
Abstract
Infections of Asian elephants (Elephas maximus) with elephant endotheliotropic herpesvirus (EEHV) can cause a rapid, highly lethal, hemorrhagic disease, which primarily affects juvenile animals up to the age of four years. So far, the majority of deaths have been attributed to infections with genotype EEHV1 or, more rarely, EEHV3 and EEHV4. Here, we report the pathological characteristics of the first fatality linked to EEHV5 infection and describe the complete viral DNA sequence. Gross post-mortem and histological findings were indistinguishable from lethal cases previously attributed to other EEHV genotypes and the presence of characteristic herpesviral inclusions in capillary endothelial cells at several sites was consistent with the diagnosis of acute EEHV infection. Molecular analysis confirmed the presence of EEHV5 DNA and was followed by sequencing of the viral genome directly from post-mortem material. The genome is 180,800 bp in size and contains 120 predicted protein-coding genes, five of which are fragmented and presumably nonfunctional. The seven families of paralogous genes recognized in EEHV1 are also represented in EEHV5. The overall degree of divergence (37%) between the EEHV5 and EEHV1 genomes and phylogenetic analysis of eight conserved genes, support the proposed classification of EEHV5 into a new species (Elephantid herpesvirus 5).
Similar content being viewed by others
Introduction
Since the first reported death attributed to a herpesvirus in an Asian elephant (Elephas maximus) in 19881, elephant endotheliotropic herpesvirus (EEHV) infection has emerged as a serious threat to captive and wild Asian elephant populations. To date, seven distinct genotypes, named EEHV1 to EEHV7, have been identified in Asian elephants and African elephants (Loxodonta africana). The spectrum of associated disease ranges from subclinical infection to rapidly fatal hemorrhagic disease2. The genotype most often involved in fatalities is EEHV1 and the complete genome sequences of the two subgenotypes (EEHV1A and EEHV1B) were resolved recently3,4. Two further genotypes, EEHV3 and EEHV4, are known to cause sporadic fatalities in young Asian elephants and EEHV2 has been attributed to a similar disease in African elephants5,6,7,8. The tropism of EEHVs for endothelium results in widespread capillary damage and subsequent lethal pericardial, thoracic and abdominal effusions and variably extensive hemorrhages in a variety of tissues7,8,9.
EEHV5 was first detected in 2007 in whole blood samples from a clinically healthy, 59 year-old, wild-born Asian elephant7. This genotype has since been associated with a small number of subclinical and clinical infections in captive Asian elephants10. Clinical findings in an EEHV5-positive herd included bilaterally swollen temporal glands, oral mucosal hyperaemia, vesicles on the tongue and generalized lethargy in an otherwise healthy adult and a one year-old calf. DNA sequence analysis revealed the presence of EEHV5 subgenotypes (EEHV5A and EEHV5B) within the herd, but no fatalities were recorded10.
Initial DNA sequence characterization of EEHV1A and EEHV1B5,11,12 was instrumental in classifying the viruses into the species Elephantid herpesvirus 1 in the genus Proboscivirus, subfamily Betaherpesvirinae, family Herpesviridae, order Herpesvirales13. Derivation of the complete genome sequences of two EEHV1A strains (EEHV1A/Kimba and EEHV1A/Raman; GenBank accession numbers KC618527 and KC462165, respectively) and one EEHV1B strain (EEHV1B/Emelia; KC462164) recently provided substantial insights into the genetic compositions of these viruses, which are characterized by numerous genes lacking counterparts in other herpesviruses3,4. In this report, we describe detailed pathological findings and a complete viral genome sequence from the first case of EEHV5-associated fatality in an Asian elephant.
Methods
Case history
The affected animal was a juvenile Asian elephant, Vijay (♂; barn name; official name Ganesh), which was born at Twycross Zoo, UK, within a herd of four adult, female Asian elephants on 6 August 2009 (http://www.elephant.se/database2.php?elephant_id=4826). This animal had a history of retarded growth and ill-health and developed an oral lesion during December 2010 and a mucocutaneous perianal lesion during March 2011, both of which resolved spontaneously over a period of 2–3 weeks. In April 2011, lethargy followed by rapidly progressive hemorrhages of mucosal membranes, edema and lingual cyanosis led to the presumptive clinical diagnosis of EEHV infection. Treatment followed protocols for extensive antiviral and supportive therapy, including drainage of the pericardial effusion under ultrasonographic guidance. Despite all efforts over a period of 6 days, the calf's health deteriorated and the decision was made to euthanize the animal. All treatments and clinical decision making in this case were conducted in accordance with the Veterinary Surgeons Act under the direct supervision of a RCVS Specialist and within the ethical policies of the zoo.
Pathology
A comprehensive post-mortem procedure following elephant necropsy protocols14 was carried out within 24 hours. For histological examination, tongue, heart, lung, spleen, liver, kidneys, stomach and all segments of intestine, urinary bladder, skeletal muscle, thyroid, adrenal and salivary glands, skin, oral mucosa and a section of trunk, pharyngeal lymphoid tissue, lymph nodes, trigeminal nerve, brain (including the pituitary gland), medulla and bone marrow were processed routinely and paraffin-embedded sections were stained with hematoxylin and eosin.
Viral diagnosis
DNA was extracted from heart, tongue, trunk mucosa, whole blood and serum samples by using a DNeasy blood and tissue kit (Qiagen, Crawley, UK). It was tested for EEHV1A and EEHV1B by using a published PCR method15 and for EEHV2 to EEHV6 by using previously unpublished PCR methods, with all assays targeting regions of the DNA polymerase gene (U38). Semi-nested PCRs for EEHV2/EEHV5/EEHV6 were performed by using forward primer 5′-GTT ACS RYB GTY ACS ACT AAC AC-3′ with reverse primers 5′-GCT ATM GSY ARA CAC GGR AAC AT-3′ in the first round and 5′-ACR GCR TTR CAY GTD AGT TTC AG-3′ in the second round. Semi-nested PCRs for EEHV3/EEHV4 were carried out by using forward primer 5′-CAT CCA GGC CTA CAA CCT CTG-3′ with reverse primers 5′-CAG TCG TTG AAG GTG TCG CAG-3′ and 5′-AGA CGG CGT TGC AGG TGA GCT-3′ in the first and second rounds, respectively. The predicted PCR product sizes for EEHV2/EEHV5/EEHV6 and EEHV3/EEHV4 were 204 and 281 bp, respectively. The tissues were also tested by using nested pan-EEHV and specific EEHV5 PCRs7, performing the first round PCRs only.
The PCRs were performed by using a fast cycling PCR kit (Qiagen), the reactions consisting of 10 μl 2 × fast cycling PCR master mix, 1 μl each of forward and reverse primers at 20 pmol/μl, 2 μl template DNA and water to a total volume of 20 μl. PCRs were initiated at 95°C for 5 min, followed by 35 cycles of 95°C for 15 s, 55°C for 30 s and 72°C for 1 min. The products were detected by agarose gel electrophoresis, purified by using a MinElute PCR purification kit (Qiagen) and sequenced by using Sanger technology.
DNA sequencing
An aliquot of DNA (240 ng) from heart tissue was sheared acoustically to an average size of 525 bp in a volume of 50 μl, using a Covaris S220 sonicator (Covaris Inc., Woburn, MA, USA). Fragment size was measured on an Agilent 2200 TapeStation (Agilent, Santa Clara, CA, USA). A KAPA library preparation kit (KAPA Biosystems, Wilmington, MA, USA) was used to prepare the sheared DNA fragments for Illumina sequencing, with the following modifications of the standard protocol. Sheared DNA was end-repaired directly (i.e., without isolation) and end-repaired DNA was purified by using 0.8 volumes of AMPureXP beads (Beckman Coulter, Indianapolis, IN, USA) in order to remove fragments smaller than the anticipated read length (250 bp) and eluted in 30 μl TE (10 mM Tris-HCl, pH 8.5), retaining the beads in the tube during all subsequent steps in order to minimize sample loss16. A 20:1 molar ratio of NEBnext Illumina adaptor (New England Biolabs, Ipswich, MA, USA) was ligated to the DNA fragments. In order to reduce PCR amplification bias and improve fidelity, the adaptor-ligated DNA was amplified by three cycles on an ABI 7500 real-time PCR cycler (Life Technologies, Grand Island, NY, USA) using a KAPA real-time library amplification kit.
A MiSeq DNA sequencer running v2 chemistry (Illumina, San Diego, CA, USA) was used to generate 15,054,115 paired-end, 250 nucleotide (nt) reads from the Vijay/EEHV5 library. The data were processed for adapter removal, quality-filtered, trimmed and subjected to read pair validation by using Trim Galore v. 0.2.2 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore), yielding 15,002,189 paired-end reads. In order to identify host data, BWA v. 0.6.2-r12617 was used in gapped, paired-end mode to map reads to the African elephant genome (Ensembl Loxafr3.0 v. 69.3), since the Asian elephant genome was not available. Unmapped read pairs were extracted by using Samtools v. 0.1.1818 and contigs were assembled de novo by using ABySS19. Scaffold_builder20 was used to produce a draft sequence by ordering the contigs against the EEHV1A/Raman genome sequence. Additional contigs were incorporated and gaps were closed by using Megamerger21, GapFiller v. 1–1122 and custom Perl scripts. The integrity of the resulting single, circular contig was verified by aligning against the original dataset using BWA and visualizing using Tablet v. 1.13.08.0523.
Identification of genome termini
The termini of the EEHV5 genome were identified essentially as described for EEHV1A/Raman and EEHV1B/Emelia4, using components described in the KAPA library preparation kit (KAPA Biosystems) and Marathon cDNA amplification kit (Clontech Laboratories, Palo Alto, CA, USA) according to the manufacturers' instructions. Briefly, aliquots of DNA extracted from heart tissue were either blunt-ended or left untreated. The former DNA was ligated to a partially double-stranded DNA adaptor that was blunt at the end designed to ligate to the genome termini and the latter DNA to a corresponding adaptor that contained an additional unpaired, redundant 3′-nucleotide (i.e., a mixture of G, A, T and C). First-round PCR was then carried out on the two ligation products by using an EEHV5-specific primer mapping close to the anticipated left or right genome terminus, plus an adaptor-specific primer (AP1). The locations of the EEHV5 termini were predicted from sequence similarities to those of EEHV1A. The EEHV5-specific primers were 5′-TCC GGG AGG GTA TAC GTC ACG GTG-3′ (left terminus) and 5′-CAG AGA TGA TTG ACG TCT ACA CGG-3′ (right terminus). Although PCR products were not detectable as visible bands by agarose gel electrophoresis, the regions containing DNA molecules of the anticipated sizes (approximately 200 and 180 bp, respectively) were purified. Nested PCR was carried out on each product by using EEHV5-specific primers 5′-TTA CTA TGA CGT CAT GAC CGG AAG-3′ (left terminus) or 5′-GCC CAC CCG CCA GCG GTA ATG ACT-3′ (right terminus), plus a second adaptor-specific primer (AP2). The PCR products (approximately 160 and 110 bp, respectively) were detected by agarose gel electrophoresis, purified by using a Geneclean turbo kit (MP Biomedicals, Solon, OH, USA) and cloned into plasmids by using the pGEM-T vector system (Promega Corporation, Madison, WI, USA) according to the manufacturer's instructions. The inserts in 12 plasmids for each of the four products were sequenced by using Sanger technology with universal primers and the termini were defined as being located at the positions in the circular sequence represented by the majority of clones.
Sequence comparisons
Overall percentages of sequence identity between EEHV genomes were assessed using Stretcher (http://emboss.bioinformatics.nl). Similarity among imputed amino acid sequences was investigated by using Needle (http://emboss.bioinformatics.nl). Phylogenetic trees were constructed by using Neighbor-Joining and Poisson model implemented in the MEGA v. 5.03 software24 with confidence values calculated from 2000 replicates, having obtained sequences from NCBI RefSeq (http://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=10292). Matrix sequence comparison plots were computed by using Compare (window = 20, stringency = 16) and Dotplot in the GCG package (http://www.accelrys.com).
Database accession
The genome sequence of EEHV5/Vijay was deposited in NCBI GenBank under accession number KF921519.
Results
Pathological findings
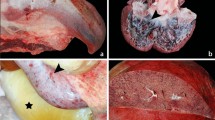
On gross post-mortem examination, Vijay presented in good body condition. No skin lesions were identified on careful examination. The tongue was enlarged and cyanotic, with multifocal (petechial to ecchymosal) mucosal and intraparenchymal hemorrhages. The head, in particular the eyelids, was swollen, with moderate to marked subcutaneous edema accompanied by few disseminated subcutaneous petechial hemorrhages. Petechial to ecchymotic hemorrhages were in addition detected within the conjunctiva, brain (including meninges), esophagus, trachea, stomach, intestine and mesentery, liver, urinary bladder and adrenal glands. Extensive atrial and ventricular hemorrhages were observed within the heart, comprising severe, confluent, irregular areas of subepicardial and subendocardial bleeding extending into the myocardium and papillary musculatures25. Severe edema and multifocal subserosal hemorrhages, typical of EEHV-induced lesions, were observed in the dorsal mediastinum (Fig. 1a). Free accumulations of approximately 500–700 ml of clear, watery fluid were present within the abdominal cavity, accompanying marked expansion of the intestinal wall, mesentery and omentum by edema. The kidneys were diffusely congested, with mottled light- to dark-red cut surfaces and retained cortico-medullary distinction. The uniformly dark-red lungs exhibited multifocal subpleural hemorrhages. Generalized diffuse reddening and mild enlargement were noted in examined visceral and subcutaneous lymph nodes.

Pathology of EEHV5 infection in Vijay.
(a) Severe edema and multifocal subserosal hemorrhages in the dorsal mediastinum. (b) Hematoxylin and eosin staining of a liver section. The hepatic architecture is disrupted by hemorrhage and edema, with multifocal fibrin deposits (arrow) and diffuse mixed cellular infiltrates. The central vessel displays denuded endothelial cells, congestion and mild granulocytostasis. Inset: Herpetic intranuclear inclusion body.
On histological examination, varying degrees of multifocal, acute hemorrhage with underlying vascular changes, including edema of the vascular wall, mild endothelial cell hypertrophy and occasional fibrinoid necrosis with thrombus formation, were present in several sections of the heart, skeletal muscle, trigeminal nerve, kidney, adrenal gland, tongue, stomach, lung, intestine, trunk, liver, oral mucosa, lymph node, pituitary, urinary bladder, cerebral medulla and cortex. Accompanying edema was most prominent within intestinal sections and lymphoid tissues. Moderate cellular infiltrates comprising neutrophils, fewer plasma cells and other lymphocytes were noted in the heart, tongue and liver, the latter of which demonstrated scattered pyknotic cells compatible with hepatocellular degeneration and necrosis. Characteristic single, variably sized, eosinophilic or amphophilic, smudged, intranuclear inclusion bodies consistent with herpesviral inclusions were detected within capillary endothelial cells of the heart, adrenal gland, lung, liver and oral mucosa (Fig. 1b). Erythrophagocytosis and mild hemosiderosis (confirmed by Perl's Prussian blue stain) were observed in lymph nodes and liver, respectively.
Preliminary molecular investigation
EEHV DNA was detected in heart, tongue, trunk mucosa, whole blood and serum in both rounds of semi-nested EEHV2/EEHV5/EEHV6 PCR (data not shown). DNA sequencing revealed that the target region was identical to that of the index EEHV5 case (EEHV5/#NAP28; GenBank accession HM060764), as were the sequences obtained using pan-EEHV and specific EEHV5 primers7. All samples proved negative for the other EEHV genotypes.
Genome structure and composition
The EEHV5/Vijay genome is 180,800 bp in size and consists of a unique sequence (U, 175,872 bp) flanked by a terminal direct repeat (TR, 2464 bp), in the arrangement TR-U-TR. The genome sizes and structures of EEHV1A/Raman and EEHV1B/Emelia are similar4. Like the genomes of EEHV1A/Raman and EEHV1B/Emelia, the EEHV5/Vijay genome contains single, unpaired C and G residues at the right and left 3′-termini, respectively. Sequence reads representing the junction between the termini amounted to approximately 10% of the mean number from the TR-U and U-TR junctions. These may have been generated from genomes that were circular, concatemeric, or contained additional copies of TR at the termini.
As calculated for TR-U (i.e. counting TR only once), 33,005 (0.11%) of the 30,004,378 processed reads for EEHV5/Vijay matched the genome sequence and coverage of the EEHV5 sequence was complete, with an average of 41 reads/nt (range 5–155). In the assembly of read data against the finished genome, there was no evidence for significant sequence variation, which indicated that Vijay had been infected by a single EEHV5 strain. The EEHV5/Vijay genotype was confirmed from comparisons with data available in GenBank for three EEHV5 strains, in each case consisting of several sequences scattered across the left half of the genome. The aggregate levels of identity for the 14,614 bp (in nine sections) from the index EEHV5 case (EEHV5/#NAP28) and the 22,143 bp (in 16 sections) from a case annotated as subgenotype EEHV5A (EEHV5A/#NAP50) are very high, at 99.6 and 99.2%, respectively. The level for the 24,190 bp (in 13 sections) from a case annotated as subgenotype EEHV5B (EEHV5B/#NAP58TW) is lower, at 94.1%. These results indicate that EEHV5/Vijay is an EEHV5A strain.
The locations of open reading frames (ORFs) in EEHV5/Vijay that encode functional proteins and fragmented ORFs that used to encode functional proteins, were predicted by using the criteria employed for EEHV1A/Raman and EEHV1B/Emelia and the names of ORFs were assigned according to the system described and justified previously for these viruses4. In this system, most EEHV ORFs that are conserved in the subfamily Betaherpesvirinae are named after their orthologs in human herpesviruses 6A and 6B26,27 and are denoted by the prefix U. ORFs that lack orthologs in other genera in the subfamily are denoted by the prefix EE. The genome of EEHV5/Vijay contains 120 ORFs (Fig. 2). The seven families of paralogous ORFs recognized in EEHV1A/Raman and EEHV1B/Emelia are represented in EEHV5/Vijay: the EE3, deoxyuridine triphosphatase-related protein (DURP), U4, seven transmembrane domain (7TM), EE20, OX-2 and EE50 families. There are five fragmented ORFs: EE44A, EE49A and EE61 by frameshifts in G:C tracts, EE62B by a frameshift in a A:T tract and EE49C by several frameshifts of other types. As indicated in Table 1, fragmented ORFs have been noted previously in EEHV1A/Raman and EEHV1B/Emelia4, and, from our analysis, are also evident in EEHV1A/Kimba. It should be noted that ORFs fragmented by frameshifts in G:C or A:T tracts are also found in members of the genus Cytomegalovirus, and, in principle, can revert to functionality via selection of length variants28. For example, in EEHV5/Vijay, the majority of reads containing the G:C tracts in EE44A and EE61 (13/17 and 11/22, respectively) contains a tract length that results in frameshifting (11 and 9 nt, respectively, as in the consensus sequence). However, a minority of reads (4/17 and 10/17, respectively) in these ORFs contains a tract length that does not result in frameshifting (10 and 10 nt, respectively), thus allowing the cognate protein to be synthesized. This analysis bears the caveat that tract length variation might result from the PCR-based processes used during sequence library preparation and not necessarily from genuine variation in the viral genome.

Map of the EEHV5/Vijay genome.
The copies of TR flanking U are shown in a thicker format. ORFs are shown by coloured arrows, with names below. Fragmented ORFs are shown in square brackets and are depicted as intact. Introns are indicated by narrow white bars. Colour shading indicates core ORFs (present in the ancestor of the subfamilies Alphaherpesvirinae, Betaherpesvirinae and Gammaherpesvirinae), betagamma ORFs (present in the subfamilies Betaherpesvirinae and Gammaherpesvirinae), beta ORFs (present only in the subfamily Betaherpesvirinae) and ORFs belonging to paralogous families. As well as being members of paralogous families, U54 and U4 are beta ORFs. A putative origin of DNA replication (ori) and a tandem reiteration (R1) are marked.
Genome comparisons
Four complete EEHV genomes have now been sequenced: those of EEHV1A/Raman, EEHV1A/Kimba, EEHV1B/Emelia and EEHV5/Vijay. They are largely colinear and have overall percentages of DNA sequence identity as follows: EEHV1A/Raman to EEHV1A/Kimba, 96.7; EEHV1A/Raman to EEHV1B/Emelia, 92.7; EEHV1A/Kimba to EEHV1B/Emelia, 92.1; EEHV1A/Raman to EEHV5/Vijay, 63.1; and EEHV1B/Emelia to EEHV5/Vijay, 63.0. Further investigation of similarities among the genomes was carried out via pairwise alignment of imputed amino acid sequences (Table 1) and via phylogenetic analysis of core genes in members of the subfamily Betaherpesvirinae (Fig. 3). As noted for EEHV1A/Raman and EEHV1B/Emelia4, divergence is particularly marked in the region near the right terminus containing the EE50 family (Fig. 4), where many EEHV5 ORFs share less than 40% identity with their EEHV1 counterparts and duplications, transpositions and losses may have occurred that make the assignment of orthologs problematic (e.g., the apparent insertion of EE49A–EE49D in EEHV5/Vijay). The EE50 family is remarkable both in its apparently rapid rate of evolution and in its inclusion of fragmented ORFs (Fig. 2 and Table 1).

Neighbour-joining phylogenetic tree of the concatenated amino acid sequences of core genes (U38, U39, U40, U41, U57, U60, U77 and U81) from members or probable members of the subfamily Betaherpesvirinae, with recognized assignments of viruses to genera indicated.
The tree is rooted at the midpoint, a position that has been shown previously to be statistically robust for this subfamily28. Confidence values are displayed as fractions. HCMV, human cytomegalovirus; CCMV, chimpanzee cytomegalovirus; GMCMV, green monkey cytomegalovirus; RhCMV, rhesus cytomegalovirus; OMCMV, owl monkey cytomegalovirus; SMCMV, squirrel monkey cytomegalovirus; GPCMV, guinea pig cytomegalovirus; MSHV, Miniopterus schreibersii herpesvirus; TuHV, tupaiid herpesvirus 1; MCMV, murine cytomegalovirus; RCMVE, rat cytomegalovirus England; RCMV, rat cytomegalovirus; HHV6A, human herpesvirus 6A; HHV6B, human herpesvirus 6B; HHV7, human herpesvirus 7; and PCMV, porcine cytomegalovirus. The scale bar shows nt differences/nt.

Matrix sequence comparison plot of the regions near the right termini of the EEHV5/Vijay and EEHV1A/Raman genomes that encompass the EE50 gene family.
The layout of ORFs in each sequence is illustrated, with shading indicating gene families provided in the key. Fragmented ORFs are shown in square brackets and are depicted as intact. The position of TR is also shown. Detectable sequence similarity is indicated by diagonal lines.
Comparisons among ORFs in the four genomes led to the following adjustments to the EEHV1A/Raman and EEHV1B/Emelia GenBank annotations. EE1A was added because it is conserved to a greater degree than the overlapping region of EE1. Indeed, it is possible either that EE1 starts downstream from EE1A, or that EE1A is located in an unidentified intron in EE1, thus eliminating the overlap of protein-coding regions. EE30A and EE32A were also added. The location of the first exon of U12 was corrected to that reported previously3 and a second exon was added to EE20 and EE32, that in the latter also having been proposed previously3. EE1, U73, U53, U52, U30, EE16 (EEHV1A/Raman only), EE19 and EE31 were truncated at their 5′-ends and EE57 was extended at its 5′-end. Several other minor improvements were also made to the ORF descriptions. It is notable that, as well as some ORFs being present only in EEHV-1 or EEHV-5 (e.g., EE3 and EE22A, respectively), some ORFs in EEHV-1B/Emilia and EEHV-5/Vijay lack counterparts in EEHV-1A (e.g., EE6), probably having been lost during evolution of the latter.
Discussion
A series of recent publications2,3,4,8,10,15,29 has demonstrated the widespread geographic distribution, prevalence and occurrence of EEHVs and enhanced our understanding of the molecular characteristics of these novel viruses via the complete genome sequencing of EEHV1A and EEHV1B. To date, EEHV5 has been associated with non-fatal clinical infection only10. However, our study demonstrates that EEHV5 must be considered with other EEHVs as a cause of fatal infection.
Vijay was reported to have suffered from ill-health of undetermined cause over a prolonged period prior to death. It is unclear whether and to what extent, this may have contributed to the development of fatal disease. Of interest in this context is the occurrence of a mucocutaneous perianal lesion. Mucosal lesions related to EEHV1 infection have been described in Asian elephants30 and a distinctive mucosal lesion in the trunk of an Asian elephant in the same group was attributed to elephant gammaherpesvirus 531. Unfortunately, further analysis of Vijay's lesion was not carried out at the time. As a result, the underlying cause remains speculative, co-infection with multiple EEHVs and elephant gammaherpesviruses being a possibility (7) and young age is probably a factor. However, PCR testing of a range of other tissues from Vijay failed to reveal the presence of genotypes of EEHV other than EEHV5 and searches of the read data obtained from heart tissue using a selection of the most extensive sequences available in GenBank for genotypes of EEHV other than EEHV5 and also for elephant gammaherpesvirus genotypes, were uniformly negative. Most elephants with fatal EEHV-related disease are juveniles and die within 24 hours of the onset of clinical signs5,9. Novel treatment approaches, such as the drainage of pericardial fluid, may have prolonged survival in this case. It is reasonable to assume that the detection of increased inflammatory infiltrates, in particular in the heart, liver and tongue, which were not a recorded feature in previous fatality cases, are a reflection of prolonged survival.
Pathological findings in fatal cases associated with recognized EEHV genotypes are typically similar and comprise extensive edema, effusions and hemorrhages. However, while the cardiovascular system appears the primary site in EEHV1 cases, fatal infection with EEHV3 has been related to renal medullary hemorrhage, retinal damage and tropism for larger veins6. Recent cases of EEHV4-associated fatality in Asia have demonstrated hemorrhages in most organs, including the gastrointestinal, respiratory and cardiovascular systems8. Widespread hemorrhages and edema were also evident in Vijay. In addition, this is the first reported detection of viral inclusion bodies in the adrenal gland and oral mucosa, with additional inclusions in the heart, lung and liver. Taken together, these findings suggest a less selective organ tropism for EEHV3, EEHV4 and EEHV5, which might reflect higher virulence of the strains involved6,8.
There is ample evidence that EEHVs are longstanding viruses of Asian elephants2,10,15,29,30 and surveys into the shedding of EEHVs in captive collections indicate that almost all animals within herds may carry and intermittently shed these viruses in the absence of overt signs10. These observations, together with the EEHV5-induced fatality reported here, have implications for herd management, where monitoring of viral shedding via PCR has become an option through the availability of sequence data and highly sensitive qPCR tests32. Thus far, however, routine investigations, in particular those into captive populations, remain focused on the detection of EEHV1 and tend to neglect other genotypes. Consequently, it is uncertain whether the sparse reporting of fatalities associated with other genotypes is a true reflection of an overall lower prevalence of these viruses, or whether it is due to the appearance of virulent variants.
The enhanced sequence comparisons possible as a result of the current study confirm that, among EEHVs as a whole, the EEHV1A strains are the most closely related to each other, with EEHV1B/Emelia somewhat more distant and EEHV5/Vijay much more distant. There is no indication that recombination has occurred between EEHV5 and EEHV1. In contrast, there are regions of the genome in which EEHV1A/Raman and EEHV1B/Emelia are more closely related to each other than is either to EEHV1A/Kimba (for example, in the amino acid sequences of EE23, EE24, EE47 and EE50; Table 1). This evidence for recombination between the EEHV1 subgenotypes is in accord with their description as being partially chimeric3. The apparent occurrence of recombination would render divisions of EEHV genotypes into subgenotypes (e.g., EEHV1A and EEHV1B and EEHV5A and EEHV5B) a potentially problematic exercise, particularly if PCR assays based on different genes form the basis of these distinctions.
Among the subfamilies of the family Herpesviridae, the genus Proboscivirus clearly has its closest genetic affinities with the subfamily Betaherpesvirinae. Since this genus forms the earliest evolutionary branch in this subfamily, it is rather divergent from the other genera. Thus, eight ORFs (U79, U54, U51, U4, U11, U12, UL34 and U14) have apparent orthologs only in the subfamily Betaherpesvirinae and two ORFs (EE7 and EE9) have orthologs only in the subfamilies Alphaherpesvirinae and Gammaherpesvirinae (Fig. 1). The phylogenetic data (Fig. 3) support the classification of EEHV5 as the founding member of a new species (Elephantid herpesvirus 5) in the genus Proboscivirus, as has been mooted on the basis of short sequences7. In addition, the current subfamily assignment of the genus Proboscivirus seems to be satisfactory and we do not think that there is a strong case for removing it from its current taxonomical position and assigning it to a new subfamily2.
The EEHVs remain an important threat to a charismatic and endangered species of major prominence, particularly juvenile animals. However, the conditions under which EEHVs cause fatal hemorrhagic disease remain unclear. Recent reports demonstrating a much higher than expected prevalence of EEHVs, including EEHV1, in healthy wild and captive Asian elephants and evidence showing that wild African elephants carry EEHV2, EEHV3, EEHV6 and EEHV7 in skin and lung nodules, make it likely that EEHVs are natural viruses of Asian and African elephants2,10,15,29,30. This case report, including determination of the viral genome sequence directly from the animal concerned, adds significantly to our knowledge of this fascinating group of viruses and will facilitate further research into the role of EEHV5 in fatal infections and the development of tests for and vaccines against, EEHVs.
References
Ossent, P. et al. Acute and fatal herpesvirus infection in a young Asian elephant (Elephas maximus). Vet. Pathol. 27, 131–133 (1990).
Hayward, G. S. Conservation: clarifying the risk from herpesvirus to captive Asian elephants. Vet. Rec. 170, 202–203 (2012).
Ling, P. D. et al. Complete genome sequence of elephant endotheliotropic herpesvirus 1A. Genome Announc. 1, e0010613. 10.1128/genomeA.00106-13 (2013).
Wilkie, G. S. et al. Complete genome sequences of elephant endotheliotropic herpesviruses 1A and 1B determined directly from fatal cases. J. Virol. 87, 6700–6712 (2013).
Richman, L. K. et al. 1999. Novel endotheliotropic herpesviruses fatal for Asian and African elephants. Science 283, 1171–1176 (2013).
Garner, M. M. et al. Clinico-pathologic features of fatal disease attributed to new variants of endotheliotropic herpesviruses in two Asian elephants (Elephas maximus). Vet. Pathol. 46, 97–104 (2009).
Latimer, E., Zong, J.-C., Heaggans, S. Y., Richman, L. K. & Hayward, G. S. Detection and evaluation of novel herpesviruses in routine and pathological samples from Asian and African elephants: identification of two new probosciviruses (EEHV5 and EEHV6) and two new gammaherpesviruses (EGHV3B and EGHV5). Vet. Microbiol. 147, 28–41 (2011).
Sripiboon, S., Tankaew, P., Lungka, G. & Thitaram, C. The occurrence of elephant endotheliotropic herpesvirus in captive Asian elephants (Elephas maximus): first case of EEHV4 in Asia. J. Zoo. Wildl. Med. 44, 100–104 (2013).
Richman, L. K. et al. Clinical and pathological findings of a newly recognized disease of elephants caused by endotheliotropic herpesviruses. J. Wildl. Dis. 36, 1–12 (2000).
Atkins, L. et al. Elephant endotheliotropic herpesvirus 5, a newly recognized elephant herpesvirus associated with clinical and subclinical infections in captive Asian elephants (Elephas maximus). J. Zoo. Wildl. Med. 44, 136–143 (2013).
Fickel, J. et al. A variant of the endotheliotropic herpesvirus in Asian elephants (Elephas maximus) in European zoos. Vet. Microbiol. 82, 103–109 (2001).
Ehlers, B. et al. Endotheliotropic elephant herpesvirus, the first betaherpesvirus with a thymidine kinase gene. J. Gen. Virol. 87, 2781–2789 (2006).
Pellett, P. E. et al. [Herpesviridae]. Virus taxonomy, ninth report of the International Committee on Taxonomy of Viruses [King, A. M. Q., Adams, M. J., Carstens, E. B. & Lefkowitz, E. J. (ed)] [111–122] (Elsevier Academic Press, London, UK 2011).
Keet, D. F. & Bengis, R. G. [A guide to post-mortem procedure and a review of pathological processes identified in the elephant]. Post-mortem Procedures For Wildlife Veterinarians And Field Biologists [Woodford, M. H. (ed)] [36–47] (Office International des Epizooties, Care for the Wild and the Veterinary Specialist Group/Species Survival Commission of the World Conservation Union (IUCN), Paris, France 2000).
Hardman, K. et al. Detection of elephant endotheliotropic herpesvirus type 1 in asymptomatic elephants using TaqMan real-time PCR. Vet. Rec. 170, 205 (2012).
Fisher, S. et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 12, R1. 10.1186/gb-2011-12-1-r1 (2011).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009).
Silva, G. G. Z. et al. Combining de novo and reference-guided assembly with scaffold_builder. Source Code Biol. Med. 8, 23. 10.1186/1751-0473-8-23 (2013).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000).
Boetzer, M. & Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 13, R56. 10.1186/gb-2012-13-6-r56 (2012)
Milne, I. et al. Tablet –next generation sequence assembly visualization. Bioinformatics 26, 401–402 (2010)
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Denk, D. et al. Fatal elephant endotheliotropic herpesvirus type 5 infection in a captive Asian elephant. Vet. Rec. 171, 380–381 (2012).
Gompels, U. A. et al. The DNA sequence of human herpesvirus-6: structure, coding content and genome evolution. Virology 209, 29–51 (1995).
Dominguez, G. et al. Human herpesvirus 6B genome sequence: coding content and comparison with human herpesvirus 6A. J. Virol. 73, 8040–8052 (1999).
Davison, A. J. et al. [Comparative genomics of primate cytomegaloviruses]. Cytomegaloviruses: From Molecular Pathogenesis To Intervention, vol.1. [Reddehase, M. J. (ed)] [1–22] (Caister Academic Press, Norwich, UK 2013).
Zachariah, A. et al. Fatal herpesvirus hemorrhagic disease in wild and orphan asian elephants in southern India. J. Wildl. Dis. 49, 381–393 (2012).
Schaftenaar, W., Reid, C., Martina, B., Fickel, J. & Osterhaus, A. D. Nonfatal clinical presentation of elephant endotheliotropic herpes virus discovered in a group of captive Asian elephants (Elephas maximus). J Zoo. Wildl. Med. 41, 626–632 (2010).
Masters, N. J., Stidworthy, M. F., Everest, D. J., Dastjerdi, A. & Bäulmer, S. Detection of EGHV-5 in a self-limiting papilloma-like lesion in the trunk of an Asian elephant (Elephas maximus). Vet. Rec. 169, 209 (2011).
Stanton, J. J., Nofs, S. A., Peng, R., Hayward, G. S. & Ling, P. D. Development and validation of quantitative real-time polymerase chain reaction assays to detect elephant endotheliotropic herpesviruses-2, 3, 4, 5 and 6. J. Virol. Methods 186, 73–77 (2012).
Acknowledgements
This work was supported by Twycross Zoo – East Midland Zoological Society, the UK Biotechnology and Biological Sciences Research Council (BB/J004243/1, BB/J004235/1 and BB/J004324/1), the UK Medical Research Council and the Zoological Society of London (ZSL). We are grateful to Erin Latimer (National Elephant Herpesvirus Laboratory, Smithsonian Conservation Biology Institute, Washington DC, USA) and Gary Hayward (Johns Hopkins School of Medicine, Baltimore, MD, USA) for their assistance during the investigation of this case. We thank Katharina Seilern-Moy for preparing DNA from Vijay's post-mortem tissues and Wai Kwong Lee and Andrew Carswell (BHF Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK) for providing Sanger DNA sequencing services used in identifying the EEHV5/Vijay genome termini.
Author information
Authors and Affiliations
Contributions
S.R. coordinated the clinical care and provided the clinical data of the case. D.D., M.F.S. and A.D. carried out the post-mortem and initial virus characterization. G.S.W., K.K., A.D. and A.J.D. carried out the full genome analysis. A.J.D. and D.D. drafted the manuscript. F.S. coordinated the project and manuscript compilation. All authors reviewed the manuscript and provided specific input.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Wilkie, G., Davison, A., Kerr, K. et al. First Fatality Associated with Elephant Endotheliotropic Herpesvirus 5 in an Asian Elephant: Pathological Findings and Complete Viral Genome Sequence. Sci Rep 4, 6299 (2014). https://doi.org/10.1038/srep06299
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06299
This article is cited by
-
Pathogenesis of hemorrhagic disease caused by elephant endotheliotropic herpesvirus (EEHV) in Asian elephants (Elephas maximus)
Scientific Reports (2021)
-
Retrospective review of 27 European cases of fatal elephant endotheliotropic herpesvirus-haemorrhagic disease reveals evidence of disseminated intravascular coagulation
Scientific Reports (2021)
-
Human cytomegalovirus multiple-strain infections and viral population diversity in haematopoietic stem cell transplant recipients analysed by high-throughput sequencing
Medical Microbiology and Immunology (2021)
-
In vivo characterization of target cells for acute elephant endotheliotropic herpesvirus (EEHV) infection in Asian elephants (Elephas maximus)
Scientific Reports (2020)
-
Molecular Evolution of Human Adenovirus (HAdV) Species C
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
