
Abstract
Cardiolipin (CL) is a mitochondria-specific phospholipid synthesized by CL synthase (CLS). We describe here a human gene for CLS and its analysis via RNAi knockdown on apoptotic progression. Although mitochondrial membrane potential is unchanged in cells containing only 25% of the normal amount of CL, free cytochrome c (cyt. c) is detected in the intermembrane space and the mitochondria exhibit signs of reorganized cristae. However, the release of cyt. c from the mitochondria still requires apoptotic stimulation. Increased sensitivity to apoptotic signals and accelerated rates of apoptosis are observed in CL-deficient cells, followed by elevated levels of secondary necrosis. Apoptosis is thought to progress via binding of truncated Bid (tBid) to mitochondrial CL, followed by CL oxidation which results in cyt. c release. The exaggerated and accelerated apoptosis observed in CL-deficient cells is matched by an accelerated reduction in membrane potential and increased cyt. c release, but not by decreased tBid binding. This study suggests that the CL/cyt. c relationship is important in apoptotic progression and that regulating CL oxidation or/and deacylation could represent a possible therapeutic target.
Similar content being viewed by others

Main
Cardiolipin (CL) is synthesized via sequential conversion of phosphatidic acid (PA) to cytidine 5′-diphosphate (CDP)-diacylglycerol, phosphatidylglycerol phosphate, phosphatidylglycerol (PG), and finally CL.1 The final step is catalyzed by cardiolipin synthase (CLS).2 CLS ablation in yeast causes CL deficiency and PG accumulation. Remarkably, mitochondrial function is not grossly perturbed, for example, cytochrome c (cyt c) oxidase levels and activity are normal, the yeast grow on nonfermentable carbon sources, and mitochondrial membrane potential is maintained.3 As CL is thought to be vital for these functions, it has been proposed that PG, which increases five-fold in the absence of CLS, can substitute for CL for all of its essential functions under normal conditions,3 although cells under stress may still need it.3, 4
CL interacts with many mitochondrial proteins including cyt. c,5 which localizes to the outer leaflet of the inner membrane.6 Binding of proteins to CL is noncovalent, usually stimulatory,2 and very specific.1 CL also plays an essential role in higher order organization (supercomplex formation) of respiratory chain homodimers of complexes III and IV7 and increases the inner membrane concentration of cyt. c.8 CL additionally mediates mitochondrial apoptotic pathways1, 9 by enabling truncated Bid (tBid) to bind to CL-rich mitochondria contact sites10, 11, 12, 13 and, after oxidation, by releasing cyt. c.14, 15
Current models propose that tBid generation and binding to CL lead sequentially to superoxide production, CL peroxidation, disengagement of cyt c. from the oxidized CL, and then release of the cyt. c from the mitochondria via pores generated by Bax and Bak.16, 17 In this study, we set out to explore the role for CL during apoptosis via a loss-of-function approach.
Results
A human CLS gene
Blast analysis using yeast CLS (Saccharomyces cerevisiae CLS, scCLS) identified a human homolog via conservation of the CDP-alcohol phosphatidyltransferase motif (CDP-OH PT, Figure 1a and Supplemental Figure 1), DG(x)2AR(x)8G(x)3D(x)3D, which catalyzes displacement of cytidine 5′-monophosphate (CMP) from CDP-alcohol.18 Human CLS (hCLS) is expressed widely with the highest levels in the liver and testis (Supplemental Figure 2). hCLS was predicted to localize to mitochondria by the program MitoProt II (probability 0.8502); this was confirmed by expressing hCLS carboxyl-terminally tagged with a c-myc epitope (hCLS-myc) in NIH3T3 cells (Supplemental Figure 3).

Human CLS structure and complentation of yeast CLS deficiency. (a) Domain structure of hCLS and galactose-inducible expression plasmids used for yeast complementation. MLS, mitochondrial localization sequence; aa, amino acid; CDP-OH PT, CDP-alcohol phosphatidyl transferase motif. hCLS is predicted to be a triple-pass membrane protein (presumably embedded in the inner membrane) with its amino-terminus oriented towards the intermembrane space (TMHMM program (http://www.cbs.dtu.dk/services/TMHMM/)). (b) CLS-lacking Saccharomyces cerevisiae yeast were transformed with galactose-inducible plasmids expressing S. cerevisiae, human, hybrid, or mutant hybrid (D169E) CLS. As controls, wild-type and CLS-lacking yeast cells were also transformed with an empty vector. The yeast strains were labeled with [3H]palmitate, induced, and processed for lipid extraction. Extracted whole-cell lipids were separated by TLC and visualized by autoradiography. The lipids indicated to the left were identified based on comigration with commercial lipid standards. CL, cardiolipin; PG, phosphatidylglycerol; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PS, phosphatidylserine; PI, phosphatidylinositol; wt, wild type; scCLS, S. cerevisiae CLS; hybrid, hCLS with its MLS replaced by the scCLS MLS
hCLS complements yeast CLS deficiency
Yeast lacking CLS are characterized by CL depletion and reciprocal PG accumulation.3 To determine if hCLS exhibits CLS activity, we transformed cls null yeast with galactose-inducible hCLS plasmids. Upon induction, CL was produced in hCLS-transformed cls null yeast cells, whereas no CL was observed in empty-vector-transformed cells (Figure 1b). However, hCLS only partially complemented the yeast deficiency (e.g. the amount of CL produced was substantially less than that characteristic of wild-type yeast cells or of null yeast cells rescued by an inducible plasmid expressing yeast CLS, and PG levels remained elevated), potentially resulting from either low activity of hCLS in yeast or reduced targeting of hCLS to yeast mitochondria. Reduced targeting appeared possible, as the scCLS and hCLS mitochondrial localization sequences (MLS) are relatively poorly conserved (Supplemental Figure 1). Accordingly, we replaced the hCLS MLS with the scCLS MLS. Transformation of cls null yeast cells with the hybrid CLS restored CL to levels comparable to wild-type yeast cells. In contrast, a mutant hybrid CLS containing a catalytic-site inactivating mutation in the CDP-OH PT motif (D169E; numbering corresponding to hCLS) did not complement. Taken together, these findings demonstrate that hCLS has bona fide CLS activity.
RNAi knockdown of hCLS decreases the level of CL in HeLa cells
CL has been implicated in apoptotic pathways involving tBid translocation and cyt. c release.2 Specifically, decreases in CL subsequent to deacylation or oxidation have been shown to correlate with and have been proposed to underlie the detachment of cyt c. from the inner membrane and subsequent egress from the mitochondria into the cytoplasm.14 However, a direct examination of lowered CL levels in mammalian apoptotic pathways has not been conducted (CL levels are decreased in Barth Syndrome (BTHS),19 but BTHS cells exhibit additional lipid profile changes as discussed later). To explore this issue, we employed RNAi. Six selected small hairpin RNAs (shRNA) substantially reduced expression of hCLS mRNA from tagged alleles after transient cotransfection in HeLa cells, and reduced endogenous levels of hCLS in pools of HeLa cells. No obvious reduction in cell viability was noted (data not shown). Two sequences were selected to generate cell lines stably expressing hCLS RNAi. Cell lines containing the shRNA sequences #1 and #2 exhibited reductions in hCLS mRNA levels to 18±3 and 13±3% of wild-type levels, respectively, in comparison to untransfected cells or to a cell line expressing a control shRNA that targeted firefly Luciferase (Luc) (Figure 2a and data not shown). No obvious clonal selection was noted during the cell line generation.

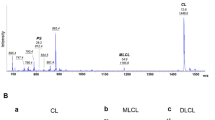
RNAi knockdown of hCLS reduces CL levels. (a) Real-time quantitative RT-PCR analysis of hCLS mRNA expression levels in control and hCLS-RNAi cells. HeLa cells stably transfected with small hairpin (sh) RNAs directed against firefly luciferase (Luc-RNAi) or hCLS (hCLS-RNAi; two different shRNA sequences were used, #1 and #2) were harvested. Total RNA was isolated, reverse-transcribed, and analyzed by real-time quantitative PCR with GAPDH or hCLS primers. The hCLS mRNA expression levels were normalized to GAPDH. Data represent mean±standard deviation of three independent experiments. (b) Phospholipid TLC analysis of hCLS-RNAi and rescue cells. Luc-RNAi, hCLS-RNAi and rescue cells (hCLS-RNAi #2 cells stably transfected with a wobble codon-mutated hCLS cDNA resistant to RNAi degradation at the site targeted in shRNA sequence #2) were labeled with [3H]palmitate and analyzed by TLC. (c) Quantitation of CL and PG levels normalized to levels of PE. Lipid spots comigrating with the CL, PG, and PE standards were scraped off the plate and scintillation counted. Data represent mean±standard deviation of three independent experiments. (d) Mass spectrometry analysis of hCLS-RNAi cells. Phospholipids extracted from control and hCLS-RNAi cells were analyzed by electrospray ionization mass spectrometry in negative mode. Internal standards (CL, 619.5 m/z, and PG, 665.5 m/z) were used but lie outside of the portion of the scan shown. Five clusters of CL peaks are observed, with the individual peaks within each cluster representing species with different degrees of saturation. Possible compositions of CL clusters would be: (C16:0)4 (m/z 672.4); (C16:0)3(C18:1) (m/z 685.5); (C16:0)2(C18:2)2 (m/z 699.4); (C16:0)(C18:1)(C18:2)2 (m/z 712.5); and (C18:0)(C18:1)(C18:2)2 (m/z 725.4). The possible composition of the PG cluster shown (m/z 712.5) would be (C16:0)(C16:1), and possible compositions of the MLCL cluster would be (C16:0)2(C18:2) (m/z 567.2); (C16:0)(C18:1)2 (m/z 581.3); and (C18:1)3 (m/z 595.5). m/z, mass-to-charge ratio. Representative of three independent experiments
To examine CL levels, the hCLS-RNAi and Luc-RNAi cells were labeled with [3H]palmitate and the lipids extracted and analyzed by thin layer chromatography (TLC), which revealed significantly decreased CL levels (to 54±13 and 25±5% of wild-type levels, respectively, in hCLS-RNAi cell lines #1 and #2) (Figure 2b and c).
RNAi targeting can generate off-target or stress responses that yield gene-nonspecific phenotypes. To address this possibility, the hCLS-RNAi cell line #2 was transfected with a plasmid encoding an hCLS cDNA allele harboring wobble codon mutations at the site targeted by the RNAi, which renders it resistant to RNAi-mediated degradation. Expression of this plasmid restored CL to a level comparable to that of control cells (Figure 2b and c), proving that the knockdown of hCLS per se yields the observed decrease in CL. An increase in PG was also observed in the hCLS-RNAi cells (to 153±22 and 315±46% of control levels in cell lines #1 and #2, respectively), supporting the interpretation of the mechanism underlying the decrease in CL (Figure 2b and c). Lipid profiles were examined using mass spectrometry (Figure 2d), which demonstrated that the drop in CL content observed by TLC was not accompanied by changes in CL fatty-acyl composition. Although the nonquantitative approach used does not permit comparison of the levels of CL between the different samples, it is evident that the amount of PG is increased in the hCLS-RNAi cell lines, particularly #2, relative to the level of CL and monolysocardiolipin (MLCL, inset), when compared to the relative levels observed in the Luc-RNAi cell line.
Taken together, these results demonstrate that hCLS-RNAi cells have significantly dereased CL, although the observation that the CL decrease is less profound than mRNA decrease suggests the presence of a negative feedback mechanism that decreases the rate of CL degradation when CL levels fall, or that CLS is not normally the rate-limiting step for CL production.
CL deficiency increases susceptibility to stimuli that elicit apoptotic cell death
CL peroxidation and decreases in the absolute level of CL have been implicated as regulators of mitochondrial cyt. c release,14, 20, 21, 22, 23 but their relative importance has not been established. Accordingly, we used our hCLS-RNAi cells to examine whether simply decreasing the level of CL increases susceptibility to cell death by treating them with anti-Fas antibody, tomor necrosis factor- alpha (TNF-α) plus cycloheximide (Chx), or staurosporine (STS) and employing two-dye flow cytometry with YOPRO-1/PI (propidium iodide) for analysis. YOPRO-1 dye passes through the plasma membrane of apoptotic cells but is not taken up by normal cells; PI only enters into necrotic cells.24
At the beginning of the analysis period (0 h, Figure 3a, left column), all three cell lines – the control Luc-RNAi cell line, hCLS-RNAi #1 which has a 46% reduction in CL, and hCLS-RNAi #2 which has a 75% reduction – exhibited equivalent viability (YOPRO-1−/PI−). However, differences became apparent 24 h after the addition of anti-Fas antibody. Although all three cell lines exhibited increased apoptosis (YOPRO-1+/PI−), hCLS-RNAi #2 exhibited an even greater fraction of cells undergoing necrotic cell death (YOPRO-1+/PI+). By 48 h, although some necrosis was observed for the Luc-RNAi cells, apoptosis still predominated. In contrast, necrosis predominated for both hCLS-RNAi cell lines, especially hCLS-RNAi #2.

Cells with reduced levels of CL are more susceptible to cell death. (a) Control and hCLS-RNAi cells were treated with anti-Fas antibody for 24 or 48 h, stained with YOPRO-1 and PI, and analyzed by two-dye flow cytometry. The histograms describe results from a representative experiment. Viable cells (YOPRO-1−/PI−), apoptotic cells (YOPRO-1+/PI−), and necrotic cells (YOPRO-1+/PI+) were tabulated. (b) The cells were treated for the indicated periods of time with 100 ng/ml TNF-α in the presence of 25 mM Chx to prevent activation of NFκB. As in (a), the number of viable cells, apoptotic cells, and necrotic cells are shown in tabulated form. Data represent mean±standard deviation of nine independent experiments. (c) Control, hCLS-RNAi, and rescue cell lines were treated with 5 mM STS for the indicated time periods and analyzed as above. (d) Control and hCLS-RNAi #2-resistant rescue plasmids were transfected into control Luc-RNAi and hCLS-RNAi cells and cultured for 48 h to permit recovery of CL levels as applicable. The cells were then retransformed with tBid-EYFP (for 4 h) to elicit apoptosis and cultured for an additional 3 h to allow cell death to occur as applicable (which takes place rapidly when tBid is employed). Cellular viability was determined using PI staining. The transfection efficiency for tBid-EYFP was approximately 75% for each cell line. Data represent mean±S.D. of five independent experiments
A similar outcome was obtained using TNF-α+Chx (Figure 3b). In this experiment, the analyses were performed every 2 h in the early part of the postinduction time period. The results indicated that viability decreased slightly faster for hCLS-RNAi #1 in comparison to the Luc-RNAi cell line, whereas hCLS-RNAi #2 exhibited a relatively rapid loss of viability (left panel). After segregation by the mechanism of cell death, it became apparent that in the early phase (the first 6 h), hCLS-RNAi #2 exhibited a more rapid entry into apoptosis – but that this then converted into a necrotic or secondary necrotic phenotype (middle and right panels).
STS-induced cell death was similar (Figure 3c). The hCLS-RNAi #2 cells were additionally transfected with the RNAi-resistant CLS rescue plasmid described in Figure 2b, which rescued hCLS-RNAi #2 such that it exhibited wild-type levels of apoptotic cell death in response to STS rather than increased levels of necrotic cell death (Figure 3c, rightmost panel).
Upon caspase-8 cleavage, Bid is converted to tBid, which then translocates to mitochondria via binding to CL.12, 13 We therefore examined whether reducing the level of CL affects tBid translocation. The control and hCLS1-RNAi #2 cell lines were transfected with tBid-EYFP and the frequency of mitochondrial decoration was quantified by confocal microscopy; no significant differences were observed (data not shown). This result is not surprising in that a significant amount of CL still exists in the hCLS-RNAi cells, and CL as a receptor for tBid has been suggested not to be easily saturable in vivo.25
The cell lines were additionally transfected with an empty vector or the #2-RNAi-resistant CLS rescue plasmid prior to the tBid transfection (Figure 3d). Substantial loss of cell viability was observed after tBid transfection when the empty vector was employed, with hCLS-RNAi #2 exhibiting a greater degree of cell death (90%) than the Luc-RNAi or hCLS-RNAi #1 cell line (55%). Forced expression of CLS using the #2-RNAi-resistant CLS rescue plasmid, however, conferred protection to the hCLS-RNAi #2 cell line, confirming that using a more direct approach CL deficiency leads to greater sensitivity to apoptotic stimuli. No protection from the apoptotic stimulus (tBid) was observed when CLS was overexpressed in wild-type control cells (Luc RNAi, Figure 3d).
Cristae reorganization is observed in mitochondria with reduced levels of CL
Mitochondria in cells lacking phosphatidylglycerophosphate synthase are characterized by reductions in both PG and CL and appear swollen with missing or disorganized cristae;26, 27 however, mitochondria in yeast lacking only CL exhibit normal morphology.3 We accordingly examined the ultrastructural morphology of mitochondria in hCLS-RNAi cells, and found that the mitochondria in hCLS-RNAi #2 cells exhibited increased electron density and disorganized cristae (Figure 4a), suggesting that CL undertakes a role in maintaining mitochondrial morphology that requires the presence of more than 25% but less than 54% of the endogenous levels.

Mitochondria in CL-deficient cells have altered ultrastructure and elevated free intermembrane cyt. c, which is released more readily during apoptosis and leads to more rapidly decreased membrane potential. (a) Control and hCLS-RNAi cells were analyzed by electron microscopy. Representative of three independent experiments. Note increased electron density and disorganized cristae in the mitochondria of hCLS-RNAi #2 cells treated with TNF-α+Chx. (b) Cyt. c release is enhanced in hCLS-RNAi cells upon TNF-α+Chx treatment. Western blot analysis of cytosolic and heavy membrane fractions from control and hCLS-RNAi cells. Untreated cells and cells treated with TNF-α+Chx were lysed, separated into cytosolic and heavy membrane fractions, and analyzed by Western blot analysis with anti-cyt. c and Omi antibodies. Anti-actin and anti-VDAC (voltage-dependent anion channel) antibodies were used for loading controls. Representative of three independent experiments. (c) Histogram of free cyt. c found in mitochondria purified from each cell line, plotted as cyt. c protein (nmol/mg) versus CL content (as % of that present in the control cells). (d) Decreased membrane potential in CL-deficient cells undergoing apoptosis. Control and hCLS-RNAi cells were treated with TNF-α+Chx, stained with PI and DiOC6(3), and analyzed by two-dye flow cytometry. (e) Increased commitment to cell death is observed for hCLS-RNAi cells after apoptotic challenge. Control and hCLS-RNAi cells were exposed to apoptotic stimuli for differing lengths of time, washed, plated at limiting dilution, and scored for colony formation after several days of subsequent culture. Gridded areas containing 1000 cells or colonies for control cultures were examined for each condition, and the colony frequency is expressed as a percentage of the control. Standard deviations averaged 4%
Enhanced release of cyt. c from hCLS-RNAi mitochondria during apoptosis
Substantial evidence suggests that cyt. c is bound to CL in unperturbed cells and becomes released into the cytoplasm upon apoptosis after CL undergoes oxidation.14, 20, 21, 28, 29 We described above accelerated progression of apoptosis in cells deficient for CL (Figure 3). A possible mechanism to explain this might involve exaggerated release of cyt. c.
Cyt. c levels in the cytosol and the heavy membrane fraction were assessed after induction of cell death using TNF-α+Chx (Figure 4b). Significant increases in the levels of cyt. c released into the cytoplasm were observed for hCLS-RNAi #2 cells in comparison to the control Luc-RNAi cells, along with a reciprocal decrease in the heavy membrane fraction. Marginally increased release of cyt. c was observed for the hCLS-RNAi #1 cells, matching the lesser degree of CL depletion. STS and anti-Fas treatment yielded similar outcomes (data not shown). Omi/HtrA2, an intermembrane space protein that does not depend on CL for membrane association but is released concomitantly with cyt. c from permeabilized mitochondria during apoptosis, also exhibited enhanced release, although not as dramatically as cyt. c. To verify the enhanced cyt. c release in the hCLS-RNAi #2 cells, we immunostained TNF-α+Chx-treated cells with anti-cyt. c antibody and observed that cyt. c became visually detectable in the cytoplasm (data not shown).
CL deficiency leads to accumulation of free cyt. c in the intermembrane space
The observation that cyt. c exhibits exaggerated release in CL-deficient cells suggested that it either disengages from the inner membrane more easily once apoptosis starts, or is already present in the free form prior to the initiation of apoptosis. To examine this, we purified mitochondria from the three cell lines, permeabilized them with Bax (to enable the free cyt. c to exit the mitochondria), and separated the membrane-bound from the free cyt. c by centrifugation. No free cyt. c was found in the wild-type Luc-RNAi control cells (Figure 4c). In contrast, the amount of free cyt. c increased as the level of CL decreased, as shown by the results obtained for the two hCLS-RNAi cell lines, indicating that free cyt. c was already present in the intermembrane space prior to the initiation of apoptosis.
CL deficiency causes accelerated loss of mitochondrial membrane potential
Induction of apoptosis decreases mitochondrial membrane potential (Δψm). To examine the kinetics of Δψm reduction in the setting of decreased CL, we performed DiOC6(3) and PI staining subsequent to TNF-α+Chx treatment. Decrease in Δψm was more prominent in the hCLS-RNAi cells, especially #2, than in the control cells (Figure 4d). Expression of the RNAi-resistant hCLS rescue cDNA in the hCLS-RNAi #2 cells restored the RNAi cells such that they exhibited a Δψm change similar to that of the control cells, confirming the specificity of the phenotype.
Yeast cells lacking cls exhibit defects in supercomplex formation of respiratory chain homodimers of complexes III and IV,30 although the components function fairly well as individual homodimers in the absence of CL,30 and abnormal respiratory chain function is observed only under stressful conditions.4 To examine this issue for mammalian cells, we assayed activities of various mitochondrial respiratory chain enzymes in control, hCLS-RNAi, and rescue cells. No significant changes were observed (Supplemental Table 1), which is not surprising given the above, and as well, that unlike the cls null yeast cells harboring an undetectable level of CL,3 the hCLS-RNAi cells examined here have CL levels at least 25% of those found in wild-type cells.
Collectively, these findings suggest that decreasing CL renders HeLa cells more susceptible to apoptotic- and necrotic-induced cell death, potentially in part via accelerating Δψm loss.
CL deficiency increases the commitment to cell death after exposure to apoptotic stimuli
To address whether CL deficiency not only accelerates cell death but in addition renders cells less likely to survive when challenged with an apoptotic stimulus, the control and RNAi cell lines were exposed to anti-Fas antibody or TNF-α plus Chx for different amounts of time, washed, plated at limiting dilution, cultured for several additional days, and subsequently scored for attachment and cell division (initiation of colony formation). A period of stimulation of 3–6 h with TNF-α+Chx, or 24–48 h with Anti-Fas antibody was used based on the results obtained in Figure 3a,b, which indicated that differences in cell viability were observed for the different cell lines with this degree of apoptotic stimulation. All of the cell lines exhibited equal and high levels of clone generation in the absence of apoptotic induction (Figure 4e). Decreased frequency of clone generation was observed for the Luc-RNAi line after 3 h of exposure to TNF-α plus Chx or 24 h of exposure to Anti-Fas antibody, and the frequency decreased further when the duration of the apoptotic stimulus was doubled. The success rate of clone generation was significantly decreased for the hCLS-RNAi #1 cell line, and dramatically decreased for the hCLS-RNAi #2 cell line at all time points. However, the ability to generate clonal populations was restored to wild-type levels for the hCLS-RNAi #2 cell line after introduction of an RNAi-resistant hCLS rescue expression plasmid, confirming the role played by CL in this process.
Discussion
We show here that HeLa cells with reduced CL are more sensitive to TNF-α-, Fas- and staurosporine-elicited cell death. In addition, we show that mitochondria with reduced CL exhibit free cyt. c in the intermembrane space, and then, upon initiation of cell death, display increased release of cyt. c and accelerated reduction in membrane potential. In contrast, changes in tBid translocation and respiratory enzyme activity are not observed. Taken together, our findings suggest that the requirement for CL in mammalian mitochondria is more rate limiting for the anchoring of cyt. c to the inner mitochondrial membrane than for tBid translocation or respiratory enzyme activity. Nonetheless, under stressful conditions, such as stimulation by apoptotic-inducing agents, cells deficient in CL are less resilient.
CL binds to most mitochondrial respiratory enzymes and is believed to be required for their function.2 Hence, it is surprising that mitochondrial respiratory enzyme activity is fairly normal in nonstressed cls null yeast cells,3, 4, 30 and our results here with hCLS-RNAi cells are similar. As one resolution for this paradox, Dowhan and co-workers have suggested that when present at sufficient levels, PG can substitute for CL for all essential functions in the mitochondria.3 The setting of CLS depletion or knockout is relevant here, as PG is the precursor to CL and increases many-fold in yeast and mammalian cells (Figure 2) in the absence or deficiency of CLS.
These findings contrast with those reported for cell lines generated from patients with BTHS, which ensues from abnormalities in CL remodeling subsequent to its biosynthesis.19 BTHS cell lines also exhibit decreases in CL, but not increased susceptibility to apoptosis.31 However, it is difficult to directly compare CLS-RNAi and BTHS cells. First, as a consequence of the different underlying biochemical mechanisms, PG levels are normal and MLCL levels are elevated in BTHS cells, whereas PG is elevated in CLS-deficient cells and MLCL is reduced. Second, CL in BTHS is characterized by altered fatty-acyl composition. Finally, the BTHS lymphoblastoid lines that were used to examine susceptibility to apoptosis were transformed by Epstein–Barr Virus,31 which suppresses apoptosis. Taken together, the cell lines described here represent a new and potentially cleaner model system for exploring the role of CL in cell viability and mitochondrial function.
Yeast cyt. c binds to CL in the quiescent state and then is released into the cytosol during apoptosis.32 However, cyt. c release was not observed for unperturbed yeast cells lacking CL in the absence of apoptosis.3, 30 We similarly did not observe noticeable release of cyt. c into the cytosol in hCLS-RNAi cells in the absence of induction of apoptosis. Dowhan and colleagues addressed this issue for yeast by suggesting that PG, which is dramatically elevated in cls null yeast cells, might substitute for CL in terms of cyt. c binding.3 This hinted that the lack of cyt. c release we found might also be attributed to the elevated level of PG in our hCLS-RNAi cells. However, as we detected free cyt. c in the intermembrane space, and induction of apoptosis triggered exaggerated cyt. c release in the RNAi cells, we would conclude that elevated levels of PG do not suffice to physiologically replace CL in this context.
Although the detailed mechanisms that regulate cyt. c release during apoptosis remain unclear, our demonstration that decreased CL levels lead to free cyt. c in the intermembrane space is a potentially interesting finding. The two-step model21 for cyt. c release during apoptosis proposes that termination of the electrostatic/hydrophobic affiliations that hold cyt. c to CL occurs in the first step, followed by mitochondrial outer membrane permeabilization which releases the cyt. c into the cytosol. Our findings could be viewed as providing support for this model, as we show that even though CL deficiency in nonapoptotic-stimulated cells liberates substantial amounts of the cyt. c from the inner membrane (step 1), cyt. c release into the cytosol still requires the permeabilization step that occurs during apoptosis. However, although mitochondria containing only 25% of the normal level of CL are relatively normal in terms of function, they are nonetheless not entirely normal, given their altered morphology, and the accelerated/exaggerated release in cyt. c and decrease in mitochondria potential, and cell viability that we observed upon apoptotic induction could ensue from multiple direct or indirect consequences of CL depletion.
Finally, our findings suggest that stabilization of CL levels might protect against apoptosis. Forced expression of CLS did not confer protection to wild-type cells (Figure 3d). However, as endogenous PG levels are quite low (Figure 2b), forced expression of CLS in wild-type cells would not be expected to elevate the levels of CL and hence does not provide an effective means to increase CL under normal circumstances. Instead therapies designed to stabilize or elevate CL levels via the inhibition of oxidative or deacylating degradation pathways might more feasibly be of use in settings where cell death is undesirable.
Materials and Methods
DNA manipulations
A human EST clone (NCBI accession number BI547549, IMAGE consortium clone number 5262376) was identified through tBlastn search with the S. cerevisiae CLS. The hCLS coding sequence was PCR-amplified from the EST clone and subcloned in-frame into the HindIII/EcoRI sites of pEGFP-N1. Human CLS(D169E) was generated using a QuickChange Site-directed Mutagenesis kit (Stratagene, La Jolla, CA, USA). A hybrid CLS was generated by PCR amplifying the residues 1–56 of scCLS (ORF YDL142C) with an appended 5′ HindIII restriction site, and residues 81–301 of hCLS with an appended 3′ SphI restriction site. The PCR products were restricted with HindIII and SphI and subcloned into pYES2 (Invitrogen). The resulting plasmids were confirmed by sequencing.
Human tissue mRNA Northern blot analysis
Random primed 32P-labeled probes were synthesized using RadPrime DNA Labeling System (Invitrogen) against the PCR-amplified carboxyl-terminal 600-bp coding region of hCLS or the 600-bp coding region of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). A human tissue Northern blot (RNWAY Laboratories, Seoul, Korea) was hybridized with the probes at 65°C in ExpressHyb Hybridization solution (Clontech) for 6 h, washed with Solution I (2 × SSC and 1% SDS) at 65°C and Solution II (0.1 × SSC and 0.5% SDS) at 55°C, and exposed to X-ray film for 2 days.
Cell culture, transfection, and immunofluorescence staining
HeLa or NIH/3T3 cells were maintained in DMEM supplemented with 10% calf serum. For transfection, cells were grown on coverslips in 35-mm dishes (3–4 × 105 cells/dish) and then switched into Opti-MEM I media (Invitrogen) before being transfected with 1 μg of DNA/dish using LipofectAMINE Plus (Invitrogen). At 2 h post-transfection, the media were replaced with fresh growth media and the cells incubated a further 20–24 h.
Cells were fixed with 3% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100, and blocked with 5% normal goat serum. The cells were then immunostained using primary antibodies against the specific proteins, followed by fluorescent dye-conjugated secondary antibodies. Stained cells were visualized using a Leica TCS SP2 confocal microscope. Images were processed using Adobe Photoshop 7.0. All experiments were performed at least three times with similar results.
CLS complementation assay in cls null yeast cells
S. cerevisiae CLS, and the human and hybrid CLS genes were subcloned into pYES2 (Invitrogen) and transformed into cls null yeast cells (MATα, cls, ura3, leu2, met). The yeast cells were then labeled with 10 μCi/ml of [3H]palmitate and protein expression was induced by galactose for 14–16 h. The cells were washed with H2O, spun down, and resuspended in 1.5 ml of chloroform/methanol/0.1 N HCl (1 : 2 : 0.8, v/v) in the presence of 500 μg of bovine heart lipid extracts (Avanti Polar Lipids) as carrier lipid. Acid-washed 0.4–0.6 mm glass beads (Sigma) were then added and cells were broken by vigorous vortexing. Chloroform (0.4 ml) and 0.1 N HCl/0.5 M NaCl (0.4 ml) were added and centrifugation was performed to achieve phase separation. The organic fraction was dried down, dissolved in chloroform, and analyzed by thin layer chromatography as described in the ‘Phospholipid Analysis’ section.
Phospholipid analysis
HeLa cells as indicated were labeled with 4 μCi/ml [3H]palmitic acid for 24 h. The cells were then harvested and homogenized with a solution of CH3OH/CHCl3/0.1 N HCl (10 : 5 : 4, v/v), and lipids were extracted by acid organic extraction (half-volumes each of CHCl3, 0.1 N HCl, and 0.5 M NaCl). The organic fraction was dried down and dissolved in chloroform. Equal amounts of radiolabeled phospholipids (ca. 1–3 million c.p.m.) along with standard phospholipids (Sigma or Avanti Polar Lipids) were applied to a 10 × 10-cm HPTLC Silica Gel 60 plate (Merck, Darmstadt, Germany). The HPTLC plate was developed in a solvent system of chloroform/methanol/acetic acid (32.5 : 14 : 4, v/v) and the standard phospholipids were visualized by exposure to iodine vapor. The plate was then sprayed with EN3HANCE autoradiography enhancer (PerkinElmer Life and Analytical Sciences, Wellesley, MA, USA) and exposed to BioMax MR X-ray film (Kodak, Rochester, NY, USA) for 2 days at −80°C. Spots of interest were scraped off of the plate and counted for radioactivity using a Beckman Coulter LS 6500 Liquid Scintillation Counter (Fullerton, CA, USA).
Generation of HeLa cells stably expressing shRNA
Two 19-nucleotide (nt) sequences (clone #1: nt 441–459 (GCTGGACTAACAGATTTGT); clone #2: nt 350–368 (GTTGTCAATGACGAGAATT)) matching human CLS mRNA (GenBank accession number NM_019095) were chosen from a larger set of sequences tested in preliminary experiments on pools of HeLa cells and in cotransfection experiments with tagged isoforms of hCLS to generate cell lines stably expressing hCLS RNAi. shRNAs were synthesized as two complementary DNA oligonucleotides, annealed, and cloned into the BglII/Acc65I sites of a modified pSUPER vector33 containing a blasticidin-resistance gene. The resulting plasmids were transfected into HeLa cells using LipofectAMINE Plus and stably transfected cells selected by blasticidin (10 μg/ml, Invitrogen). An siRNA sequence corresponding to nucleotides 1235–1253 (GATTTCGAGTCGTCTTAAT) of the Photinus pyralis (firefly) luciferase mRNA (GenBank accession number M15077) was used as a negative control.
Quantitative real-time PCR
Total RNA was isolated from HeLa cells as indicated using an RNeasy Mini Kit (QIAGEN, Valencia, CA, USA) and reverse transcribed into single-stranded cDNA using the first Strand cDNA Synthesis Kit for RT-PCR (AMV) (Roche Diagnostics, Indianapolis, IN, USA). Quantitative real-time PCR analysis was performed using the LightCycler system (Roche) and FastStart DNA Master SYBR Green I (Roche). The results were analyzed with LightCycler Software 3.5 (Roche). Calculated concentration of hCLS cDNA was normalized with respect to the expression level of GAPDH for each sample. Primers used for real-time PCR were human GAPDH, 5′-GAAGGTGAAGGTCGGAGTC-3′ (sense) and 5′-GAAGATGGTGATGGGATTTC-3′ (antisense); hCLS, 5′-CCCAGTTCTGGGCTATTTG-3′ (sense) and 5′-TCAAGAGCACTTCCCAAAGC-3′ (antisense).
Mass spectrometry
Lipids were extracted from HeLa cells by CHCl3/CH3OH(1 : 1, v/v), dried down under a stream of nitrogen at 50°C, dissolved in CHCl3/CH3OH/0.1 N HCl (50 : 45 : 5, v/v), and injected into a Quattro II triple-quadrupole mass spectrometer (Micromass, Manchester, UK) set in the negative electrospray ionization mode. Nitrogen was used as nebulizing gas, and argon as collision gas at a pressure of 2.5 × 10−3 mBar. The capillary voltage was set to 3 kV, the source temperature was maintained at 80°C, and the optimal cone voltage energy for CL and MLCL was 30 V. For CL and MLCL analysis, mass spectra were obtained by continuous scanning in the range of 400–1000 m/z with a scan rate of 200 Da/s in a time window of 2.5 min, during which both types of compound elute from a column. Daughter fragments of CL and MLCL molecular species were obtained using the optimal collision energy of 40 eV as described previously.34
In a separate analysis, single-ion recording was adopted for a semiquantitative measurement of selected CL and MLCL.31 The area of each peak corresponding to CL or MLCL or added internal standard (IS; a CLS IS of 619.5 m/z and a PG IS at 665.5 m/z were used) was quantified using MassLynx 3.3 (Micromass). If appropriate, the peak areas were corrected for the contribution of naturally occurring isotopes. To compare individual samples, the ratio of a CL or MLCL peak area to an IS peak area was also corrected for protein content. As this method has not been validated for different CL or MLCL molecular species and we did not determine if the response of IS was equal to that of the quantified CL or MLCL, we showed these values as arbitrary units, normalized per milligram of protein content.
Cell death induction, flow cytometry, and clonogenic analysis
Cells were treated for the induction of cell death with anti-Fas antibody (0.5 μg/ml, Clone CH-11, Immunotech, France) or staurosporine (5 μM, Sigma) in the absence or presence of 25 μM Chx (25 μM, Sigma), stained with the indicated dyes and then subjected to flow cytometry using a FACScalibur 4C (Becton Dickinson, San Jose, CA, USA). The resulting fluorescence was captured and analyzed by the Cell Quest software (BD Biosciences, San Jose, CA, USA). The light-scatter and forward-scatter channels were set at a linear gain and the fluorescence channels at a logarithmic scale. Unless stated otherwise, at least 104 cells were analyzed in each experiment. Cell size and density (or granularity) were assessed with forward versus side light scatters (FSC versus SSC).
To distinguish apoptosis from necrosis, 106 cells were stained with YOPRO-1 and PI using a Vybrant Apoptosis Assay Kit (Molecular Probes) as per the manufacturer's instructions and analyzed by flow cytometry with FL1 channel for YOPRO-1 and FL3 channel for PI. For the analysis of Δψm versus cellular viability, cells were stained with a Δψm indicator, 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3), 40 nM) or tetramethylrosamine ethyl ester (TMRE, 20 nM) (both from Molecular Probes), incubated for 20 min at room temperature, labeled again with a cellular viability dye, PI (2 μg/ml) or TOPRO-3 (2 μg/ml), and analyzed by flow cytometry. To determine the correlation among superoxide anion, Δψm, and cellular viability, 106 cells were first double stained with DiOC6(3) (40 nM) and hydroethidine (0.5 μM), superoxide anion indicator, for 20 min at room temperature, washed with phosphate-buffered saline (PBS), counterstained with TOPRO-3 (2 μg/ml) and subjected to flow cytometry. Fluorescence was captured via FL1 channel for DiOC6(3), FL2 channel for hydroethidine, and FL4 channel for TOPRO-3.
For clonogenic assays, the cells were washed after exposure to apoptotic stimuli for the times indicated, replated at low density, cultured for several additional days, and colony formation scored by microscopy by examining a gridded area that in control plates contained approximately 1000 cells or colonies. Colonies are expressed as a percentage of the total cells plated.
Subcellular fractionation and Western blotting
Control and hCLS-RNAi cells left untreated or treated with anti-Fas antibody and Chx for 7 h were washed in PBS and broken open using a Dounce homogenizer. Nuclei and unbroken cells were separated at 120 × g for 5 min. The resulting supernatant was centrifuged at 10 000 × g for 10 min to collect the heavy membrane pellet. Subsequently, the resulting supernatant was centrifuged at 100 000 × g for 30 min to yield the light membrane pellet and final cytosolic fraction. Heavy membrane and cytosolic fractions were mixed with Laemmli's buffer (60 mM Tris HCl (pH 6.8), 10% (v/v) glycerol, 2% (w/v) SDS, 5% (v/v) β-mercaptoethanol, and 0.002% (v/v) Bromophenol Blue), boiled for 5 min, subjected to 15% SDS-PAGE, and transferred to a nitrocellulose membrane. The blot was then probed with indicated primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Fluorescent signals were detected with an enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ, USA). Primary antibodies used are as follows: anti-actin and anti-cytochrome c (Pharmingen, La Jolla, CA, USA) and anti-VDAC (Calbiochem, La Jolla, CA, USA).
Transmission electron microscopy
Cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) for 1 h at 4°C, followed by 1% OsO4 for 1 h at 4°C. Subsequently, cells were ‘en bloc’ contrasted in 1% uranyl acetate, dehydrated through a graded series of acetonitrile and embedded in epoxy resin. Thin sections (100 nm) were cut on a Reichert Ultracut E ultramicrotome, placed on 100-mesh copper grids, contrasted in uranyl acetate and lead citrate and viewed under a Hitachi H600 electron microscope (Tokyo, Japan).
Detection of loosely bound or free cyt. c and measurement of mitochondrial CL content
Mitochondria were purified from HeLa cells and resuspended in KCl buffer (150 mM KCl/5 mM Tris-HCl, pH 7.4). Mitochondria in 1 mM EGTA were energized with 5 mM succinate in the presence of rotenone to maintain pyridine in its reduced state. The presence of EGTA and total absence of inorganic phosphate keep the mitochondrial permeability pore closed, that is, blocking cyt. c release. Respiring mitochondria were incubated with recombinant Bax (10 μg/ml, gift from Professor J.C. Martinou, University of Geneva, Switzerland) for 8–10 min with stirring and then centrifuged. Cyt. c content in the resulting supernatant was determined by HPLC using a 5 mm C4 reverse-phase column (150 × 4.6 mm) on an HP series 1100 HPLC chromatograph. A gradient of 20% acetonitrile in water with trifluoroacetic acid (0.1% v : v) to 60% acetonitrile in water with trifluoroacetic acid (0.1% v : v) over 12 min with a flow rate of 1 mL/min was used. Absorption was measured at 393 nm. The amount of cyt. c is expressed in nmol/mg protein.
Measurement of mitochondrial CL content
CL content was determined by HPLC. Lipids were extracted from HeLa cell mitochondria with chloroform/methanol and the phospholipids separated by HPLC with an Altex ultrasil-Si column (4.6 × 250). The chromatographic system was programmed for gradient elution using two mobile phases: solvent A, hexane/2-propanol (6 : 8, v/v) and solvent B, hexane/2-propanol/water (6 : 8 : 1.4, v/v). The percentage of solvent B in solvent A was increased from 0 to 100% over a 15 min time period. Flow rate was 2 ml/min and absorbance was measured at 206 nm.
Cell respiration and mitochondrial enzyme activities
Respiration and mitochondrial substrate oxidation with malate/glutamate and with succinate were polarographically studied in cell suspensions of digitonin-permeabilized HeLa cells as described previously for fibroblasts.35, 36 The activity of the following respiratory chain complexes were spectrophotometrically measured on freeze–thaw permeabilized HeLa cells in cell suspension as described previously for lymphoblasts:35, 36 NADH-ubiquinone reductase (NQR, complex I), succinate-cytochrome c reductase (SCCR, complex II–III), quinol cytochrome c reductase (QCCR, complex III), cytochrome c oxidase (COX, complex IV), mitochondrial ATP synthetase (ATPase, complex V), and citrate synthase (CS).
Abbreviations
- CDP:
-
cytidine 5′-diphosphate
- Chx:
-
cycloheximide
- CL:
-
cardiolipin
- CLS:
-
cardiolipin synthase
- CMP:
-
cytidine 5′-monophosphate
- cyt. c:
-
cytochrome c
- DHB:
-
2,5-dihydroxybenzoic acid
- MLS:
-
mitochondrial localization sequence
- PA:
-
phosphatidic acid
- PG:
-
phosphatidylglycerol
- PGP:
-
phosphatidylglycerophosphate
- STS:
-
staurosporine
- tBid:
-
truncated Bid
- TLC:
-
thin layer chromatography
- TNF-α:
-
tumor necrosis factor-α
References
Hatch GM . Cell biology of cardiac mitochondrial phospholipids. Biochem Cell Biol 2004; 82: 99–112.
Schlame M, Rua D, Greenberg ML . The biosynthesis and functional role of cardiolipin. Prog Lipid Res 2000; 39: 257–288.
Chang SC, Heacock PN, Mileykovskaya E, Voelker DR, Dowhan W . Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J Biol Chem 1998; 273: 14933–14941.
Zhong Q, Gohil VM, Ma L, Greenberg ML . Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J Biol Chem 2004; 279: 32294–32300.
Tuominen EK, Wallace CJ, Kinnunen PK . Phospholipid–cytochrome c interaction: evidence for the extended lipid anchorage. J Biol Chem 2002; 277: 8822–8826.
Spooner PJ, Watts A . Cytochrome c interactions with cardiolipin in bilayers: a multinuclear magic-angle spinning NMR study. Biochemistry 1992; 31: 10129–10138.
Zhang M, Mileykovskaya E, Dowhan W . Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 2002; 277: 43553–43556.
Robinson NC . Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr 1993; 25: 153–163.
Wright MM, Howe AG, Zaremberg V . Cell membranes and apoptosis: role of cardiolipin, phosphatidylcholine, and anticancer lipid analogues. Biochem Cell Biol 2004; 82: 18–26.
Gonzalvez F, Bessoule JJ, Rocchiccioli F, Manon S, Petit PX . Role of cardiolipin on tBid and tBid/Bax synergistic effects on yeast mitochondria. Cell Death Differ 2005; 12: 659–667.
Gonzalvez F, Pariselli F, Dupaigne P, Budihardjo I, Lutter M, Antonsson B et al. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ 2005; 12: 614–626.
Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X . Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol 2000; 2: 754–761.
Lutter M, Perkins GA, Wang X . The pro-apoptotic Bcl-2 family member tBid localizes to mitochondrial contact sites. BMC Cell Biol 2001; 2: 22.
Orrenius S, Zhivotovsky B . Cardiolipin oxidation sets cytochrome c free. Nat Chem Biol 2005; 1: 188–189.
Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem 1999; 274: 1156–1163.
Epand RF, Martinou JC, Fornallaz-Mulhauser M, Hughes DW, Epand RM . The apoptotic protein tBid promotes leakage by altering membrane curvature. J Biol Chem 2002; 277: 32632–32639.
Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2002; 2: 55–67.
Williams JG, McMaster CR . Scanning alanine mutagenesis of the CDP-alcohol phosphotransferase motif of Saccharomyces cerevisiae cholinephosphotransferase. J Biol Chem 1998; 273: 13482–13487.
Barth PG, Valianpour F, Bowen VM, Lam J, Duran M, Vaz FM et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A 2004; 126: 349–354.
Ostrander DB, Sparagna GC, Amoscato AA, McMillin JB, Dowhan W . Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J Biol Chem 2001; 276: 38061–38067.
Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S . Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 2002; 99: 1259–1263.
Iverson SL, Orrenius S . The cardiolipin–cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys 2004; 423: 37–46.
Nakagawa Y . Initiation of apoptotic signal by the peroxidation of cardiolipin of mitochondria. Ann N Y Acad Sci 2004; 1011: 177–184.
Wronski R, Golob N, Grygar E, Windisch M . Two-color, fluorescence-based microplate assay for apoptosis detection. Biotechniques 2002; 32: 666–668.
Hu X, Han Z, Wyche JH, Hendrickson EA . Helix 6 of tBid is necessary but not sufficient for mitochondrial binding activity. Apoptosis 2003; 8: 277–289.
Kawasaki K, Kuge O, Chang SC, Heacock PN, Rho M, Suzuki K et al. Isolation of a Chinese hamster ovary (CHO) cDNA encoding phosphatidylglycerophosphate (PGP) synthase, expression of which corrects the mitochondrial abnormalities of a PGP synthase-defective mutant of CHO-K1 cells. J Biol Chem 1999; 274: 1828–1834.
Ohtsuka T, Nishijima M, Suzuki K, Akamatsu Y . Mitochondrial dysfunction of a cultured Chinese hamster ovary cell mutant deficient in cardiolipin. J Biol Chem 1993; 268: 22914–22919.
Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 2005; 1: 223–232.
Shidoji Y, Hayashi K, Komura S, Ohishi N, Yagi K . Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem Biophys Res Commun 1999; 264: 343–347.
Zhang M, Mileykovskaya E, Dowhan W . Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem 2005; 280: 29403–29408.
Valianpour F, Mitsakos V, Schlemmer D, Towbin JA, Taylor JM, Ekert PG et al. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J Lipid Res 2005; 46: 1182–1195.
Ludovico P, Madeo F, Silva M . Yeast programmed cell death: an intricate puzzle. IUBMB Life 2005; 57: 129–135.
Brummelkamp TR, Bernards R, Agami R . A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296: 550–553.
Valianpour F, Wanders RJ, Barth PG, Overmars H, van Gennip AH . Quantitative and compositional study of cardiolipin in platelets by electrospray ionization mass spectrometry: application for the identification of Barth syndrome patients. Clin Chem 2002; 48: 1390–1397.
Chretien D, Benit P, Chol M, Lebon S, Rotig A, Munnich A et al. Assay of mitochondrial respiratory chain complex I in human lymphocytes and cultured skin fibroblasts. Biochem Biophys Res Commun 2003; 301: 222–224.
Darin N, Kadhom N, Briere JJ, Chretien D, Bebear CM, Rotig A et al. Mitochondrial activities in human cultured skin fibroblasts contaminated by Mycoplasma hyorhinis. BMC Biochem 2003; 4: 15.
Acknowledgements
We thank D. Bogenhagen, W. Dowhan, E.Y. Kim, J. Martinou, U. Moll, A. Neiman, R. Sternglanz, A. Sutton, S. Tsirka, R. Houtkooper, and members of the Frohman lab for technical advice and assistance, reagents, and critical discussions. We thank G. Petrosillo and G. Paradies (University of Bari, Italy) for their advice and discussion concerning RP-HPLC determination of CL and cyt. c levels. The work was supported by NIHDDK 64166 and NIHGM71520 to MAF, a fellowship award to SYC provided by the United Mitochondrial Disease Foundation, an NRSA T32 fellowship to GMJ, and by AFM (Association Française contre les Myopathies) grant no.11557 (2005) and support from the CNRS (Centre National de la Recherche Scientifique) to PXP.
While this manuscript was under review, the identification of the hCLS gene product was reported elsewhere: Houtkooper et al. 2006. FEBS Lett. 580: 3059–3064; Lu et al. 2006. J Lipid Res. 47: 1140–1145; Chen et al. (2006). Biochem. J. in press.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by D Vaux
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Choi, SY., Gonzalvez, F., Jenkins, G. et al. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ 14, 597–606 (2007). https://doi.org/10.1038/sj.cdd.4402020
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402020
Keywords
This article is cited by
-
In situ metabolite and lipid analysis of GluN2D−/− and wild-type mice after ischemic stroke using MALDI MSI
Analytical and Bioanalytical Chemistry (2020)
-
Tibolone Preserves Mitochondrial Functionality and Cell Morphology in Astrocytic Cells Treated with Palmitic Acid
Molecular Neurobiology (2017)
-
Cardiolipin or MTCH2 can serve as tBID receptors during apoptosis
Cell Death & Differentiation (2016)
-
Altered Cytoskeleton as a Mitochondrial Decay Signature in the Retinal Pigment Epithelium
The Protein Journal (2016)
-
Multifaceted role of prohibitin in cell survival and apoptosis
Apoptosis (2015)
