
Abstract
Porcine epidemic diarrhea virus (PEDV) is a coronavirus that infects pigs and can have mortality rates approaching 100% in piglets, causing serious economic impact. The 3C-like protease (3CLpro) is essential for the coronaviral life cycle and is an appealing target for the development of therapeutics. We report the expression, purification, crystallization and 2.10 Å X-ray structure of 3CLpro from PEDV. Analysis of the PEDV 3CLpro structure and comparison to other coronaviral 3CLpro’s from the same alpha-coronavirus phylogeny shows that the overall structures and active site architectures across 3CLpro’s are conserved, with the exception of a loop that comprises the protease S2 pocket. We found a known inhibitor of severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro, (R)-16, to have inhibitor activity against PEDV 3CLpro, despite that SARS-3CLpro and PEDV 3CLpro share only 45.4% sequence identity. Structural comparison reveals that the majority of residues involved in (R)-16 binding to SARS-3CLpro are conserved in PEDV-3CLpro; however, the sequence variation and positional difference in the loop forming the S2 pocket may account for large observed difference in IC50 values. This work advances our understanding of the subtle, but important, differences in coronaviral 3CLpro architecture and contributes to the broader structural knowledge of coronaviral 3CLpro’s.
Similar content being viewed by others

Introduction
The causative virus of porcine epidemic diarrhea in pigs was first described in England in 19781, though the disease was first observed in 1971 among English feed pigs2. Subsequently termed porcine epidemic diarrhea virus (PEDV), it was classified as a coronavirus as a result of its appearance in electron microscopes and its polypeptide structure3,4,5. Before its occurrence in the United States, there were numerous reports of PEDV in various European countries, as well as in Korea, China, Japan, the Philippines and Thailand6. The first four reports of PEDV in the United States occurred within 10 days of each other on four farms in Iowa in late April of 20137. The disease is characterized by porcine epidemic diarrhea with concomitant vomiting and dehydration, resulting in high fatality rates especially in piglets where mortality rates approach 100%8. The origin of transmission of PEDV to the United States is still unknown; however, in the past two years, it has resulted in a sizeable and negative impact on the US economy. The estimated net annual loss for the US economy as a result of PEDV, summed across all effects, is reported to range from $900 million to $1.8 billon as of June, 20149.
Coronaviruses are enveloped, single-stranded, positive-sense zoonotic RNA viruses with genomes ranging from 27 to 32 kilobases in length, where the genes encoding for the structural proteins are located at the 3′ end and the replicase genes are located at the 5′ end of the genome. Two long ORFs encode for the replicase proteins, ORF1a and ORF1b, which are connected by a ribosomal frame-shift and produce the coronaviral polyproteins pp1a and pp1ab upon translation10. These viral polyproteins are processed at 14 cleavage sites by two or three cysteine proteases producing 16 non-structural proteins (nsps). The papain-like protease (PLP1 and PLP2 or PLpro) cleaves the polyprotein at 3 sites, whereas the 3C-like protease (3CLpro), also known as the main-protease (Mpro) or nsp5, cleaves the polyprotein at 11 sites. The function of both proteases is essential for viral replication and therefore they are attractive targets for the development of anti-coronaviral therapeutics. In this study, we report the expression, purification, crystallization and X-ray structure of PEDV-3CLpro in its unbound state to 2.10 Å. We tested PEDV-3CLpro for inhibition by (R)-16, a compound known to inhibit severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro and found (R)-16 to inhibit PEDV-3CLpro with an IC50 of 25.4 ± 1.4 μM. The structural information provided by the PEDV-3CLpro X-ray crystal structure adds new details to our general understanding of the subtle differences between phylogenetically related 3CLpro’s and provides key insights that can be used to inform and guide the design of inhibitors against PEDV-3CLpro.
Results and Discussion
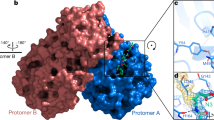
PEDV-3CLpro crystallized in space group P212121 as a dimer in the asymmetric unit (Table 1). As reported for both human coronavirus 229E 3CLpro and SARS-3CLpro, PEDV-3CLpro is a homodimer containing three domains in each monomer (Fig. 1)11,12. The active site of PEDV-3CLpro, which contains a catalytic dyad formed from residues Cys144 and His41, is located in a cleft between domains I and II (Fig. 1a). Domain III is involved in monomer dimerization, which is ultimately responsible for forming the active protease (Fig. 1a)12,13. In the absence of substrate, water and solvent molecules (MPD, DMSO and IPA) reside in the active site of PEDV-3CLpro, which is solvent exposed on one side (Fig. 1a,b). Interestingly, different solvent molecules are found in the respective active sites of the PEDV-3CLpro monomers composing the PEDV-3CLpro dimer, where the a single DMSO molecule resides in the Chain A active site and the Chain B active site houses both isopropanol (IPA) and 2-methyl-2,4-pentanediol (MPD) (Fig. 1b). The presence of different solvent molecules in each of the active sites of the dimer supports the assignment of one biological dimer in the asymmetric unit.

The X-ray crystal structure of PEDV-3CLpro determined to 2.10 Å in its unbound state (PDB entry 5HYO).
(a) The crystallographic dimer of PEDV-3CLpro is shown with the backbone represented as a ribbon overlaid on a surface representation, where Chain A is colored in orange and Chain B is colored in deep slate. The active site catalytic dyad residues, His41 and Cys144, are represented as sticks in each monomer. The catalytic dyad is explicitly labeled in Chain B and domains I-III are labeled in Chain A. (b) Zoom-in of both Chain A and B active sites. The residues of the catalytic dyad are represented as sticks, colored by atom and labeled accordingly. Waters are shown as non-bonded spheres and are colored red. Solvent molecules are shown as blue sticks, colored by atom, with surrounding electron density omit map (Fo − Fc) shown in grey mesh and contoured to +3.0σ (DMSO = dimethyl sulfoxide, MPD = 2-methyl-2,4-pentanediol, IPA = isopropanol).
Least-squares (LSQ) superposition of PEDV-3CLpro and the unbound form of human coronavirus 229E 3CLpro (PDB entry 1P9S)12, which are both from the same alpha-coronavirus phylogenetic group and share 69.3% sequence identity, resulted in an all-atom root-mean-square deviation (RMSD) value of 1.69 Å and a C-alpha RMSD value of 1.28 Å (Fig. 2a). The LSQ superposition of 229E-3CLpro and PEDV-3CLpro shows that the overall architectures of both the 229E-3CLpro and PEDV-3CLpro active sites in their unbound states are structurally very similar and the active site residues of the catalytic dyad, residues Cys144 and His41 in both PEDV-3CLpro and 229E-3CLpro, are located in almost identical structural space within the active site cavity, which is solvent exposed on one side (Fig. 2b). In the absence of substrate, both water and non-water solvent molecules (dioxane in 229E-3CLpro and MPD, DMSO and IPA in PEDV-3CLpro) are found in the active site (Fig. 2b).

Superimpositions of PEDV-3CLpro, 229E-3CLpro (PDB entry 1p9s) and FIPV-3CLpro (PDB entry 4zro).
(a) Superimposition of PEDV-3CLpro and 229E-3CLpro, where each enzyme is represented as a ribbon. PEDV-3CLpro is colored in orange and 229E-3CLpro is colored light pink. (b) Zoom-in of PEDV-3CLpro:229E-3CLpro superimposition Chain A active site, where catalytic dyad residues and solvent are shown in stick, colored according to atom and to which protein they belong. (c) Superimposition of PEDV-3CLpro and FIPV-3CLpro, where each enzyme is represented as a ribbon. PEDV-3CLpro is colored in orange and 229E-3CLpro is colored magenta. (d) Zoom-in of PEDV-3CLpro:FIPV-3CLpro superimposition Chain A active site, where catalytic dyad residues and solvent are shown in stick, colored according to atom and to which protein they belong. The covalent FIPV-3CLpro inhibitor, 6, is represented as ball-and-stick and colored according to atom.
In order to better understand the features of the PEDV-3CLpro active site that are important in inhibitor and substrate binding, we generated an LSQ superposition of PEDV-3CLpro and an inhibitor-bound form of feline infectious peritonitis virus 3CLpro (FIPV-3CLpro, PDB entry 4ZRO), which belongs to the same alpha-coronavirus lineage as PEDV-3CLpro and has 61.9% sequence identity (Fig. 2c)14. LSQ superposition resulted in an all-atom RMSD value of 2.11 Å and a C-alpha RMSD value of 1.69 Å. We found the overall active site architectures of the unbound PEDV-3CLpro and the inhibitor-bound form of FIPV-3CLpro to be remarkably similar with the catalytic dyad residues (Cys144 and His41 in both FIPV- and PEDV-3CLpro) in nearly identical orientations despite Cys144 of FIPV-3CLpro being covalently modified by the inhibitor, compound 6 (Fig. 2d).
Interestingly, in both the superimpositions of PEDV-3CLpro with 229E-3CLpro and FIPV-3CLpro, the loops comprising the protease subsites of the 3CLpro active site are in nearly identical structural locations, with the exception of the loop that comprises the S2 subsite, the S2 loop. The S2 loop forms the outer boundary of the S2 binding pocket and shows positional variability across the X-ray structures of 229E-, FIPV- and PEDV-3CLpro, which may lead to differences in the size of the S2 subsites across 3CLpro’s (Fig. 2b,d).
Our observations of the overall conserved structural features surrounding the PEDV-3CLpro catalytic dyad, but subtle differences in the overall active site architecture, made us curious as to whether one of the inhibitors we developed for SARS-3CLpro would also inhibit PEDV-3CLpro15. SARS-3CLpro belongs to a different phylogenetic lineage than PEDV-3CLpro and shares lower sequence identity (45.4%) with PEDV-3CLpro; however, we reasoned that the similar tertiary structure and conserved active site architecture of 3CLpro’s would allow for inhibition by the same molecule. We therefore tested the inhibition of PEDV-3CLpro by (R)-16, which was developed as a non-covalent inhibitor against SARS-3CLpro with potential broad-spectrum activity (Fig. 3a)15. We found (R)-16 to inhibit PEDV-3CLpro with an IC50 value of 25.4 ± 1.4 μM, where the representative curve for (R)-16 inhibition of PEDV-3CLpro is shown in Fig. 3a and the data for (R)-16 inhibition of SARS-3CLpro has been previously published15. The previously reported IC50 of (R)-16 against SARS-3CLpro is 1.5 ± 0.3 μM15, which indicates a ~17-fold weaker interaction of (R)-16 with PEDV-3CLpro. The inhibition of PEDV-3CLpro by (R)-16, though weak, is significant because it indicates that the development of non-covalent broad-spectrum inhibitors of 3CLpro’s may be possible.

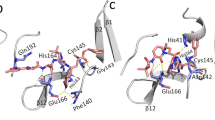
Superimposition of the X-ray structures of unbound PEDV-3CLpro (colored in orange) and (R)-16 bound SARS-3CLpro (colored in mauve, PDB entry 3v3m).
(R)-16 is shown as stick representation in deep violet and colored according to atom type. (a) Chemical structure of (R)-16 with representative IC50 data for (R)-16 against PEDV-3CLpro displayed. (b) Superimposition of PEDV-3CLpro and SARS-3CLpro, where both are displayed in ribbon with the residues of the catalytic dyad shown as sticks. (c) (R)-16 interactions with the SARS-3CLpro residues where hydrogen-bonding interactions are represented as sticks and the distances between heteroatoms are labeled in angstroms (Å). (d) Overall active site architecture of PEDV-3CLpro and SARS-3CLpro, where the catalytic dyad and residues involved in (R)-16 binding are shown in sticks.
To gain structural insights into how (R)-16 may bind to PEDV-3CLpro, a structural alignment of the X-ray structure of SARS-3CLpro:(R)-16 complex with PEDV-3CLpro was generated (Fig. 3b). LSQ superposition of unbound PEDV-3CLpro and inhibitor-bound SARS-3CLpro (PDB entry 3V3M)15, where PEDV-3CLpro is from the alpha-coronavirus phylogenetic group and SARS-3CLpro is from the beta-coronavirus phylogenetic subgroup 2b, resulted in an all-atom RMSD value of 5.18 Å and a C-alpha RMSD value of 4.98 Å. The structural alignment shows that, similarly to 229E-3CLpro and FIPV-3CLpro, the overall active site architectures of SARS-3CLpro and PEDV-3CLpro are largely similar despite their lower sequence identity (Fig. 3b–d) and the residues directly involved in binding to (R)-16 via hydrogen-bonding interactions are all conserved (Gly142 and His162 in PEDV-3CLpro and Gly143 and His163 in SARS-3CLpro, Fig. 3c,d).
The active sites of both SARS-3CLpro and PEDV-3CLpro are solvent exposed on one side with the residues of their catalytic dyads, His41 and Cys144 (for PEDV-3CLpro) or Cys145 (for SARS-3CLpro), located in almost identical structural space (Fig. 3c,d). The superposition shows that (R)-16 similarly occupies the S2-S1′ subpockets in each of the 3CLpro active sites, where the tert-butyl amide resides in the channel leading to the active site, the tert-butylanilido group (P2) sits in the S2 pocket, the 3-pyridyl group (P1) resides in the S1 region and the tetrahydrofuran (P1′) occupies the S1′ subsite (Fig. 3c,d). As shown in the SARS-3CLpro 3V3M structure, the 3-pyridyl nitrogen of (R)-16 acts as a hydrogen-bond acceptor for SARS-3CLpro His163, with a distance of 2.8 Å between heteroatoms (Fig. 3d). This interaction is likely conserved in the inhibition of PEDV-3CLpro by (R)-16 as His162 of PEDV-3CLpro is in an almost identical structural location as that of SARS-3CLpro His163. Additionally, the bifurcated interaction between the furan ring oxygen and the amide carbonyl oxygen of (R)-16 and the backbone amide NH of SARS-3CLpro Gly143 is mimicked by PEDV-3CLpro Gly142 (Fig. 3d).
Though (R)-16 apparently binds in the same orientation in the PEDV-3CLpro active site as it does in the SARS-3CLpro active site and likely utilizes the same hydrogen-bonding interactions, the IC50 of (R)-16 against PEDV-3CLpro is 17-fold higher than against SARS-3CLpro. We were therefore curious if the difference in position of the S2 loop between PEDV- and SARS-3CLpro may be important for (R)-16 binding. A structural alignment of the ligand-bound structures of FIPV- and SARS-3CLpro (4ZRO and 3V3M, respectively) shows that compounds 6 and (R)-16 bind to the respective 3CLpro’s by positioning their sterically bulky, hydrophobic groups in the S2 subsite (leucine and t-butylanilido, respectively; Fig. 4a). This suggests that hydrophobic interactions between the inhibitor or substrate and residues of the S2 loop are important for their binding.

Analysis of S2 loops in coronaviral 3CLpro’s.
Where SARS-3CLpro is colored in mauve and FIPV-3CLpro in magenta and displayed in ribbon representation (PDB entries 3v3m and 4zro, respectively). Inhibitors 6 and (R)-16 are shown in sticks and colored according to atom type. (a) Superposition of inhibitor bound forms of FIPV- and SARS-3CLpro, where 6 is shown in magenta and (R)-16 in deep violet. The regions of the S2 loop that are comprised by residues 47 and 48 in each FIPV- and SARS-3CLpro are indicated by a cyan dashed line. (b) Analysis of S2 loops from various coronaviral 3CLpro’s.
We then analyzed the reported X-ray structures and sequences for other alpha-coronavirus 3CLpro’s in the PDB databank including the enzymes from the following coronaviruses: FIPV, transmissible gastroenteritis virus (TGEV) and human coronaviruses NL63 and 229E12,13,14,15,16,17,18,19,20. A sequence comparison of the S2 loops of FIPV-, TGEV-, NL63-, 229E-, PEDV- and SARS-3CLpro shows that the SARS-3CLpro S2 loop is one residue longer than those from the alpha-coronaviruses and shares no sequence identity with FIPV-, TGEV-, NL63-, 229E- and PEDV-3CLpro (Fig. 4b). Additionally, the S2 loop of SARS-3CLpro contains anionic amino acids, Glu and Asp, at positions 47 and 48, respectively. These variations in the S2 loop between the alpha-coronavirus 3CLpro’s and SARS-3CLpro may explain the large observed difference in the IC50 value of (R)-16 against PEDV-3CLpro as compared to SARS-3CLpro (25.4 ± 1.4 μM vs. 1.5 ± 0.3 μM, respectively). Furthermore, the combination of the increase in length and charge properties of the S2 loop of SARS-3CLpro relative to that of the alpha-coronavirus 3CLpro’s likely changes the size and shape of the S2 binding pocket and therefore may allow for increased variability at the P2 site of inhibitor and substrate molecules.
To further investigate the variation in the S2-subsite and relationship to inhibitor and substrate binding, we examined the recognition cleavage sequences in polyprotein 1ab of both PEDV- and SARS-3CLpro (Fig. 5)21,22. The S2-subsite residue preference is identical for PEDV- and SARS-3CLpro in most all cleavage sites, with the exception of nsp5/6 and nsp12/13. SARS-3CLpro can accommodate a larger P2-phenylalanine, e.g. at the nsp5/6 cleavage site, while PEDV-3CLpro recognizes residues only as large as a P2-leucine at this position in the polyprotein. At the nsp12/13 cleavage site, PEDV polyprotein 1ab has a serine at the P2 site, while SARS polyprotein 1ab has a P2-leucine. A larger size of the S2 pocket in SARS-3CLpro is consistent with the SARS polyprotein 1ab having a phenylalanine residue at the P2 position of the nsp5/6 cleavage site. This larger S2-pocket in SARS-3CLpro may allow for better binding of (R)-16, allowing the bulky t-butylanilido group to take full advantage of hydrophobic interactions within the S2 subsite and position (R)-16 to optimize hydrogen-bonding with SARS-3CLpro Gly143 and His163.

PEDV and SARS polyprotein 1ab recognition cleavage sites.
(a,b) Sequence logos showing amino acid conservation for the eleven polyprotein 1ab cleavage sites of PEDV- and SARS-3CLpro, respectively, generated using the WebLogo server. (c) The full eleven polyprotein 1ab recognition sequences for PEDV- and SARS-3CLpro.
Conclusions
In this study, we have determined the X-ray crystal structure of an unbound form of PEDV-3CLpro to 2.10 Å resolution. We found the structure of PEDV-3CLpro to be similar to the X-ray structure of the unbound human coronavirus 229E 3CLpro, which belongs to the same coronaviral phylogenetic subgroup as PEDV-3CLpro. To investigate the role of inhibitor binding on the overall architecture of the catalytic site, we generated a superimposition of PEDV-3CLpro with FIPV-3CLpro (PDB entry 4zro), which belongs to the same alpha-coronavirus phylogenetic group as PEDV-3CLpro and shares 61.9% sequence identity, in complex with the covalent inhibitor 6. We observed little difference between the active site architectures of unbound PEDV-3CLpro and the inhibitor-bound form of FIPV-3CLpro, except for differences in the position and composition of a loop comprising the S2 subsite, which was also observed in comparison to the 229E-3CLpro structure.
We tested PEDV-3CLpro for inhibition by a known SARS-3CLpro inhibitor, compound (R)-16 and found (R)-16 to be capable of inhibiting PEDV-3CLpro although the IC50 value was roughly 17-fold higher than the reported IC50 for (R)-16 against SARS-3CLpro. Structural comparison of SARS-3CLpro bound with (R)-16 and PEDV-3CLpro in its unbound form revealed that the residues that directly interact with (R)-16 in SARS-3CLpro are conserved in PEDV-3CLpro. Additional analysis of the S2 loop across alpha-coronavirus 3CLpro’s and SARS-3CLpro proved that the sequence identity of the S2 loop is not conserved across alpha- and beta-coronaviral 3CLpro’s and the SARS-3CLpro S2 loop is one residue longer than that of the alpha-coronaviral 3CLpro’s, which likely increases the effective volume of the SARS-3CLpro S2 protease subsite relative to the alpha-coronaviral 3CLpro S2 subsites. These findings provide a potential explanation for the roughly 17-fold weaker inhibition of PEDV-3CLpro by (R)-16 compared to SARS-3CLpro.
This work advances our understanding of the subtle, but important, structural differences between 3C-like proteases from different coronaviral phylogenetic groups and contributes to the broader structural knowledge of coronaviral 3CLpro’s. Small structural changes have been shown to be essential in enzymatic catalysis and an understanding of these such differences is also very pertinent for the design of both broad-spectrum and selective coronaviral 3CLpro inhibitors for the treatment of coronaviral infection23.
Methods
Protein expression and purification
The gene encoding the 3CLpro of PEDV (residues 2998–3299 in the Porcine epidemic diarrhea virus polyprotein AHA38151.1)24 was codon optimized for expression E. coli and cloned into pET-11a expression vector with an N-terminal (His)6-tag followed by nsp4-/5 auto-cleavage site by BioBasic Inc. This pET-11a PEDV-3CLpro construct was used because it results in the expression of PEDV-3CLpro without an N-terminal or C-terminal extension.
E. coli BL21(DE3) cells were transformed with the pET-11a PEDV-3CLpro plasmid and then grown at 25 °C for 24 hours in 500 mL Super LB media (3 g potassium phosphate monobasic, 6 g sodium phosphate dibasic, 20 g tryptone, 5 g yeast extracts, 5 g sodium in 1 L water, pH 7.2 adjusted with 1 M NaOH) that was also supplemented with 1 mL 100 mg mL−1 carbenicillin, 25 mL 8% lactose, 10 mL 60% glycerol and 5 mL of 10% glucose per 1 L of expression culture. The cells were harvested by centrifugation (8,400 g for 20 min) to yield 14.5 g L−1 of cells. The cell pellet was then re-suspended in Buffer A (50 mM Tris pH 7.5, 0.2 M ammonium sulfate, 0.05 mM EDTA, 5 mM BME) containing 1 mg mL−1 lysozyme, where 5 mL of Buffer A was used per 1 g cell pellet. After the cells were homogenized, they were lysed via sonication for a total of 10 minutes with 10 s pulses and 20 s delays at 50% amplitude using a Branson Digital Sonifier.
The cell lysate was clarified by pelleting the cell debris via centrifugation (28,960 g, 4 °C, 20 minutes).
The resultant supernatant was loaded onto a 60 mL Phenyl Sepharose 6 Fast Flow HiSub (GE Healthcare) column equilibrated with Buffer A. Protein was eluted with a linear gradient to 100% Buffer B (20 mM Tris pH 7.5, 0.05 mM EDTA, 5 mM BME) over five column volumes (300 mL) collecting 5 mL fractions. Fractions containing PEDV-3CLpro enzymatic activity were pooled and loaded onto a 60 mL DEAE Sepharose Fast Flow (GE Healthcare) column equilibrated with Buffer B. Protein was eluted with a linear gradient to 50% Buffer C (50 mM Tris pH 7.5, 1 M sodium chloride, 0.05 mM EDTA, 5 mM BME, 10% glycerol) over five column volumes (300 mL) collecting 5 mL fractions. Fractions containing pure PEDV-3CLpro, as judged by SDS-PAGE and specific activity, were pooled, dialyzed into storage buffer (25 mM HEPES, pH 7.5, 2.5 mM DTT, 10% glycerol) and concentrated using a spin concentrator (Millipore) with a 10 kDa molecular weight cutoff membrane. The protein was flash-frozen in liquid nitrogen and then stored at −80 °C in a freezer until further use (Fig. 1a). Prior to crystallization, PEDV-3CLpro was concentrated to 4.0 mg mL−1 using a spin concentrator (10 kDa cutoff membrane, Millipore) and loaded onto a 24 mL Superdex 75 (GE Healthcare) size exclusion chromatography column equilibrated with 25 mM HEPES pH 7.5 and 2.5 mM DTT. Peak fractions containing PEDV-3CLpro, as determined via visualization of SDS-PAGE and confirmed by protein activity assay, were pooled and concentrated to protein concentrations of 1.5, 2.8 and 4.0 mg mL−1 for crystallization.
Crystallization
The PEGS II Suite (Qiagen) sparse-matrix screen was used to screen for initial PEDV-3CLpro crystallization conditions. Crystallization trials (150 nL protein and 150 nL of crystallization solution) were set up in a 96-well sitting drop tray (Intelli-Plate 96) using a TTP LabTech Mosquito® liquid robotics system at PEDV-3CLpro concentrations of 1.5, 2.8 and 4.0 mg mL−1 in buffer containing 25 mM HEPES pH 7.5 and 2.5 mM DTT. An initial hit of clusters of needle-like crystals was obtained at 20 °C from the PEGS II Suite condition No. 50 consisting of 10% (w/v) PEG-4000 and 20% (w/v) isopropanol. Crystals were also obtained at each of the other PEDV-3CLpro concentrations. Optimization of this condition was achieved by varying the concentration of both isopropanol and PEG-4000 between 15–30% and 7–22% respectively, where diffraction quality crystals grew at a variety of conditions from this round of optimization. The best crystals grew at 20 °C in 25% isopropanol and 22% PEG-4000 at a protein concentration of 1.5 mg mL−1 and appeared after 24–72 hours (Fig. 4). Crystals were transferred using 0.05–0.1 μm nylon loops to small drops containing the crystallization solution plus the cryo-protectant, which was 15% 2-methyl-2,4-pentanediol (MPD). After cryo-protection, the crystals were remounted into the nylon loop and rapidly flash-cooled in liquid nitrogen.
Data collection and structure refinement
X-ray diffraction data were collected on LS-CAT beamline 21-ID-F at the Advanced Photon Source (APS) at Argonne National Laboratory, Argonne, Illinois, USA. Data were indexed, integrated and scaled using HKL-200025 and the resulting structure factor amplitudes were used for molecular replacement (MR). The program Phaser-MR (simple interface) module of PHENIX26 was used to perform MR and the X-ray structure of the human coronavirus 229E-3CLpro in complex with the peptidomimetic compound EPDTC (PDB entry 2zu213) with ligands and waters removed was used as a search model. Iterative rounds of manual building and structural refinement were completed using PHENIX and manual inspection, rebuilding and the addition of water molecules were accomplished using the programs Coot27 and the refinement module of PHENIX. The final data collection and refinement statistics are summarized in Table 1.
Inhibitor characterization
The compound (R)-16 was synthesized according to Jacob et al.15. Inhibition of PEDV-3CLpro by (R)-16 at a concentration of 100 μM was first tested in an enzymatic assay containing the following buffer (50 mM HEPES, 0.1 mg/mL BSA, 0.01% TritonX-100, 1 mM DTT). The assays were carried out in triplicate using Costar 3694 EIA/RIA 96-Well half-area, flat bottom, black polystyrene plates from Corning Incorporated. 1 μL of 100X inhibitor stock in DMSO was added to 79 μL of enzyme in assay buffer and the enzyme-inhibitor mixture was incubated for 10 minutes. The reaction was initiated by the addition of 20 μL of 10 μM UIVT3 substrate, a custom synthesized Förster resonance energy transfer peptide substrate with the following sequence: HilyteFluorTM488-ESARLQSGLRKAK-QXL520TM-NH2, producing final concentrations of 100 nM for the 3CLpro enzyme and 2 μM UIVT3 substrate. The increase in fluorescence intensity of the reaction was then measured over a period of 10 minutes as relative fluorescence units (RFUt). An excitation wavelength of 485 (bandwidth of 20 nm) and an emission wavelength at 528 (bandwidth of 20 nm) was used to monitor the reactions using a BioTek Synergy H1 multimode microplate reader. The percent inhibition of the PEDV-3CLpro was determined using the following equation (1):

The IC50 value of (R)-16 against PEDV-3CLpro was determined at ambient temperature using 100 μL assays conducted in the following buffer: 50 mM HEPES, 0.1 mg/mL BSA, 0.01% TritonX-100, 1 mM DTT. Kinetic assays were carried out in triplicate and in Costar 3694 EIA/RIA 96-Well Half Area, flat bottom, black polystyrene plates from Corning Incorporated. (R)-16 was tested at concentrations of 0.313, 0.652, 1.25, 2.5, 5.0, 10, 20, 40, 60, 80, 100 and 120 μM by adding 1 μL of 100X inhibitor stock in DMSO to 79 μL of enzyme in assay buffer and then incubating the enzyme-inhibitor mixture for 10 minutes. The reaction was initiated by the addition of 20 μL of 10 μM UIVT3 substrate, producing final concentrations of 100 nM for 3CLpro and 2 μM for the UIVT3 substrate. The increase in fluorescence intensity over time during the reaction was then measured.
The average percent inhibition of PEDV-3CLpro was then calculated from triplicate data and the final averaged data with standard deviation were then plotted as a function of inhibitor concentration. The data were fit to the following equation (2) to determine the IC50:

where %Imax is the percent maximum inhibition of 3CLpro. The errors in the IC50 and %Imax values are the errors in the fitted parameters resulting from fitting of the equations to the data.
Additional Information
Accession codes: Model coordinates and structure factor amplitudes for the X-ray crystal structure of unbound PEDV-3CLpro were deposited in the Protein Data Bank (acc. #: 5HYO).
How to cite this article: St. John, S. E. et al. X-Ray Structure and Inhibition of 3C-like Protease from Porcine Epidemic Diarrhea Virus. Sci. Rep. 6, 25961; doi: 10.1038/srep25961 (2016).
References
Debouck, P. & Pensaert, M. Porcine Rotavirus–Literature-Review and Isolation in Belgium. Vlaams Diergen Tijds 47, 144–155 (1978).
Wood, E. N. An apparently new syndrome of porcine epidemic diarrhoea. The Veterinary record 100, 243–244 (1977).
Hofmann, M. & Wyler, R. Propagation of the Virus of Porcine Epidemic Diarrhea in Cell-Culture. J Clin Microbiol 26, 2235–2239 (1988).
Egberink, H. F., Ederveen, J., Callebaut, P. & Horzinek, M. C. Characterization of the Structural Proteins of Porcine Epizootic Diarrhea Virus, Strain Cv777. Am J Vet Res 49, 1320–1324 (1988).
Utiger, A., Tobler, K., Bridgen, A. & Ackermann, M. Identification of the Membrane-Protein of Porcine Epidemic Diarrhea Virus. Virus Genes 10, 137–148, doi: 10.1007/Bf01702594 (1995).
Song, D. & Park, B. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis and vaccines. Virus Genes 44, 167–175, doi: 10.1007/s11262-012-0713-1 (2012).
Stevenson, G. W. et al. Emergence of Porcine epidemic diarrhea virus in the United States: clinical signs, lesions and viral genomic sequences. J Vet Diagn Invest 25, 649–654, doi: 10.1177/1040638713501675 (2013).
Zhang, Q. et al. Occurrence and investigation of enteric viral infections in pigs with diarrhea in China. Archives of virology 158, 1631–1636, doi: 10.1007/s00705-013-1659-x (2013).
Paarlberg, P. Updated estimated economic welfare impacts of porcine epidemic diarrhea virus (PEDV). Purdue University, Department of Agricultural Economics, Working Papers 14–4, 1–38 (2014).
Kocherhans, R., Bridgen, A., Ackermann, M. & Tobler, K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes 23, 137–144, doi: 10.1023/A:1011831902219 (2001).
Barrila, J., Bacha, U. & Freire, E. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 45, 14908–14916, doi: 10.1021/bi0616302 (2006).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767, doi: 10.1126/science.1085658 (2003).
Lee, C. C. et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J Biol Chem 284, 7646–7655, doi: 10.1074/jbc.M807947200 (2009).
St John, S. E. et al. X-ray structure and inhibition of the feline infectious peritonitis virus 3C-like protease: Structural implications for drug design. Bioorg Med Chem Lett 25, 5072–5077, doi: 10.1016/j.bmcl.2015.10.023 (2015).
Jacobs, J. et al. Discovery, synthesis and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. Journal of Medicinal Chemistry 56, 534–546, doi: 10.1021/jm301580n (2013).
Anand, K. et al. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. Embo J 21, 3213–3224 (2002).
Yang, H. T. et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. Plos Biology 3, 1742–1752, doi: ARTN e324, doi: 10.1371/journal.pbio.0030324 (2005).
Kim, Y. et al. Broad-Spectrum Antivirals against 3C or 3C-Like Proteases of Picornaviruses, Noroviruses and Coronaviruses. Journal of Virology 86, 11754–11762, doi: 10.1128/Jvi.01348-12 (2012).
Hsu, M. F. et al. Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem 280, 31257–31266 (2005).
Xu, T. et al. Structure of the SARS coronavirus main proteinase as an active C-2 crystallographic dimer. Acta Crystallogr F 61, 964–966, doi: 10.1107/S1744309105033257 (2005).
UniProt-Consortium. Porcine epidemic diarrhea virus strain CV7777 Replicase polyprotein 1ab. UniProt. Available at: http://www.uniprot.org/uniprot/P0C6Y4 (Accessed 3rd March 2016).
UniProt-Consortium. Human SARS coronavirus Replicase polyprotein 1ab R1AB_CVHSA. Available at: http://www.uniprot.org/uniprot/P0C6X7 (Accessed 3rd March 2016).
Mesecar, A. D., Stoddard, B. L. & Koshland, D. E. Orbital steering in the catalytic power of enzymes: Small structural changes with large catalytic consequences. Science 277, 202–206, doi: 10.1126/Science.277.5323.202 (1997).
Lawrence, P. K., Bumgardner, E., Bey, R. F., Stine, D. & Bumgarner, R. E. Genome sequences of porcine epidemic diarrhea virus: in vivo and in vitro phenotypes. Genome announcements 2, doi: 10.1128/genomeA.00503-14 (2014).
Otwinowski, Z. & Minor, W. In Methods in Enzymology Vol. 276 Macromolecular Crystallography (eds Carter, Jr. C.W. & Sweet, R. M. ) 307–326 (Academic Press, 1997).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D 66, 213–221, doi: 10.1107/S0907444909052925 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, doi: 10.1107/S0907444910007493 (2010).
Acknowledgements
We wish to thank Sakshi Tomar for designing the PEDV-3CLpro expression plasmid. This work was supported in part by grants (AI085089 and AI26603) to ADM from the National Institutes of Health via the National Institute of Allergy and Infectious Diseases. ADM also wishes to also acknowledge partial support from the Walther Cancer Foundation. Crystallization and DNA sequencing were partially supported by the Purdue Center for Cancer Research Macromolecular Crystallography and DNA Sequencing Shared Resources which are partially supported by an NIH grant (P30 CA023168). Finally, the authors would like to acknowledge the LS-CAT beamline staff for their help in acquiring X-ray data. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Author information
Authors and Affiliations
Contributions
S.E.S.J. and A.D.M. outlined the experiments, S.E.S.J. and B.J.A. conducted the experiments, S.E.S.J. and A.D.M. analyzed the data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
St. John, S., Anson, B. & Mesecar, A. X-Ray Structure and Inhibition of 3C-like Protease from Porcine Epidemic Diarrhea Virus. Sci Rep 6, 25961 (2016). https://doi.org/10.1038/srep25961
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25961
This article is cited by
-
In silico analysis of echinocandins binding to the main proteases of coronaviruses PEDV (3CLpro) and SARS-CoV-2 (Mpro)
In Silico Pharmacology (2021)
-
Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
