.Bo. designed the experiment, took part in data analysis and wrote the final manuscript. A.S. and Z.Ba. conducted bioinformatics analysis. D.S. performed PacBio sequencing. M.S conceived and designed the experiments. D.T. performed PacBio and MinION sequencing, analysed the data and wrote the manuscript. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Boldogkői, Z., Szűcs, A., Balázs, Z. et al. Transcriptomic study of Herpes simplex virus type-1 using full-length sequencing techniques. Sci Data 5, 180266 (2018). https://doi.org/10.1038/sdata.2018.266
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.266
This article is cited by
-
Meta-analytic approach for transcriptome profiling of herpes simplex virus type 1
Scientific Data (2020)
