
Abstract
In Europe, concordant geographical distribution among genetic lineages within different species is rare, which suggests distinct reactions to Quaternary ice ages. This study aims to determine whether such a discrepancy also affects a pair of sympatric species, which are morphologically and taxonomically closely related but which have slight differences in their ecological habits. The phylogeographic structures of two European rodents, the Yellow-necked fieldmouse (A. flavicollis) and the woodmouse (Apodemus sylvaticus) were, therefore, compared on the basis of mitochondrial DNA cytochrome b (mtDNA cyt b) sequences (965 base pairs) from 196 specimens collected from 59 European localities spread throughout the species distributions. The results indicate that the two species survived in different ways through the Quaternary glaciations. A. sylvaticus survived in the Iberian Peninsula from where it recolonized almost all Europe at the end of the last glaciation. Conversely, the refuge from which A. flavicollis recolonized Europe, including northern Spain, during the Holocene corresponds to the Italo-Balkan area, where A. sylvaticus suffered a serious genetic bottleneck. This study confirms that even closely related species may have highly different phylogeographic histories and shows the importance of ecological plasticity of the species for their survival through climate change. Finally, it suggests that phylogeographic distinctiveness may be a general feature of European species.
Similar content being viewed by others
Introduction
Phylogeography has become a powerful field of research revealing the principles and processes determining the geographic distribution of genealogical lineages, within and among closely related species (Avise, 2000). An extension of the phylogeography of single species is to compare patterns in multiple codistributed species. The main goal of this ‘comparative phylogeography’ has been to search for concordant geographical distribution among lineages within different species, which would indicate the influence of common historical factors. Recent comparative phylogeographic studies have found unanticipated biogeographic patterns, which suggest that vicariance has played a more important role in the appearance of modern biotic assemblages than current taxonomy would indicate (Arbogast and Kenagy, 2001).
Examining the mitochondrial DNA (mtDNA) polymorphism in various animal species, including fishes, turtles, birds and invertebrates, Avise (2000) reports the existence of similar phylogeographic patterns in South-Eastern USA, suggesting the influence of common historical factors in structuring the geographical distribution of the genetic variability of these taxa. Concordance between phylogeographic patterns was also observed in other regions, such as central America, in several rodent species (ie Bermingham and Martin, 1998; Riddle et al, 2000; Sullivan et al, 2000), southern America in various bat, rodent and marsupial species (ie da Silva and Patton, 1993,1998; Ditchfield, 2000) as well as in Australian birds, reptiles (Joseph et al, 1995) and snails (Hugall et al, 2002). Furthermore, Lessa et al (2003) detected a strong concordance of demographic expansion in North American mammals after the last ice age, 10 000 years ago. Finally, in contrast to previous paleontological studies (Graham et al, 1996), recent fossil analyses strongly suggest that many North American species shifted their ranges in similar ways during the Quaternary climate changes (Lyons, 2003).
However, some other examples show that such a concordance should not be considered a general trend, incongruences appearing in the phylogeographic structure of different species, namely among different plants and animals from north western North America (Brunsfeld et al, 2001) or among various small mammals in Southern America (Costa, 2003). The most striking situation is found in Europe. Indeed, although some general trends can be identified concerning refuge regions, postglacial recolonization routes or location of contact zones (Bilton et al, 1998; Taberlet et al, 1998; Hewitt, 2001), important differences are detected between phylogeographic patterns throughout Europe suggesting that the taxa responded differently to the Quaternary ice ages (Taberlet et al, 1998; Hewitt, 1999,2001).
This important lack of congruence between the phylogeographic histories of European species is probably linked to the particular topography of Europe. Indeed, strong biogeographic barriers such as the Mediterranean and the Black Seas or the Pyrenees, the Alps and the Carpathians Mountains run across Europe in an east–west orientation. They probably limited the southward movements of fauna and flora during the ice ages and the northward expansion from the Mediterranean peninsulas during interglacial periods. Depending on their ecological ability to cross these barriers as well as to stochastic effects (ie important environment changes or diseases over very short periods, which probably led to the disappearance of many populations, even in the potential refuges), the European species probably reacted to the Quaternary glaciations and survived them in many different ways.
This study aims to determine whether such incongruence between the phylogeographic patterns also occurs in closely related sympatric species with similar behavior and morphology but with some differences in their ecological habits: (1) Did they survive in the same regions during the Quaternary glaciations? (2) Did they expand rapidly in northern European regions from these refuges at the end of the last glaciation (like North American mammals)? (3) Did they recolonize these northern regions by the same routes?
In other terms, do these two species have a similar phylogeographic structure and is it possible to discover the influence of common historical factors or, on the contrary, do they have independent phylogeographic histories as a consequence of different ecological habits?
In order to answer these questions, we compared the well-known phylogeographic structure of the woodmouse (Apodemus sylvaticus) (Michaux et al, 1996,1998,2003) to that of the yellow-necked fieldmouse (Apodemus flavicollis). These species appear to be an interesting model, because they are phylogenetically and taxonomically closely related (Michaux et al, 2002), both have been present in Europe for at least 3 My (Michaux and Pasquier, 1974), their ecological preferences are slightly different but overlap, they are sympatric and even syntopic over a great part of their distribution, and their phenotypes are often so similar that species discrimination in the field is often problematic (Niethammer, 1978; Michaux et al, 2001).
Materials and methods
A total of 98 A. flavicollis coming from 39 localities sampled throughout their European range have been analyzed (Table 1 and Figure 1). These were compared with 102 A. sylvaticus from 40 localities and already described in Michaux et al (2003).

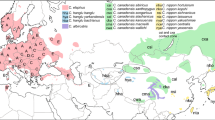
Geographical distribution of the Apodemus flavicollis (a) and A. sylvaticus (b) samples. The shaded zones correspond to the distribution area of each species in Europe respectively (as described by Schilling et al, 1986; Mezhzherin, 1997; Zagorodnyuk et al, 1997). Thick lines correspond to the main biogeographic barriers: the Pyrenees and the Alps. In (a), squares, triangles and circles correspond to clades A, B and C, respectively. White stars and diamond shape correspond to localities where haplotypes from the clades A and C or from B and C are mixed, respectively. See Table 1 for locality symbols. In (b), squares, triangles, white stars and circles represent localities where the clades 1a and 1b, 2a and 2b were found, respectively (see Michaux et al, 2003).
DNA was extracted from ethanol-preserved tissue following Sambrook et al (1989). Tissues were taken from the Apodemus tissue collection of JR Michaux (JRM-numbers) and the mammal tissue collection housed at the Institut des Sciences de l'Evolution de Montpellier (Catzeflis, 1991; T-numbers).
A large portion of cytochrome b (965 bp) was amplified using the same primers and protocols previously used for A. sylvaticus (see Michaux et al, 2003).
Sequence alignment
The published cytochrome b sequences for A. flavicollis (n=3: AB032853, AF159392 and AF127539), A. sylvaticus (n=2: AJ311148 and AJ311149), and A. mystacinus (n=2: AJ311146 and AJ311147) were downloaded from GenBank (see Table 1) and aligned to the new sequences using the ED editor (MUST package, Philippe (1993).
Analyses
The aligned sequences were analyzed by distance (neighbor joining, NJ; Saitou and Nei, 1987) and maximum parsimony (MP) (Kluge and Farris, 1969) methods. We used PAUP 4.0b8 (Distance: ME criterion, Kimura's 2 parameter (K2P) and general time reversible (GTR) models; MP: heuristic search, TBR branch swapping option, Maxtrees=1000 (Swofford, 1998)). The distance analysis was performed assuming a gamma distribution for substitution rates across sites, where the parameter alpha (Yang, 1996) and the proportion of invariant sites (I) was estimated by the maximum-likelihood method assuming the GTR phylogeny using the PAUP 4.0b8. The robustness of inferences was assessed by bootstrap resampling (BP) (Felsenstein, 1985) (1000 random repetitions). A minimum spanning network was constructed using the program TCS (Clement et al, 2000). Nucleotide diversity, π, was estimated using the DNASP program (Rozas and Rozas, 1997). A ‘mismatch distribution’ of substitutional differences between pairs of haplotypes was calculated within the main genetic lineages (clades A, B and C) and compared with a fit to the Poisson model using DNASP (Rozas and Rozas, 1997). This method can be used to assess the demographic history (patterns of population growth or stability) (Emerson et al, 2001) of the main clades. Moreover, a coalescent approach was also developed to validate the results revealed by the mismatch distributions (Emerson et al, 2001). This latter method is explained more extensively in Michaux et al (2003).
An approximate timing of divergence between the observed mtDNA lineages was calculated as explained in Michaux et al (2003). For this procedure, two calibration points derived from paleontological data were used for this analysis. First, the divergence time between the ‘big’ A. mystacinus and all the ‘small’ Sylvaemus (A. flavicollis, A. sylvaticus, A. alpicola, A. uralensis) was estimated from fossil data (cranial and dental characters) at approximately 7 My (Aguilar and Michaux, 1996; Michaux et al, 1997). Secondly, the same methods allowed an estimate of the divergence between A. sylvaticus and A. flavicollis at approximately 4 My (Michaux and Pasquier, 1974; Jacques Michaux, personal communication). Relative-rate tests were conducted according to Robinson et al (1998).
Results
Haplotype variation
We identified 79 haplotypes among the A. flavicollis cytochrome b sequences (GenBank Accession Numbers AJ605600–AJ605665, AJ605672–AJ605675, AJ631968–AJ631970, AJ605686, AJ605687, AJ605691, AJ605692+published sequences: ABO32853, AF159392). The complete data matrix comprised these 79 haplotypes as well as two A. sylvaticus and two A. mystacinus sequences used as outgroups. This matrix provided 965 bp of which 298 sites (31%) were variable and 189 (19%) were parsimony informative, respectively. The mean transition to transversion ratio was 3.49. The nucleotide frequencies were 0.272, 0.290, 0.314 and 0.123 for C, T, A and G, respectively.
Phylogenetic analyses
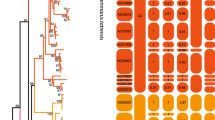
The A. flavicollis NJ tree (Figure 2a) is characterized by a weak genetic and geographic structure. However, three clades can be observed: the first one (clade A) is well supported (BP of 82%) and characterized by specimens from northern Europe (Russia, Estonia, Lithuania, Belarus, Sweden) and the Balkan region (Greece, Romania and European Turkey). The second one (B) (BP: 99%) corresponds to animals from southern Russia, Romania and Yugoslavia and appears more closely related to the first lineage than to the third one, which is less well supported (BP: 61%) and comprises individuals from the whole Western Palearctic region including Western (France, Belgium), Northern (Sweden) and Eastern Europe up to Ukraine as well as the three Mediterranean peninsulas (Iberian, Apennine and Balkan). The genetic divergence between these clades is low, varying between 1.3 and 1.9% K2P distance. The MP analyses yielded one most parsimonious tree for each species (L=472 and 678 steps; CI=0.65 and 0.58; RI=0.90 and 0.82, respectively) identical to the NJ tree.

Neighbor-joining tree for the 79 A. flavicollis (a) and 102 A. sylvaticus (b) mtDNA haplotypes (labelled from 1 to 79 and 1 to 102). Numbers above the branches correspond to bootstrap support above 50% obtained in the NJ and MP analyses, respectively. Geographic origins (see Table 1 and Figure 1) are shown in parentheses.
The network calculated using the TCS program (Clement et al, 2000) (Figure 3a) show similar results: almost all the haplotypes are connected together in a huge star-like topology (subclade C), whereas the clade A is separated from this group by three mutational steps. Between the clades A and B, there are nine further mutational steps.

Networks constructed using mitochondrial cytochrome b gene sequences of A. flavicollis (a) and A. sylvaticus (b). In (a), the numbers correspond to the 79 haplotypes obtained in the study (for their geographic origins, see Figure 2). The black dots correspond to the mutational steps between haplotypes obtained using the TCS program (Clement et al, 2000). The number 9 in italics correspond to the mutational steps observed between clades A and B. In (b) the results of Michaux et al (2003) on A. sylvaticus are shown. Geographic origins of haplotypes are indicated (for more informations, see Michaux et al, 2003). Numbers in italics correspond to the mutational steps observed between haplotypes.
Nucleotide diversity and genetic structure
To assess whether genetic diversity was higher within the potential refuge regions as compared to northern populations, the samples were separated into different subgroups corresponding to these regions (Italy+Balkans, Iberian Peninsula+southern France, Western+Central Europe+Northern Europe, Ukraine+Russia) (Figure 1), then nucleotide and haplotype diversities were calculated for every group (see Table 2). The Italian and Balkan populations were combined as they were not genetically differentiated (mean K2P distance between the Balkan and Italian populations versus within the Balkan group=1.0/1.2%). The results indicate that the Italo-Balkan region (Figure 1) has the highest nucleotide diversity (P<0.05, t-test) as compared to the populations from all the other regions. The animals from southwestern Europe were the least genetically diversified. Conversely, the Russian population is characterized by a higher level of nucleotide and haplotypes diversities, corresponding to the presence of two different clades (A and B) occurring syntopically in the region. For comparison, data obtained on A. sylvaticus (Michaux et al, 2003) are also included in Table 2.
A signature of population growth (ie a bell-shaped distribution) is clearly evident in the distribution of pairwise distribution within clade C (Figure 4). Moreover, the coalescence analysis rejects the null hypothesis of a stable population for this clade (Table 3). On the other hand, the clades A and B are bimodal suggesting constant population size for these groups. This is corroborated by the coalescence test that did not reject the constant population model. However, this result must be treated cautiously as the sample size for these groups is very low (seven individuals for each).

Mismatch distribution for mtDNA types from the three major A. flavicollis genetic clades observed in Europe. The expected frequency is based on a population growth-decline model (for the clades A, B and C, respectively: θ initial=0, 2.4 and 0, θ final=1000, τ=4.4, 9.2 and 2.9), determined using the DNASP v3.5 program (Rozas and Rozas, 1997) and is represented by a continuous line. The observed frequency is represented by a dotted line.
A population within a given area is the actual (resident) population. Thus, to test expansion of an actual population, it is obligatory to include its individuals, irrespective of genetic clades. Following this idea, we performed other mismatch distribution analyses considering different geographic populations of A. flavicollis (Italy+Balkans, Iberian Peninsula+southern France, Western+Central Europe, Northern Europe, Ukraine+Russia) and not genetic clades. The results (data not shown) evidence that the Italian–Balkan population is characterized by a bimodal curve suggesting constant population size, whereas the Western+Central European population is characterized by a bell-shaped curve (signature of population growth). More heterogeneous distribution appears in the other populations suggesting that they are more stable. However, this result was obtained on a very small sampling (maximum seven individuals for each population) and should be, again, considered carefully.
Divergence time
The relative rate test indicates no significant rate heterogeneity (both Ks and Ka) for cytochrome b between the different A. flavicollis clades and the Apodemus specimens.
The mean K2P distance between A. mystacinus and A. flavicollis/A. sylvaticus that diverged 7 My ago is 18.2%. The distance between A. flavicollis and A. sylvaticus that diverged 4 My ago is 11.4%. These values give a rate of 2.6–2.85% K2P distance per million years. Therefore, taking into account the correction for ancestral mtDNA polymorphism, the separation time between clades A–B and C and between clades A and B took respectively 0.6–0.5 and 0.4 My ago. For comparison, the separation time between clades 1 and 2 of A. sylvaticus dates back to 1.5–1.6 My (Michaux et al, 2003).
Discussion
Comparison of the genetic structure of A. sylvaticus and A. flavicollis
The distance, maximum parsimony and network analyses clearly show that the two Apodemus species have distinct genetic structure (Figure 2 and 3).
The A. flavicollis populations are split into three main genetic lineages separated by a low level of nucleotide divergence (between 1.3 and 1.9% K2P distance) and overlapping in a part of their geographical distribution in the Balkan region. The divergence time estimated on the basis of two paleontological calibration points (Aguilar and Michaux, 1996; Michaux et al, 1997) indicates a separation time of 0.6–0.5 My between the ancestral haplotypes that have led to these three lineages. Therefore, it appears that the intraspecific structure of A. flavicollis developed during this period. This result is consistent with the hypotheses of Avise (2000),Hewitt (2001) and Seddon et al (2001) who postulated that the majority of the intraspecific differentiation in vertebrates occurred during the Quaternary.
The European continental A. sylvaticus populations are split into two main genetic lineages nonoverlapping in their geographical distributions and separated by a high level of genetic divergence (5.4% of K2P genetic distance) (Figures 2b and 3b; Michaux et al, 2003): the first is widespread from southern Spain in the south to Sweden in the north and Ukraine in the East; the second one is limited to Italy and the Balkans (Figure 1b). As already proposed for several other European species (Taberlet et al, 1998; Hewitt, 2001; Seddon et al, 2001), this structure could be explained by the isolation of two woodmice groups in two different refugia (Iberian peninsula and Italy–Balkans) during one of the first Quaternary glacial periods. This hypothesis is corroborated by the molecular clock analysis that estimates the separation time between the ancestral haplotypes at 1.5–1.6 My ago. This isolation was probably reinforced by the presence of the Alps that played and probably continue to play the role of a biogeographic barrier for the woodmouse (Michaux et al, 1996). These two factors likely have prevented genetic exchanges between both groups for a long time and have led to the strong genetic differences observed within this species. In contrast, the frequent drops in the level of the Adriatic Sea during the Quaternary ice periods (Strahler and Strahler, 1989) apparently allowed the Italian and Balkan woodmice populations to remain in contact. This could also explain the great similarity observed between the A. flavicollis from Italy and the Balkans.
Refuge regions and postglacial recolonizations
As predicted by the expansion/contraction model (Nichols and Hewitt, 1994; Santucci et al, 1998), the analysis of nucleotide diversity (Table 2) indicates that the Italo-Balkan region was a refuge for A. flavicollis. Indeed, these populations are characterized by a genetic diversity significantly higher (π=0.015, P<0.05) than in northern, eastern or western populations. This result is also observed for each of the three genetic lineages analyzed separately and is corroborated by the mismatch distribution analysis performed on different geographic populations of A. flavicollis. Indeed, this analysis suggests the existence of a stable population in the Italo-Balkan region. The three genetic lineages probably survive to the last Quaternary glaciations in three independent Balkan refuges where they diverged separately. They invaded later the entire Western Palearctic region during interglacial periods. These results rather correspond to the hypothesis of Hewitt (1999) and Taberlet et al (1998) which proposed a South to North expansion of the European species, in contrast to the hypothesis of Bilton et al (1998), suggesting a colonization of Europe from Eastern regions (Russia, Asia). Indeed, in this last case, the Eastern populations of A. flavicollis would be characterized by a higher level of genetic diversity as compared to the Balkan ones. However, despite their low number (seven individuals), the Russian–Ukrainian fieldmice are characterized by a relatively high level of nucleotide diversity and genetic divergence as well as by an important number of haplotypes (6). As high genetic diversity also arises when diverse clades occur syntopically, this result would be explained by the presence of two A. flavicollis clades (A and B) in this region. Nevertheless, without a larger sample for this region, the hypothesis of another Eastern refuge cannot be precluded.
Conversely, the population of the southwestern potential refuge (Iberian Peninsula and southern France) has the lowest level of nucleotide diversity and, in contrast to A. sylvaticus, does not possess specific haplotypes absent from other regions. This can be explained by a recent colonization of this region from the Balkan refuge population. Indeed, as proposed by Hewitt (1999) and Avise (2000), a rapid expansion from refugial populations involves serial bottlenecks with progressive loss of allelic diversity resulting in less genetic diversity among populations living in the more recently colonized places. This hypothesis is corroborated by the intermediate level of nucleotide diversity characterizing the populations from Central and Western Europe as well as from Northern Europe.
Therefore, it seems that: (i) the Iberian peninsula did not constitute a refuge for this species during the last glaciation. This is corroborated by paleontological data, which attest the presence of A. flavicollis in this region only from the end of the Lower to the beginning of the Upper Pleistocene (Sese, 1994); (ii) A. flavicollis did not survive up to the last glaciation in Western, Eastern and Northern Europe. This hypothesis is also confirmed by paleontological data (Cordy, 1984; Aguilar and Michaux, unpublished data); (iii) Three genetic lineages of the Yellow-necked fieldmouse surviving in the Balkan region recolonized the main part of the Western Palearctic region after the last ice age, 10 000 years ago. This scenario of expansion is corroborated by a bell-shaped curve in the mismatch distribution analysis at least for the main genetic lineage (C) (Figure 4) (Luikart et al, 2001), the results from the coalescent analysis as well as from allozyme data (Filippucci et al, 2002). Moreover, the star-like topology characterizing the main genetic lineage (clade C) suggests that this expansion was probably very rapid and explosive for this group, which recolonized almost all Europe as compared to the other lineages, and took place after a bottleneck, which probably lowered its ancestral genetic diversity (Avise, 2000).
For A. sylvaticus, the results strongly suggest that southwestern Europe (the Iberian Peninsula and southern France) was a refuge region (Michaux et al, 2003). Indeed, these populations are characterized by a genetic diversity significantly higher (π=0.0087, P<0.05) than in the northern populations (Table 2). Therefore, it is probable that woodmice recolonized and expanded throughout the main part of the Western Palearctic region from southwestern Europe at the end of the last ice age. This scenario of expansion is supported by a bell-shaped aspect of the curve obtained in the mismatch distribution analysis for the main European lineage (Luikart et al, 2001; Michaux et al, 2003). The Pyrenees were therefore not an effective barrier to the northward expansion of the woodmouse (Michaux et al, 1998). In contrast to the southwestern group, the Italo-Balkan populations are characterized by a lower genetic diversity (π=0.0052, P<0.05). This observation is somewhat surprising as paleontological (Aguilar and Michaux, unpublished data) and paleoclimatological (Tzedakis et al, 1997) data attest that Italy and the Balkans were a refuge region for A. sylvaticus during the Quaternary glaciations. This low genetic variability could be explained by a genetic bottleneck that appeared during one of the last ice ages. This hypothesis is corroborated by three results: the very short branch lengths between haplotypes within this group in the distance analysis (Figure 2b); the star-like topology in the minimum spanning network (Figure 3b) suggesting a rapid expansion from a relative small number of founder animals and a low value of haplotype (h) and nucleotide (π) diversity (Table 2) (Avise, 2000). The factors that could explain this genetic bottleneck are discussed in details in Michaux et al (2003). It could also explain why the Italo-Balkan populations did not contribute to the postglacial colonization of the northern regions, as expansion could only have occurred after population growth. During this time, the western group rapidly expanded towards Northern and Central Europe, thereby preventing a northern colonisation by the Italo-Balkan animals. Indeed, once established, resident rodents often aggressively exclude newcomers (Granjon and Cheylan, 1989). Moreover, the presence of topographic obstacles such as the Alps might slow down a postglacial expansion of the Italo-Balkan group.
This study also evidenced that Sicily is a refuge for A. sylvaticus and that this region is characterized by a genetically well-differentiated and stable population, which is separated from the Italian animals since 0.8/0.9 Myr (for more details, see Michaux et al, 2003). Moreover, this species invaded North Africa recently, probably via the Gibraltar Strait and anthropogenic introductions (Libois et al, 2001; Michaux et al, 2003). This wider distribution range of the woodmouse in Western Europe and North Africa is probably associated to its greater ecological plasticity as compared to A. flavicollis, enabling it to colonize regions covered by habitats other than forests (eg Mediterranean scrub and open fields with small groves).
Conclusions
Although A. sylvaticus and A. flavicollis are closely related from a taxonomical point of view and that they have very similar morphology and distribution, they reacted and survived to the Quaternary glaciations in totally different ways. Indeed, from paleontological and genetic data, A. flavicollis did not survive, at least during the last glaciation (22 000–16 000 years ago), in the Iberian Peninsula, whereas A. sylvaticus persisted and recolonized almost all Europe from there at the end of the last glaciation. Conversely, the refuge region from which A. flavicollis recolonized Europe during the Holocene corresponds to the Balkan area where A. sylvaticus seems to have suffered a severe genetic bottleneck. This result corresponds to the Gleasonian model (Graham et al, 1996), which predicts that species respond in an individualistic manner to climate changes, and expand and contract their ranges independently from each other.
The differences between the two Apodemus species or could be explained by stochastic events or interspecific competition: As these species often live in the same habitats, high interspecific competition during the Quaternary ice ages may have played a role in the observed phylogeographic patterns. However, behavioral studies (Hoffmeyer, 1973; Montgomery, 1978,1985; J Cassaing, unpublished data) showed that the overall level of interaction is weak and neither species can exclude the other. Moreover, in interspecific contests, A. sylvaticus is always dominated by A. flavicollis. Therefore, this hypothesis would better explain the decrease of the genetic variability of the Italo-Balkan A. sylvaticus than the disappearance of A. flavicollis in the Iberian region. Another explanation could be related to the evolutionary history of these species. Indeed, according to the paleontological (Michaux and Pasquier, 1974) and molecular (Michaux et al, 2002) data, the differentiation between A. flavicollis and A. sylvaticus is probably the result of an allopatric speciation: the ancestors of A. sylvaticus were isolated in Spain and southern France, whereas those of A. flavicollis rather lived in Central Europe and in the Balkans. Each species could be more adapted to the environment of its ancestral region and, therefore, have had more chances of surviving there. However, as the differentiation between the two species is very old (4 My), both had time to establish across the European regions during the Quaternary (Aguilar and Michaux, unpublished data) and they probably had time enough to adapt themselves to the different European environments. Moreover, the differences between Western and Eastern European environments are probably not sufficient (Blondel, 1996) to support this hypothesis, particularly for such species characterized by strong ecological adaptability (Renaud and Michaux, 2003).
Several ecological (eg Kaustuv et al, 2001; Holt, 2003) and paleontological (Barnosky et al, 2001) studies have suggested the importance of ecological factors in the changes of species ranges, particularly during climate variation. A. sylvaticus is characterized by a greater ecological plasticity than A. flavicollis which is more associated with forested habitat with a high canopy cover (Marsh and Harris, 2000). Thus, the fragmentation of forests during the last glaciation in the Iberian peninsula (Blondel, 1996) may have played a role in its extinction in this region. Moreover, every climate change involves important habitat reorganizations and new biological interactions (Graham et al, 1996). Therefore, a more adaptable species like A. sylvaticus was probably more resistant to these factors than A. flavicollis. Finally, A. flavicollis shows poorer winter survival than A. sylvaticus (Montgomery, 1980; Marsh and Harris, 2000). Therefore, the harsher climatic conditions that characterized the Iberian peninsula during the last glaciation as compared to the Balkan region (Tzedakis et al, 1997) could also explain the extinction of the Yellow-necked fieldmouse in Western Europe during this period.
The time of their respective intraspecific differentiation can be compared under the assumption that cytochrome b gene evolves at the same rate in the two species (Michaux et al, 2002) and that their ancestral effective population size was the average of the current population sizes (Edwards and Beerli, 2000). A. sylvaticus is then estimated to have diverged during Early and Middle Pleistocene (1.5–1.6 My ago for the two continental lineages) (0.8–0.9 My between the Sicilian and Italian lineages) whereas A. flavicollis diverged more recently (0.4–0.6 My ago). This result could be explained by the disappearance of ancient A. flavicollis lineages in southwestern Europe during the Early Quaternary ice ages, which is corroborated by paleontological data (Sese, 1994). In contrast, the better ecological plasticity of the woodmouse probably enabled it to better survive the Quaternary glaciations, even if its populations were periodically fragmented leading to different and deep genetic lineages.
In conclusion, the present results confirm that different species, even if they are closely related, can have strongly different phylogeographic histories and suggest that the phylogeographic exception may be a general feature of European species. This shows the importance of ecological plasticity of the species, for their survival to climate changes. Finally, the present results evidence that the search of the influence of common historical factors to explain the phylogeographic structure of the European species turns out to be very complex.
References
Aguilar J-P, Michaux J (1996). The beginning of the age of Murinae (Mammalia: Rodentia) in southern France. Acta Zool Cracov 39: 35–45.
Arbogast B, Kenagy GJ (2001). Comparative phylogeography as an integrative approach to historical biogeography. J Biogeogr 28: 819–825.
Avise JC (2000). Phylogeography. The History and Formation of Species, Harvard University Press, Cambridge, MA.
Barnosky AD, Hadly EA, Bell CJ (2001). Mammalian response to global warming on varied temporal scales. J Mammal 84: 354–368.
Bermingham E, Martin AP (1998). Comparative phylogeography of neotropical freshwater fishes: testing shared history to infer the evolutionary landscape of lower Central America. Mol Ecol 7: 499–517.
Bilton DT, Mirol PM, Mascheretti S, Fredga K, Zima J, Searle JB (1998). Mediterranean Europe as an area of endemism for small mammals rather than a source for northwards postglacial colonization. Proc Roy Soc Lond Biol Sci 265: 1219–1226.
Blondel J (1996). Biogéographie: approche écologique et évolutive, Masson: Paris.
Brunsfeld SJ, Sullivan J, Soltis DE, Soltis PS (2001). Comparative phylogeography of Northwestern North America: a synthesis. In: Silvertown J, Antonovics J (eds) Integrating Ecological and Evolutionary Processes in a Spatial Context, Blackwell Science: Oxford. pp 319–339.
Catzeflis F (1991). Animal tissue collections for molecular genetics and systematics. TREE 6: 168.
Clement M, Posada D, Crandall KA (2000). TCS: a computer program to estimate gene genealogies. Mol Ecol 9: 1657–1659.
Cordy JM (1984). Evolution des faunes quaternaires en Belgique. In: Cahen D, Haesaerts P (eds) Peuples chasseurs de la Belgique préhistorique dans leur cadre naturel, Bruxelles: Belgium. pp 67–77.
Costa LP (2003). The molecular phylogeographic bridge between the Amazon and the Atlantic forest: a case study with small mammals. J Biogeogr 30: 71–86.
da Silva MN, Patton JL (1993). Amazonian phylogeography: mtDNA sequence variation in arboreal echimyid rodents (Caviomorpha). Mol Phyl Evol 2: 243–255.
da Silva MN, Patton JL (1998). Molecular phylogeography and the evolution and conservation of Amazonian mammals. Mol Ecol 7: 475–486.
Ditchfield AD (2000). The comparative phylogeography of neotropical mammals: patterns of intraspecific mitochondrial DNA variation among bats contrasted to nonvolant small mammals. Mol Ecol 9: 1307–1318.
Emerson B, Paradis E, Thébaud C (2001). Revealing the demographic histories of species using DNA sequences. TREE 16: 707–716.
Edwards SV, Beerli P (2000). Perspective: gene divergence, population divergence and the variance in coalescence time in phylogeographic studies. Evolution 54: 1839–1854.
Felsenstein J (1985). Confidence limits on phylogenies with a molecular clock. Syst Zool 34: 152–161.
Filippucci M-G, Macholan M, Michaux JR (2002). Genetic variation and divergence among Apodemus species. Biol J Linn Soc 75: 395–420.
Graham RW, Lundelius EL, Graham MA, Schroeder EK, Toomey RS, Anderson E et al (1996). Spatial response of mammals to Late Quaternary Environmental Fluctuations. Sciences 272: 1601–1606.
Granjon L, Cheylan G (1989). Le sort de rats noirs (Rattus rattus) introduits sur une île, révélé par radiotracking. CR Acad Sci, Paris 309: 571–575.
Hewitt GM (1999). Post-glacial recolonisation of European biota. Biol J Linn Soc 58: 87–112.
Hewitt GM (2001). Speciation, hybrid zones and phylogeography – or seeing genes in space and time. Mol Ecol 10: 537–549.
Hoffmeyer I (1973). Interaction and habitat selection in the mice Apodemus flavicollis and Apodemus sylvaticus. Oikos 24: 108–116.
Holt RD (2003). On the evolutionary ecology of species' ranges. Evol Ecol Res 5: 159–178.
Hugall A, Moritz C, Moussalli A, Stanisic J (2002). Reconciling paleodistribution models and comparative phylogeography in the Wet Tropics rainforest land snail Gnarosophia bellendenkerensis (Brazier 1875). Proc Natl Acad Sci, USA 99: 6112–6117.
Joseph L, Moritz C, Hugall A (1995). Molecular support for vicariance as a source of diversity in rainforest. Proc R Soc Lond B 260: 177–182.
Kaustuv R, Jablonski D, Valentine JW (2001). Climate change, species range limits and body size in marine bivalves. Ecol Let 4: 366–370.
Kluge AG, Farris JS (1969). Quantitative phyletics and the evolution of anurans. Syst Zool 18: 1–32.
Lessa EP, Cook JA, Patton JL (2003). Genetic footprints of demographic expansion in North America, but not Amazonia, during the Late Quarternary. Proc Natl Acad Sci USA 100: 10331–10334.
Libois RM, Michaux JR, Ramalhinho MG, Maurois C, Sarà M (2001). On the origin and systematics of the North African wood mouse (Apodemus sylvaticus) populations. A comparative study of mtDNA restriction patterns. Can J Zool 79: 1503–1511.
Luikart G, Gielly L, Excoffier L, Vigne J-D, Bouvet J, Taberlet P (2001). Multiple maternal origins and weak phylogeographic structure in domestic goats. Proc Natl Acad Sci USA 98: 5927–5932.
Lyons SK (2003). A quantitative assessment of the range shifts of Pleistocene mammals. J Mammal 84: 385–402.
Marsh ACW, Harris S (2000). Partitioning of woodland habitat resources by two sympatric species of Apodemus: lessons for the conservation of the Yellow-necked mouse (Apodemus flavicollis) in Britain. Biol Cons 92: 275–283.
Mezhzherin SV (1997). Revision of mice genus Apodemus (Rodentia, Muridae) of Northern Eurasia. Vestnik Zool 31: 29–41 (in Russian, with English abstract).
Michaux J, Aguilar J-P, Montuire S, Wolff A, Legendre S (1997). Les Murinae (Rodentia, Mammalia) néogènes du Sud de la France: évolution et paléoenvironnements. Geobios 20: 379–385.
Michaux J, Pasquier L (1974). Dynamique des populations de mulots (Rodentia, Apodemus) en Europe durant le Quaternaire. Premières données. Bull Soc Géol France 7: 431–439.
Michaux JR, Filippucci M-G, Libois RM, Fons R, Matagne RF (1996). Biogeography and taxonomy of Apodemus sylvaticus (the woodmouse) in the Tyrrhenian region: enzymatic variations and mitochondrial DNA restriction pattern analysis. Heredity 76: 267–277.
Michaux JR, Chevret P, Filippucci M-G, Macholan M (2002). Phylogeny of the genus Apodemus with a special emphasis to the subgenus Sylvaemus using the nuclear IRBP gene and two mitochondrial markers: cytochrome b and 12S rRNA. Mol Phyl Evol 23: 123–136.
Michaux JR, Kinet S, Filippucci M-G, Libois R, Besnard A, Catzeflis F (2001). Molecular characterization of three sympatric species of wood mice (Apodemus sylvaticus, A. flavicollis, A. alpicola) in Europe (Muridae: Rodentia). Mol Ecol Notes 1: 260–263.
Michaux JR, Libois R, Ramalhinho M-G, Maurois C (1998). On the mtDNA restriction patterns variation of the Iberian wood mouse (Apodemus sylvaticus). comparison with other west Mediterranean populations. Hereditas 129: 187–194.
Michaux JR, Magnanou E, Paradis E, Nieberding C, Libois RM (2003). Mitochondrial phylogeography of the woodmouse (Apodemus sylvaticus) in the Western Palearctic region. Mol Ecol 12: 685–697.
Montgomery WI (1978). Intra- and interspecific competition of Apodemus sylvaticus (L.) and A. flavicollis (Melchior) under laboratory conditions. Ann Behav 26: 1247–1254.
Montgomery WI (1980). Population structure and dynamics of sympatric Apodemus species (Rodentia, Muridae). J Zool Lond 192: 351–377.
Montgomery WI (1985). Interspecific competition and the comparative ecology of two congeneric species of mice. In: Cook LM (eds) Case Studies in Population Biology, Manchester University Press: Manchester. pp 126–187.
Nichols RA, Hewitt GM (1994). The genetic consequences of long distance dispersal during colonization. Heredity 72: 312–317.
Niethammer J (1978). Apodemus flavicollis (Melchior, 1834) – Gelbhalsmaus; Apodemus sylvaticus (Linnaeus, 1758) – Waldmaus. In: Niethammer J, Krapp F (eds) Handbuch der Säugetiere Europas, Akademische Verlagsgesellschaft: Wiesbaden. pp 325–358.
Philippe H (1993). MUST, a computer package for Management Utilities for Sequences and Trees. Nucleic Acids Res 21: 5264–5272.
Renaud S, Michaux JR (2003). Are shape changes associated with latitude a new adaptive trend? Evidences from the woodmice. J Biogeogr 30: 1617–1628.
Riddle BR, Hafener DJ, Alexander LF (2000). Comparative phylogeography of Bailey's pocket mouse (Chaetodipus baileyi) and the Peromyscus eremicus species group: historical vicariance of the Baja California Peninsular desert. Mol Phyl Evol 17: 161–172.
Robinson M, Gouy M, Gautier C, Mouchiroud D (1998). Sensitivity of the relative-rate test to taxonomic sampling. Mol Biol Evol 15: 1091–1098.
Rozas J, Rozas R (1997). DNASP, version 2.0: a novel software package for extensive molecular population genetic analysis. Comp Appl Biosci 13: 307–311.
Saitou N, Nei M (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425.
Sambrook J, Fritsch EF, Maniatis T (1989). Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory: Cold Spring Harbor, NY.
Santucci F, Emerson BC, Hewitt GM (1998). Mitochondrial DNA phylogeography of European hedgehogs. Mol Ecol 7: 1163–1172.
Schilling D, Singer D, Diller H (1986). Guide des Mammifères d'Europe, Delachaux and Niestlé: Paris.
Seddon JM, Santucci F, Reeve NJ, Hewitt GM (2001). DNA footprints of European hedgehogs, Erinaceus europaeus and E. concolor: Pleistocene refugia, postglacial expansion and colonization routes. Mol Ecol 10: 2187–2198.
Sese C (1994). Paleoclimatical interpretation of the Quaternary small mammals of Spain. Geobios 27: 753–767.
Strahler AH, Strahler AN (1989). Elements of Physical Geography, 3rd edn. John Wiley & Sons: New York.
Sullivan J, Arellano D, Rogers DS (2000). Comparative phylogeography of Mesoamerican Highlands Rodents: concerted versus independent response to past climatic fluctuations. Am Natur 155: 755–768.
Swofford DL (1998). PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0b Sinauer Associates: Sunderland, MA.
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998). Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7: 453–464.
Tzedakis PC, Andrieu V, de Beaulieu JL, Crwhurst S, Follieri M, Hooghiemstr H (1997). Comparison of terrestrial and marine records of changing climate of the last 500 000 years. Earth Planet Sci Lett 150: 171–176.
Yang Z (1996). Among-site rate variation and its impact on phylogenetic analyses. TREE 11: 367–372.
Zagorodnyuk IV, Boyeskorov GG, Zykov OE (1997). Variation and taxonomic status of the steppe forms of genus Sylvaemus ‘sylvaticus’ (falzfeini–fulvipectus–hermonensis–arianus). Vestnik Zool 31: 37–56 (in Russian, with English abstract).
Acknowledgements
We thank Drs Janice Britton-Davidian and Léon Dirick for their helpful comments on the manuscript. We thank all those people who provided tissue samples of Apodemus: L Balciauskas, E Bellinvia, J Cassaing, F Catzeflis, S Escutenaire, R Fons, D Frynta, B Krystufek, P Lymberakis, G Lauters, M Macholan, S Morand, D Murariu, S Naidenko, V Orlov, MG Ramalhinho, M Sará, D Sirugue, H Tegelström, G Tikhonova, P Trontelj, V Vohralik and R Wolf. This work was supported by a Belgian FNRS fellowship to JR Michaux (mandat ‘collaborateur scientifique’) and financial grants of the Belgian FNRS (crédits aux chercheurs, crédits pour brefs séjours à l'étranger to JMR and RL).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Michaux, J., Libois, R. & Filippucci, MG. So close and so different: comparative phylogeography of two small mammal species, the Yellow-necked fieldmouse (Apodemus flavicollis) and the Woodmouse (Apodemus sylvaticus) in the Western Palearctic region. Heredity 94, 52–63 (2005). https://doi.org/10.1038/sj.hdy.6800561
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800561
Keywords
This article is cited by
-
Phylogeography and demographic history of the subterranean rodents belonging to the Ctenomys talarum phylogenetic group (Rodentia: Ctenomyidae)
Mammalian Biology (2023)
-
Bifidobacterium castoris strains isolated from wild mice show evidence of frequent host switching and diverse carbohydrate metabolism potential
ISME Communications (2022)
-
Idiosyncratic responses to drivers of genetic differentiation in the complex landscapes of Isthmian Central America
Heredity (2021)
-
Population structure of Apodemus flavicollis and comparison to Apodemus sylvaticus in northern Poland based on RAD-seq
BMC Genomics (2020)
-
Comparative phylogeography of two hemipteran species (Geisha distinctissima and Megacopta cribraria) in the Zhoushan Archipelago of China reveals contrasting genetic structures despite concordant historical demographies
Heredity (2020)
