
Abstract
Catabolism of galactose by Streptococcus pneumoniae alters the microbe’s metabolism from homolactic to mixed acid fermentation, and this shift is linked to the microbe’s virulence. However, the genetic basis of this switch is unknown. Pyruvate formate lyase (PFL) is a crucial enzyme for mixed acid fermentation. Functional PFL requires the activities of two enzymes: pyruvate formate lyase activating enzyme (coded by pflA) and pyruvate formate lyase (coded by pflB). To understand the genetic basis of mixed acid fermentation, transcriptional regulation of pflA and pflB was studied. By microarray analysis of ΔpflB, differential regulation of several transcriptional regulators were identified, and CcpA, and GlnR’s role in active PFL synthesis was studied in detail as these regulators directly interact with the putative promoters of both pflA and pflB, their mutation attenuated pneumococcal growth, and their expression was induced on host-derived sugars, indicating that these regulators have a role in sugar metabolism, and multiple regulators are involved in active PFL synthesis. We also found that the influence of each regulator on pflA and pflB expression was distinct in terms of activation and repression, and environmental condition. These results show that active PFL synthesis is finely tuned, and feed-back inhibition and activation are involved.
Similar content being viewed by others


Introduction
Microbes commonly use multiple pathways for metabolism of the same nutrient, very likely due to operation of different pathways, that require distinct environmental conditions1,2. The presence of functionally similar pathways gives rise to the emergence of different phenotypes. However, the environmental and genetic factors giving rise to different metabolic outcomes, and hence phenotypes, are poorly understood. The important human pathogen Streptococcus pneumoniae is a good example for studying this case. It can exist as a commensal in the nasopharynx but also, for incompletely understood reasons, it can be a pathogen, causing diseases with high morbidity and mortality, such as pneumonia, bacteraemia, otitis media and meningitis3. It has been demonstrated by several studies that the pneumococcus exhibits different phenotypes in different tissues that enable the microbe to colonize mucosal surfaces or enhance its virulence. Changes in operation of its metabolic machinery under the influence of tissue-specific environmental conditions and nutrients will be a major contributing factor4,5.
Energetic metabolism has a fundamental influence on pneumococcal colonization and virulence1,2. S. pneumoniae produces its energy exclusively through fermentative breakdown of sugars because it lacks a complete set of genes required for respiration6. The pneumococcus has the capacity to utilize 32 different sugars7, which may provide a selective advantage to the microbe when it encounters different sugars in host tissues. The two most abundant sugars for the pneumococcus are glucose in the blood and galactose in the respiratory tract1,2,8. Fermentation of each leads to formation of pyruvate, from glycolysis for glucose, and the Leloir or tagatose pathways for galactose1. When growing on glucose, homolactic fermentation occurs, with pyruvate being converted to lactic acid by lactate dehydrogenase, thus replenishing NAD+. On galactose, however, there is a different metabolic outcome. In this instance, pyruvate can be converted to acetyl-CoA and formate by pyruvate formate lyase (PFL)2,9. The formation of acetyl-CoA gives the possibility of formation of an additional ATP via the action of acetate kinase or regeneration of two molecules of NAD+ with the formation of ethanol.
We show that utilization of galactose leads to the generation of mixed acids, and that pyruvate formate lyase (PFL), converting pyruvate to formate and acetyl-CoA, is the key enzyme for mixed acid fermentation2. Mutation of either pflB, which codes for PFL, or pflA, which codes for the pyruvate formate lyase activating enzyme (PFL-AE), being responsible for posttranslational activation of inactive PFL, results in abrogation of mixed acid fermentation on galactose, and interestingly leads to a decrease in pneumococcal virulence2. Despite the importance of PFL activity for pneumococcal metabolism and virulence, transcriptional regulation of the genes responsible for active PFL synthesis is not known in detail, neither in pneumococcus nor in other Gram-positive bacteria. So far, only the catabolite control protein A (CcpA) has been implicated in regulation of pflB10. However, the mechanism of CcpA-exerted pflA and pflB regulation remains to be investigated. Moreover, it is reasonable to assume that alternative regulatory systems that are operative independently of CcpA, may also have effect on the regulation of pflA and pflB11.
In this study, our objective was to identify the transcriptional regulators (TR) modulating the expression of genes responsible for active PFL synthesis in S. pneumoniae. We found that multiple regulators control the transcription of pflA and pflB. We gathered evidence showing that some of these regulators are induced by galactose, their control over pflA and pflB is influenced by sodium formate, they exert regulatory influence on each other, and are required for pneumococcal colonization and virulence.
Results
Transcriptome analysis of ΔpflB
To identify transcriptional regulators of pflB, the transcriptional profile of ΔpflB was obtained during growth on galactose relative to the wild type D39 strain. This analysis revealed differential expression of 113 genes in the mutant relative to the wild type (Table 1). The microarray results were validated for selected genes by quantitative real time PCR, and a similar trend of expression was obtained (Table 2). Out of these 113 genes, 62 were down-regulated and 51 were up-regulated in ΔpflB. The notable gene classes with differential expression were consistent with PFL’s role in galactose metabolism, pyruvate dissimilation, and catabolic and anabolic reactions, and included those coding for galactose hydrolysis and sugar uptake (n = 27), genes implicated in cell shape (n = 6), and fatty acid biosynthesis and acetate dissimilation (n = 13). Interestingly, the expression of loci containing Leloir pathway genes (n = 7) decreased in the mutant, whereas tagatose pathway genes were not affected, implying that the two galactose catabolic pathways are governed by distinct regulatory processes.
The expression of seven genes annotated as transcriptional regulators was also either significantly up- or down-regulated in the mutant relative to the wild type (Table 1). These were, as annotated in the D39 genome (http://www.ncbi.nlm.nih.gov), MarR (SPD_0379), GlnR (SPD_0447), GntR (SPD_1524), SPD_1594, PlcR (SPD_1745), CcpA (SPD_1797), and NmlR (SPD_1637). These regulators are known to be involved in diverse metabolic functions in bacteria, from the control of sugar metabolism (CcpA), fatty acid synthesis (MarR), control of glutamine metabolism (GlnR), and virulence gene expression (PlcR), to sensing environmental and nutritional cues (GntR)12. Differential expression of the seven transcriptional regulators, including CcpA, led to the hypothesis that some of these regulators mediate the expression of pflA or pflB. We could not test our hypothesis for SPD_1594 because, despite repeated attempts, this gene could not be mutated, implying that it is likely to be essential13.
CcpA, GlnR, and GntR interact with the putative promoters of pflA and pflB
The direct interaction of transcriptional regulators with the putative promoters (P) of pflA (PpflA) and pflB (PpflB), was determined by electrophoretic mobility shift assay (EMSA). For this, recombinant CcpA, PlcR, NmlR, GntR, GlnR, and MarR were purified (Supplementary Fig. S1), and their interactions with PpflA and PpflB were studied. The results showed that, CcpA and GlnR bound to both PpflA and PpflB. Interaction of transcriptional regulators with their target DNA was concentration-dependent and specific, as the regulators could not bind to a gyrB-specific probe (Fig. 1a–d). It should be also noted that CcpA binding to PpflA and PpflB occurred without the addition of HPr[Ser-P], which is reported to be essential for CcpA-DNA interaction in other lactic acid bacteria14,15. GntR could also form complexes with PpflB (Supplementary Fig. S2) but its inducibility with different sugars, and its involvement in regulation of pflA and pflB could not be established with subsequent reporter assays. In addition, the other transcriptional regulators did not interact either with PpflA or PpflB (data not shown).

EMSA analysis showing the direct interaction of CcpA (a and b), GlnR (c and d), with PpflB or PpflA, respectively. Each lane contains approximately 30 ng PpflB or PpflA. CcpA and GlnR were used between 0.1 to 0.5 μM. The coding sequence of gyrB (30 ng) was used as a negative control. Gels were stained with SYBR Green EMSA for visualizing DNA.
PFL activity leads to the generation of up to 35 mM formate when the pneumococcus is propagated on 55 mM galactose2. Hence, we tested whether sodium formate would alter the binding affinity of CcpA, and GlnR for PpflB. For this, the recombinant proteins were preincubated with 10 mM sodium formate before addition of the DNA probe. The results showed that formate enhanced the binding of CcpA to PpflB such that with sodium formate Kd was 0.35 ± 0.09 μM, and without it was 1.27 ± 0.1 μM (p < 0.01) (Fig. 2 and Supplementary Fig. S3). The effect of formate was specific because the addition of 10 mM Tris-HCl did not have any effect on binding (data not shown). On the other hand, formate reduced GlnR (Kd: 0.79 ± 0.1 μM) affinity for the pflB probe relative to without formate (Kd: 0.31 ± 0.09 μM) (p < 0.01) (Fig. 2).

Sodium formate enhanced CcpA- and decreased GlnR affinity for PpflB (b and d, respectively) compared to CcpA (a), and GlnR (c) interaction without sodium formate. Each lane contains approximately 30 ng PpflB; 0.2–1.5 μM of CcpA, or GlnR with or without 10 mM sodium formate. The experiment was repeated three times, and a representative image is shown.
CcpA and GlnR bind to promoters containing cre sites
After establishing the interaction of CcpA and GlnR with PpflA and PpflB, we set out to determine the binding site of these regulators. Initially, we mapped the putative binding sites on PpflA and PpflB (Supplementary Fig. S4), and identified multiple sites resembling a cre consensus sequence, the known binding site for CcpA from Bacillus subtilis (TGWAARCGYTWNCW, where N is any base, W is A or T, R is A or G, Y is C or T)16,17. At each putative promoter site, cre1 had the highest level of homology to the consensus sequence, 12 out of 14 nucleotides being similar. Therefore, EMSA was performed to determine whether cre1 had any role in binding of CcpA, and GlnR. For this, a promoter probe that excluded the cre1 sequence was produced and designated as PpflA(cre1−) and PpflB(cre1−), which was similar in size, approximately 100 bp, to the PpflA and PpflB.
EMSA analysis showed that CcpA and GlnR could not bind to PpflB(cre1−), indicating that the 14 nucleotide cre sequence located in PpflB contains the binding site for these regulators (Fig. 3a). Conversely, the results show that CcpA and GlnR bind to both PpflA and PpflA(cre1−) (Fig. 3b), However, they had higher binding affinities for PpflA than for PpflA(cre1−), which has an additional putative cre-like sequence.

EMSA analysis showing sequence specificity of CcpA, and GlnR for the cre1 sites in (a) PpflB and (b) PpflA. Each lane contains approximately 30 ng of promoter probes, and 0.5 μM of CcpA or GlnR.
Host-derived sugars induce the expression of transcriptional regulators
The inducibility of the regulators by different sugars was determined using transcriptional reporter assays in anaerobically grown cultures. For this, the putative promoter region of each transcriptional regulator was fused to a promoterless lacZ in the wild type D39 background, and β-galactosidase activity in resulting reporter strains, PccpA::lacZ-wt, and PglnR::lacZ-wt, was determined in the presence of glucose, galactose, mannose, or N-acetyl glucosamine (Table 3). All sugars induced the transcriptional regulator-driven β-galactosidase activity relative to expression in the absence of sugar (p < 0.01), but the highest inducer was found to be galactose.
In addition, the responsiveness of the regulators to 10 mM sodium formate, which does not affect pneumococcal growth, was also determined. For this, the reporter strains were grown in the presence of glucose because formate production was found to be below the detection limit on glucose2,18, hence endogenous formate would not interfere with the assay. The results showed that the addition of sodium formate, but not Tris-HCl, induced β-galactosidase activity in PccpA::lacZ-wt by 2.4-fold, but decreased the activity in PglnR::lacZ-wt by 4.3-fold compared to the activity on glucose alone (Table 3) (p < 0.01). These data indicate that ccpA and glnR expression are responsive to formate, and that the absence of formate must be partly responsible for the transcriptional profile of ΔpflB.
CcpA, and GlnR have different roles in expression of pflA and pflB
To test the regulatory roles of CcpA, and GlnR on pflA and pflB, PpflA::lacZ and PpflB::lacZ fusions were introduced into both wild type D39, and the mutants ΔccpA, and ΔglnR. The resulting strains PpflA::lacZ-wt, PpflA::lacZ-ΔccpA, PpflA::lacZ-ΔglnR, PpflB::lacZ-wt, PpflB::lacZ-ΔccpA, and PpflB::lacZ-ΔglnR were tested during anaerobic growth in CDM supplemented with 55 mM of galactose. The results show that β-galactosidase activity in PpflA::lacZ-ΔccpA and PpflA::lacZ-ΔglnR increased by 2.8- and 2.2-fold, respectively, compared to the activity in PpflA::lacZ-wt (Fig. 4a) (p < 0.0001), suggesting that CcpA and GlnR repress the transcription of pflA. On the other hand, β-galactosidase activity in PpflB::lacZ-ΔglnR increased by 7.2-fold, whereas in PpflB::lacZ-ΔccpA it decreased by 28.7-fold compared to the wild type (p < 0.0001) (Fig. 4b), indicating that GlnR repress, and that CcpA activates pflB on galactose. On the other hand, on glucose both CcpA and GlnR were found to repress both pflA and pflB, (Fig. 4c and d) (p < 0.001). To rule out the possibility that the expression data was influenced by lacZ fusion, pflA, and pflB expression was also determined by real time quantitative reverse transcriptase PCR (qRT-PCR) in ΔccpA and ΔglnR grown on galactose relative to wild type. The results obtained with qRT-PCR were consistent with those of the transcriptional reporter assays, ruling out any interference by lacZ (Supplementary Table S1).

Expression levels (in Miller Units) of pneumococcal transcriptional lacZ-fusions to the promoters of pflA (a and c) and pflB (b and d) in different backgrounds grown anaerobically in CDM supplemented with 55 mM of galactose (a and b) or glucose (c and d). The activity is expressed as nmol p-nitrophenol/min/ml. Error bars show the standard error of the mean for three individual measurements each with three replicates. ***p < 0.001, ****p < 0.0001.
Next, we investigated whether CcpA and GlnR could play a regulatory role in each other’s expression. This hypothesis was tested by use of EMSA and LacZ reporter assays. EMSA showed that both CcpA and GlnR could bind to PccpA, indicating that ccpA regulates its own transcription, and that GlnR is involved in ccpA expression (Fig. 5). This binding was specific as CcpA and GlnR could not bind to the putative promoter region of gyrB (lane 2 in Fig. 1a–d). The interaction between CcpA and GlnR was further investigated by LacZ reporter assays. It was shown that β-galactosidase activity in PccpA::lacZ-ΔccpA and PccpA::lacZ-ΔglnR decreased both on glucose, 2.5- and 10.3-fold, and on galactose, 30.7- and 1.4-fold, respectively, compared to the activity of PccpA::lacZ-wt (Table 4) (p < 0.001). This result shows that both on glucose and galactose CcpA increases its own expression, and GlnR activates ccpA, particularly on glucose.

(a) Schematic representation showing the analysis of predicted promoter region and binding sites of PccpA, and (b) EMSA analysis showing the direct interaction of CcpA and GlnR with PccpA. (a) The core promoter region containing the −10 and −35 elements is indicated. The putative cre sequences are indicated in red. F: indicates forward primer while R: refers to the reverse primer used for amplifying the promoter probe. T: potential terminator structure. The black arrow presents the direction of transcription. (b) Each lane contains approximately 30 ng PccpA; 0.5 μM of CcpA or GlnR as appropriate.
CcpA’s role in regulation of glnR was also determined. CcpA could not bind to PglnR, which was consistent with the lack of a discernible cre sequence in the promoter region. The reporter assay results show that β-galactosidase activity in PglnR::lacZ-ΔccpA decreased by 1.7- and 3.8-fold on glucose and galactose, respectively, compared to PglnR-lacZ-wt. This result suggests that CcpA increases glnR expression in the presence of both glucose and galactose, and GlnR does not effect its own expression, since β-galactosidase activity in PglnR::lacZ-ΔglnR on glucose (89.7 MU ± 4.4, n = 3) and galactose (572.3 MU ± 4.2, n = 3) was similar to that of PglnR::lacZ-wt (with glucose 93.4 MU ± 1.8, and with galactose: 565.0 MU ± 7.1, n = 3 for both) (Table 4). Moreover, the results showed that GlnR activates ccpA expression both on glucose and galactose (Table 4).
Growth and end product analysis
To further investigate CcpA, and GlnR’s role in pneumococcal sugar metabolism, the growth profiles and fermentative end-products of ΔccpA, and ΔglnR were determined. Moreover, to evaluate the added impact of CcpA and GlnR mutation on pneumococcal metabolism, a ΔccpAΔglnR mutant was also constructed and tested. On glucose, ΔccpA, ΔglnR, and ΔccpAΔglnR had significantly reduced growth rates (0.36 h−1 ± 0.01, 0.47 h−1 ± 0.01, and 0.31 h−1 ± 0.01 respectively, n = 3) compared to wild type D39 (0.67 h−1 ± 0.01) (p < 0.0001 for all strains). Despite a low rate of growth, ΔccpA growth yield (max change in OD500), was similar to that of wild type, while ΔglnR had a lower yield than the wild type. In addition, ΔccpAΔglnR had a lower growth rate and yield than each single mutant (p < 0.01) (Fig. 6a). The reason for growth attenuation of strains with a glnR mutation, ΔglnR and ΔccpAΔglnR, is not known but it may be linked to GlnR’s involvement in glucose transport and metabolism.

(a) shows growth in 55 mM glucose and (b) in galactose. (c) and (d) show fermentation end product profiles after growth on glucose and galactose, respectively. Error bars show the standard error of the mean for three individual measurements each with three replicates. Significant differences were seen comparing the growth rates, and the fermentative profile of mutant strains to the wild type D39 using ANOVA followed by Dunnett’s multiple comparison test. **p < 0.01 and ****p < 0.0001.
On galactose, the growth rate and yield of all strains decreased approximately to half or less than that on glucose (Fig. 6b). ΔccpA, ΔglnR, and ΔccpAΔglnR had significantly reduced growth rates (0.24 h−1 ± 0.01, 0.26 h−1 ± 0.01, and 0.07 h−1 ± 0.01, respectively, n = 3) compared to the wild type strain (0.35 h−1 ± 0.01, n = 3) (p < 0.01). ΔglnR had an extended lag phase and started to grow after 11 h, which may indicate its involvement in sensing of galactose. Moreover, there was significant difference in the rate of growth between ΔccpAΔglnR and each single mutant (p < 0.001), and the growth yield (max change in OD500) of the double mutant (0.21 ± 0.02, n = 3) was significantly lower than that of the single mutants (0.79 ± 0.07 for ΔccpA, 0.74 ± 0.06 for ΔglnR, n = 3) (p < 0.01).
In the presence of glucose the wild type, and ΔglnR strains displayed typical homolactic behaviour with lactate as the main fermentation product, and minor amounts of formate and acetate (Fig. 6c). Loss of CcpA in ΔccpA and ΔccpAΔglnR caused a shift from homolactic to mixed-acid fermentation. In ΔccpA, a 4.4-fold increase in the yield of formate and 23.5-fold in acetate was detected relative to the wild type. On galactose, the wild type, and ΔglnR displayed a mixed-acid fermentation pattern with lactate, formate and acetate as the end products. However, ΔccpA and ΔccpAΔglnR had a homolactic product pattern with lactate production in ΔccpA (29.1 mM ± 0.9, n = 3) being significantly higher than in the wild type (p < 0.0001), whereas formate and acetate production in these strains was significantly decreased compared to the wild type (p < 0.0001) (Fig. 6d). Therefore, the results showed that both transcriptional regulators are important for growth on both glucose and galactose, and CcpA has a major impact on the composition of the mixed acid profile.
Virulence studies
In vitro assays indicated CcpA and GlnR are involved in pflA and pflB regulation, and they have a regulatory influence on each other’s expression. Therefore, we determined the contribution of CcpA and GlnR to nasopharyngeal colonization, and virulence in mouse models of pneumococcal infection. The results show that the median survival time of mice infected intranasally with ΔccpA, ΔglnR, and ΔccpAΔglnR (168 h ± 17.3, 168 h ± 13.0 and 168 h ± 0.0 respectively, n = 10) was significantly longer than the wild type infected group (35 h ± 13.2, n = 10) (p < 0.001 for all strains) (Fig. 7a). The introduction of intact copies of ccpA and glnR into ΔccpA and ΔglnR, respectively, reconstituted the virulence of these strains with the median survival times of mice infected with ΔccpAComp (37 h ± 2.3, n = 10) and ΔglnRComp (45 h ± 19.5, n = 10), being not significantly different from the wild type infected cohort (p > 0.05), eliminating the possibility of a polar effect of the mutations (Fig. 7a).

(a) Survival time of mice infected intranasally with approximately 2 × 106 CFU pneumococci. Each dot represents the survival time of individual animal, and the horizontal bars mark the median survival times derived from 10 animals. (b) Progression of bacteraemia in mice infected intranasally with ΔccpA, ΔglnR and their derivatives at 24 h and 36 h post-infection. Each point is the mean of data from ten mice. (c) Pneumococcal strains defective in ccpA and glnR were less able to colonize nasopharynx. Mice were infected approximately with 1 × 105 CFU pneumococci. At day 0 and day 7, five mice were culled, and bacterial CFU/mg were determined by serial dilutions of nasopharyngeal homogenates. Each column represents the mean of data from five mice. Error bars show the standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
The progression of bacteraemia in mice intranasally infected with the pneumococcal strains was also determined. The concentrations of pneumococci retrieved from the blood are shown in Fig. 7b. The bacterial load in the ΔccpA infected group was significantly lower at 24 and 36 h post-infection (log10 0.18 ± 0.18 CFU/ml and log10 0.35 ± 0.2 CFU/ml, respectively, n = 10) compared to the wild type (24 h: log10 4.71 ± 0.6 CFU/ml, 36 h: log10 5.8 ± 0.3 CFU/ml, n = 10) (p < 0.0001). On the other hand, no bacterial growth could be detected in the blood of ΔglnR or ΔccpAΔglnR infected animals at 24 and 36 h of infection (p < 0.0001). Moreover, both complemented strains, ΔccpAComp (24 h: log10 4.97 ± 0.5 CFU/ml, 36 h: log10 5.69 ± 0.6 CFU/ml, n = 10) and ΔglnRComp (24 h: log10 3.05 ± 0.8 CFU/ml, 36 h: log10 3.6 ± 0.9 CFU/ml, n = 10), had significantly higher loads in the blood of infected mice at 24- and 36 h of post infection compared to ΔccpA and ΔglnR (p < 0.0001) (Fig. 7b).
We also evaluated the contribution of CcpA and GlnR in nasopharyngeal colonization. One hour after infection of the nasopharynx, the pneumococcal load in the nasopharyngeal tissue for all strains (log10 2.38 ± 0.2 CFU/mg, log10 2.31 ± 0.4 CFU/mg, log10 2.24 ± 0.6 CFU/mg, log10 2.55 ± 0.5 CFU/mg and log10 2.30 ± 0.3 CFU/mg, for ΔccpA, ΔglnR, ΔccpAΔglnR, ΔccpAComp, and ΔglnRComp, respectively, n = 5) was similar to that of wild type (log10 2.44 ± 0.3 CFU/mg, n = 5) (p > 0.05) (Fig. 7c). On the other hand, at 7 days post-infection the colony counts for ΔccpA, ΔglnR, and ΔccpAΔglnR (log10 1.24 ± 0.2 CFU/mg, log10 1.52 ± 0.1 CFU/mg and log10 0.58 ± 0.1 CFU/mg respectively, n = 5) were significantly lower than the counts of wild type strain (log10 2.88 ± 0.2 CFU/mg, n = 5) (p < 0.01, p < 0.05 and p < 0.0001 for ΔccpA, ΔglnR. and ΔccpAΔglnR, respectively) (Fig. 7c). No significant differences were seen in the bacterial load of the complemented strains, ΔccpAComp and ΔglnRComp (log10 2.96 ± 0.4 CFU/mg and log10 2.74 ± 0.3 CFU/mg respectively, n = 5) compared to the wild type (p > 0.05). These results show that CcpA and GlnR contribute both to pneumococcal virulence and colonization of nasopharynx.
Discussion
This study provides new insights into how the important human pathogen S. pneumoniae regulates its galactose metabolism, which is known to have a major effect on pneumococcal survival in vivo6. Pyruvate formate lyase is a key enzyme for pyruvate dissimilation in S. pneumoniae. In addition to its role in pneumococcal energetics, the products of the PFL-catalysed reaction, formate, and acetyl CoA, play a key role in anabolic reactions. For example, while formate is essential for serine biosynthesis by hydroxymethylation of glycine, acetyl-CoA is important for fatty acid biosynthesis2,19. Until this study, very little was known about the transcriptional regulation of genes involved in active PFL synthesis.
Our results show that multiple regulators modulate the expression of pflA and pflB. While CcpA and GlnR are involved in regulation of both pflA and pflB, GntR could only bind to putative promoter region of pflB. The other regulators identified to be differentially expressed in ΔpflB could bind neither to PpflA nor to PpflB, presumably their differential expression in ΔpflB was due to indirect effect of pflB mutation. CcpA was found to repress pflA and pflB on glucose, and activate pflB and repress pflA expression on galactose. This opposing regulatory influence of CcpA on pflA and pflB could indicate that constitutive pflA expression sufficiently activates PFL synthesis. This is supported with the fact that the association rate between PFL-AE and PFL is low for biological interactions because of large conformational changes when these two enzymes interact. Hence a high level pflA expression would be wasteful because of a low association rate20. Moreover, intracellular calculations in E. coli revealed that PFL-AE is almost entirely in the PFL-AE/AdoMet PFL pyruvate complex, and its activation requires reduction by flavodoxin to initiate catalysis. Therefore, despite its constitutive expression, the existing PFL-AE can be promptly activated in this complex when needed. It was also noticed that induction level of PpflA is lower than PpflB on galactose (Fig. 4). The expression data are very likely reflecting the amount of PFL-AE required to activate PFL, because PFL is completely activated by 0.01 equivalents of PFL-AE in 100 minutes21. The opposing effects of transcriptional regulators on pflA and pflB must be an important pneumococcal adaptation mechanism in tissues with different sugar profiles and oxygen concentrations. For example, in an oxygenated environment active PFL is not critical as it is sensitive to oxygen. On the other hand, in tissues with high galactose content but limited oxygen concentration, the pneumococcus requires mechanisms to induce pflB expression.
In other Gram positive bacteria CcpA binding requires HPr-[Ser-P], while the pneumococcal CcpA bound to a cre sequence in the absence of HPr-[Ser-P]22. However, we found that 10 mM sodium formate enhanced CcpA affinity for PpflB, consistent with its induction by sodium formate in the transcriptional reporter assays. Although the inducibility of ccpA expression by different carbon compounds had been shown previously11,23, this is the first demonstration of the responsiveness of this master regulator to formate. It is not clear how formate enhances CcpA binding and stimulates the expression, but given that formate is a potent reducing agent, with a redox potential of −420 mV24, it is very likely due to its effect on the conformation or multimerization of protein structure. Addition of formate increased ccpA-, and decreased glnR transcription, indicating the responsiveness of S. pneumoniae to formate. Formate’s role as a transcriptional signal also shows that the absence of formate is partly responsible for the transcriptional profile of ΔpflB. In addition, we previously demonstrated that fatty acid composition of S. pneumoniae changes in ΔpflB, which cannot produce formate, also indicating the importance of formate on transcription and protein synthesis2. Formate was reported to have a major influence on bacterial gene expression, and acts as a diffusible signal to induce Salmonella enterica serovar Typhimurium invasion25. In Staphylococcus aureus, it was suggested that formate production is necessary for formyl-tetrahydrofolate synthesis, which is required for protein and purine synthesis18. Further work is required to understand the mechanism by which formate impacts on the pneumococcus. We did use galactose similarly to formate to determine its effect on the affinity of transcriptional regulators to PpflA and PpflB. However, no difference could be detected.
The cre1 within PpflB has been identified to be the binding site for GlnR. GlnR bound two cre sites in PpflA but here it had a higher binding affinity for cre1 than cre2. GlnR binding affinity for pflB was shown to decrease in the presence of 10 mM sodium formate, consistent with a decrease in PglnR driven β-galactosidase activity with sodium formate. This is the first demonstration of interaction of proteins, other than CcpA and HPr, with cre. Given that cre sites are plentiful in Gram-positive bacteria, it is plausible that some of these targets are regulated by multiple regulators, either independently or in concert with CcpA.
Interestingly, although CcpA plays a role in glnR regulation, as determined by the LacZ reporter assay, PglnR does not have an identifiable cre-like sequence. Therefore, it is possible that CcpA-mediated glnR regulation requires the involvement of another, unknown, protein(s). It is known that several genes with no cre or un-functional cre have been reported to be indirectly regulated by CcpA26,27,28. Our results showed that both CcpA and GlnR have a significant role in pneumococcal virulence and colonization. The results obtained in this study on CcpA’s impact on pneumococcal colonization and virulence are in line with previous reports11,23. However, our in vivo results for GlnR differ from the study of Hendriksen et al., who could not attribute a role for GlnR in pneumococcal virulence and colonization29. One reason for this discrepancy can be linked to the difference in dose-preparation, for example, while in this study we did not passage the bacterial inoculum through peritoneum before infection, Hendriksen’s study used passaged bacteria. In vivo passaging before infection can introduce mutations to a bacterial population due to strong within-host selective pressure30.
The GntR family of transcriptional regulators is a group of transcriptional repressors distributed among diverse bacterial groups, and mediate bacterial response to nutritional and/or environmental signals31. In addition to CcpA and GlnR, GntR could also bind to PpflB. However, GntR could not bind to PccpA and PpflA, even though both have a cre-like sequence. Unlike cre1 in PpflB (TGAAATCGGTTACT, conserved residues are underlined), which is categorised as a high-affinity cre box because it contains all four strongly conserved residues, G2, C7, G8, and C13, the cre sequences in PpflA, and PccpA contain only 2 or 3, respectively, conserved residues16,17. Therefore, it is likely that GntR only recognizes highly conserved cre boxes. PgntR-driven β-galactosidase activity was not induced by any of the tested sugar nor ΔgntR is attenuated in growth on galactose (data not shown). This suggests that in addition to sugar dependent regulation, sugar independent regulation of pflA and pflB may take place.
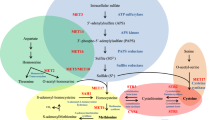
Based on the available data we propose the model for pflA and pflB regulation shown in Fig. 8. Growth on galactose increases PFL activity, which leads to increased production of formate, and also induces glnR expression. When the formate level reaches a certain threshold, GlnR repression over pflA and pflB expression is removed, and GlnR then binds to cre, up-regulating ccpA transcription. In addition, the increased formate production also increases ccpA expression as well as CcpA affinity for PpflB, leading to an increase in pflB transcription. However, CcpA and GlnR also bind to PpflA, and repress pflA expression to fine-tune the level of pflA transcript. On the other hand, on glucose, GlnR increases ccpA expression, and CcpA activates its own expression. This, in turn, results in repression of both pflA and pflB, and pyruvate is dissimilated mainly to lactate by lactate dehydrogenase activity1,2.

On galactose, formate production by PFL activity decreases GlnR affinity for PpflB and GlnR increases ccpA transcription. The increased formate production increases CcpA affinity for PpflB, and CcpA either alone or after interacting with GlnR binds to PpflB and increases pflB transcription. However, CcpA also binds to PpflA and represses its expression to fine-tune the level of active PFL. Increased formate production then creates a positive feed back loop, and CcpA self-regulates its own expression. Glu: glucose, Gal: galactose, SF: sodium formate. The green arrow indicates increase in expression, whereas the red arrow is for decreased expression. *Posttranslational activation of PFL by PFL-AE has been previously reported9,47.
Galactose is found in high concentration on host glycoconjugates in the nasopharynx, where the pneumococcus resides, and the microbe is able to cleave and utilize galactose from these sources8. However, galactose is not efficiently catabolized by the pneumococcus, possibly due to the possession of low-affinity galactose importers6, and/or to a slow association rate between nascent PFL and PFL-AE for the formation of active PFL32. In addition, at the transcriptional level the expression of pflA and pflB is down-regulated by the regulators identified in this study, with the exception of CcpA, in the presence of galactose. Taken together, these data indicate that the pneumococcus has evolved to catabolize galactose at a lower rate than other sugars, such as glucose. It can be speculated that low galactose catabolic rate allows the microbe to maintain a stable relationship with its host. This can be linked to galactose catabolism and its role in pneumococcal virulence. For example, the pneumococcus produces more capsule, an essential virulence determinant, on galactose than it does on glucose10.
This study demonstrates that PFL synthesis is fine-tuned at the transcriptional level by multiple regulators and various environmental conditions. PFL activity is known to be crucial for galactose catabolism, which is linked to pneumococcal colonization and virulence. Detailed knowledge of regulation of pneumococcal sugar metabolism may allow identification of targets for new antiinfectives against this important pathogen.
Materials and Methods
DNA manipulations
Standard methods were used for chromosomal DNA isolation, restriction enzyme digestion, cloning, transformation, agarose gel electrophoresis, and sodium dodecyl sulphate polyacrylamide gel electrophoresis33. Plasmids were extracted using QIAprepSpin Miniprep Kit (Qiagen, UK), and PCR products were purified using the Wizard® SV Gel and PCR Clean-Up System (Promega, UK).
Bacterial strains, plasmids and growth conditions
The strains and plasmids used and constructed in this study are listed in Supplementary Table S2. Pneumococcal strains were grown microaerobically at 37 °C as described previously2. For anaerobic growth, an anaerobic cabinet was used. Pneumococci were also grown in chemically defined medium with selected sugars (CDM)1. Growth was monitored by measuring the optical density at 500 nm (OD500). Growth rates (μ) were calculated through linear regressions of the plots of ln(OD500) versus time during the exponential growth phase. When required, the pneumococcal growth medium was supplemented with spectinomycin (100 μg/ml), tetracycline (15 μg/ml), and kanamycin (250 μg/ml).
Escherichia coli strains, used for cloning and protein expression, were grown in Luria Bertani (LB) broth with shaking at 37 °C or on LB agar plates. For E. coli cultures, ampicillin and kanamycin at 100- and 50 μg/ml, respectively, were used when required.
Microarray analysis
S. pneumoniae D39 and its isogenic mutant strain ΔpflB, deficient in pyruvate formate lyase (PFL) activity, were grown anaerobically in CDM supplemented with 55 mM galactose as the main carbon source. The experiments were repeated with four biological replicates. The MicroPrep software package was used to obtain the microarray data from the slides. CyberT implementation of a variant of t-test (http://bioinformatics.biol.rug.nl/cybert/index.shtml) was performed and false discovery rates (FDRs) were calculated27. For differentially expressed genes, p < 0.001 and FDR < 0.05 were taken as standard. For the identification of differentially expressed genes a Bayesian p-value of < 0.001 and a fold change cut-off twofold were applied. All other procedures for the DNA microarray experiments and data analysis were performed as described before34.
Gene splicing by overlap extension (gene SOEing)
Allelic replacement mutagenesis was achieved by transformation with an in vitro mutagenized SOEing construct. To do this, spectinomycin (SpecR) or kanamycin (KanR) resistance gene cassettes35,36 were amplified using the primes Spec-F and Spec-R or Kan-F and Kan-R primers (Supplementary Table S3). The primers LF-SOEX-F/LF-SOEX-R and RF-SOEX-F/RF-SOEX-R (where X indicates the gene code) were used to amplify the left and right flanking regions of each target gene, generating PCR products of approximately 600 bp in length. The amplified antibiotic resistance cassette and the PCR products flanking the target gene were then fused using LF-SOEX-F and RF-SOEX-R primers. The fused product was gel-purified after electrophoresis (Qiagen) and transformed into S. pneumoniae37. The transformants were selected on blood agar base supplemented with 5% (v/v) defibrinated horse blood containing the appropriate antibiotic and the mutations were confirmed by PCR and sequencing. The double mutant was constructed by transformation of amplicons representing the mutated region of glnR into the ccpA mutant.
Genetic complementation of mutants
To eliminate the possibility of polar effects, the mutant strains were genetically complemented using plasmid pCEP36. The wild type copy of mutated genes with their upstream sequence were amplified with X-Comp-F and X-Comp-R primers (where X indicates the gene code) (Supplementary Table S3). The amplicons were digested with NcoI-BamHI or SphI-BamHI and were ligated into similarly digested pCEP. A sample of ligation mixture was transferred into One Shot® TOP10 competent E. coli cells (Invitrogen, UK). The transformants were selected on kanamycin-containing LB agar plates, and successful cloning was confirmed by PCR using Comp-Seq-F and Comp-Seq-R primers. The recombinant plasmid was purified, and a portion was transformed into the appropriate mutant strains. The complemented strains were designated as ΔccpAComp, and ΔglnRComp.
Quantification of extracellular metabolites
Samples (2 ml) of anaerobic cultures growing in CDM containing 1% (w/v) glucose or galactose were collected at late exponential phase during growth, centrifuged (6700 × g, 10 min, 4 °C), filtered (Millex-GN 0.22 μm filters) and the supernatant solutions were stored at −20 °C until analysis with commercial kits for lactate, formate, and acetate detection (Megazyme, Ireland). The amount of organic acids produced by different strains was normalized to 1 × 108 cells. The spent culture supernates were obtained at late exponential phase and this corresponded to the following OD500s for the different strains grown on glucose and galactose, respectively: for D39 1.9 and 1.1; for ΔccpA 1.8 and 0.7; for ΔglnR 1.2 and 0.6; for ΔccpAΔglnR 0.7 and 0.2.
Purification of recombinant proteins
The genes of interest were amplified and cloned into the hexa histidine-tagged, ampicillin resistant plasmid pLEICS-01 (PROTEX, University of Leicester) using the primers (Indicated with C-F/R in Supplementary Table S3) containing 15 nucleotides complementary to the cloning site in pLEICES-01. Recombinant plasmids were transformed into E. coli BL21 (DE3) pLysS (Agilent Tech, USA) for protein expression at 18 °C and 0.1 M of IPTG (Isopropyl β-D-1-thiogalactopyranoside) was used for induction of protein expression. When the OD600 reached 1.5–1.6, the culture was centrifuged and the pellet was resuspended with the binding buffer (20 mM Tris, 150 mM NaCl, pH 7.45) prior to sonication at an amplitude of 8 microns for 15 sec with 45 sec intervals on ice. The lysate was then centrifuged and filtered through a 0.45 μm acrodisc (Fisher Scientific, UK). Proteins were purified using PD-10 columns (GE Healthcare Life Sciences, UK) containing 2 ml of TALON Metal Affinity Resin (Clontech, USA) and finally eluted using different concentrations of imidazole elution buffer (20 mM Tris, 150 mM NaCl, pH 7.45). Protein fractions were dialysed against the binding buffer using Amicon Ultra-15 Centrifugal Filter Units (Millipore, UK) and the concentrations were determined using the Bradford protein assay38.
Electrophoretic mobility shift assay (EMSA)
In silico analysis, using the bacterial promoter prediction tool (BPROM)39 and the Motif-based sequence analysis tools (MEME), was used to predict the presence of regulatory elements in the putative promoter regions of target genes40. Based on in silico analysis, primers (indicated by EMSA-F/R in Supplementary Table S3) were designed to amplify 95–100 bp DNA fragment representing putative promoter sites upstream of each gene.
EMSA was performed according to the protocol described previously41 using Molecular Probes fluorescence-based EMSA kit (Invitrogen). Briefly, 5X FY binding buffer (20 mM Tris-HCl pH 7.5, 30 mM KCl, 1 mM DTT, 1 mM EDTA pH 8.0 and 10% v/v glycerol) was prepared to incubate the promoter probes and proteins. The binding reaction was set up by mixing a constant amount of target promoter probes (~30 ng), and increasing amounts (0.1–0.5 μM) of purified and dialysed His-tagged proteins. The binding reaction was incubated at room temperature for 20 min in a total volume of 20 μl, and then analysed on an 8% w/v non-denaturing polyacrylamide gel. After electrophoresis, gels were stained with SYBR® Green EMSA gel stain (Invitrogen) and visualized using a Typhoon Trio+ scanner (GE Healthcare Life Sciences) with a 526 nm short-pass wavelength filter.
Construction of lacZ-fusions
Chromosomal transcriptional lacZ-fusions to the target promoters were constructed via double crossover in the bgaA gene by using the integration plasmid pPP142. As the extracellular BgaA enzyme is responsible for removal of galactosides from complex carbohydrates, inactivation of the bgaA gene will not change the growth of S. pneumoniae in media and will not compromise regulatory studies42. The putative promoter regions were amplified using the primers modified to incorporate SphI and BamHI sites (indicated with Fusion-F/R in Supplementary Table S3). The amplicons were digested with SphI and BamHI and were ligated into similarly digested pPP1. All plasmid constructs were confirmed by sequencing.
β-galactosidase assays
β-galactosidase activity was measured as described before43 using cells grown anaerobically in CDM at 37 °C supplemented with 55 mM of the selected sugars, or 10 mM sodium formate, and harvested in the mid-exponential phase of growth.
Total RNA purification and quantitative RT-PCR
The extraction of RNA was done by the Trizol method using mid-exponential phase cultures, as described previously44. Before use the RNA was treated with DNase using a TURBO DNA-free™ Kit (Ambion, UK), and subsequently purified with an RNeasy Mini Kit (Qiagen).
First strand cDNA was synthesized using approximately 1 μg of DNase-treated total RNA, immediately after isolation, random hexamers and 200 U of SuperScript III reverse transcriptase (Invitrogen) at 42 °C for 55 min as described previously2. Three independent RNA preparations were used for qRT-PCR analysis. The transcription level of specific genes was normalized to gyrB transcription, which was amplified in parallel using the primers in Supplementary Table S4 (indicated with RT-F/R tags). cDNA samples obtained from individual biological samples were analysed in triplicates. The results were analysed by the comparative threshold cycle CT method45. Differences in expression of twofold or greater relative to control were considered as significant.
In vivo virulence studies
To evaluate the virulence of pneumococcal strains, nine to ten-week-old female MF1 outbred mice (Charles River, UK) were lightly anesthetized with 3% v/v isoflurane and oxygen mixture, and an inoculum of 50 μl containing approximately 2 × 106 CFU in PBS was administered dropwise into the nostrils. Disease signs in infected animals were monitored regularly2,46. When the mice become lethargic, they were culled by cervical dislocation, “survival time” was defined as the time to reach the lethargic state. Mice that were alive 7 days after infection were deemed to have survived the infection. To monitor the development of bacteraemia, approximately 20 μl of venous blood was obtained from each mouse at predetermined time points after infection, and viable counts were determined, as described above. Colonization experiments were done principally as described previously, using 5 × 105 CFU of S. pneumoniae in 10 μl PBS, given intranasally8. Survival data were analysed by the Mann-Whitney test. Colonization and bacteraemia data were analysed by analysis of variance followed by Tukey’s multiple comparisons test. Statistical significance was considered to be a p-value of < 0.05.
Ethics statement
Mouse experiments at the University of Leicester were performed under appropriate project (permit no. 60/4327) and personal (permit no. 80/10279) licenses according to the United Kingdom Home Office guidelines under the Animals Scientific Procedures Act 1986, and the University of Leicester ethics committee approval. The protocol was approved by both the U.K. Home Office and the University of Leicester ethics committee. Where indicated, the procedures were carried out under anesthetic with isoflurone. Animals were housed in individually ventilated cages in a controlled environment, and were frequently monitored after infection to minimize suffering. Every effort was made to minimize suffering and in bacterial infection experiments mice were humanely culled if they became lethargic.
Data Availabilty
Microarray data have been submitted to GEO (Gene Expression Omnibus) database under the accession number GSE67668 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ypkhqogmvhwrvst&acc=GSE67668).
Additional Information
How to cite this article: Al-Bayati, F. A. Y. et al. Pneumococcal galactose catabolism is controlled by multiple regulators acting on pyruvate formate lyase. Sci. Rep. 7, 43587; doi: 10.1038/srep43587 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Gaspar, P., Al-Bayati, F. A. Y., Andrew, P. W., Neves, A. R. & Yesilkaya, H. Lactate dehydrogenase is the key enzyme for pneumococcal pyruvate metabolism and pneumococcal survival in blood. Infect. Immun. 82, 5099–109 (2014).
Yesilkaya, H. et al. Pyruvate formate lyase is required for pneumococcal fermentative metabolism and virulence. Infect. Immun. 77, 5418–5427 (2009).
Kadioglu, A., Weiser, J., Paton, J. & Andrew, P. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6, 288–301 (2008).
Sandrini, S., Alghofaili, F., Freestone, P. & Yesilkaya, H. Host stress hormone norepinephrine stimulates pneumococcal growth, biofilm formation and virulence gene expression. BMC Microbiol. 14, 180 (2014).
Oggioni, M. R. et al. Switch from planktonic to sessile life: a major event in pneumococcal pathogenesis. Mol. Microbiol. 61, 1196–210 (2006).
Paixão, L. et al. Host glycan sugar-specific pathways in streptococcus pneumonia: Galactose as a key sugar in colonisation and infection. PLoS One 10 (2015).
Bidossi, A. et al. A functional genomics approach to establish the complement of carbohydrate transporters in Streptococcus pneumoniae. PLoS One 7, e33320 (2012).
Terra, V. S., Homer, K. A., Rao, S. G., Andrew, P. W. & Yesilkaya, H. Characterization of novel beta-galactosidase activity that contributes to glycoprotein degradation and virulence in Streptococcus pneumoniae. Infect Immun 78, 348–357 (2010).
Melchiorsen, C. R. et al. Synthesis and posttranslational regulation of pyruvate formate-lyase in Lactococcus lactis . J. Bacteriol. 182, 4783–8 (2000).
Carvalho, S. M., Kloosterman, T. G., Kuipers, O. P. & Neves, A. R. CcpA ensures optimal metabolic fitness of streptococcus pneumoniae. PLoS One 6 (2011).
Iyer, R., Baliga, N. & Camilli, A. Catabolite control protein A (CcpA) contributes to virulence and regulation of sugar metabolism in Streptococcus pneumoniae . J. Bacteriol. 187, 8340–8349 (2005).
Lanie, J. A. et al. Genome sequence of Avery’s virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 189, 38–51 (2007).
van Opijnen, T., Bodi, K. L. & Camilli, A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–72 (2009).
Hueck, C., Kraus, A. & Hillen, W. Sequences of ccpA and two downstream Bacillus megaterium genes with homology to the motAB operon from Bacillus subtilis. Gene 143, 147–148 (1994).
Asanuma, N., Yoshii, T. & Hino, T. Molecular characterization of CcpA and involvement of this protein in transcriptiorial regulation of lactate dehydrogenase and pyruvate formate-lyase in the ruminal bacterium Streptococcus bovis. Appl. Environ. Microbiol. 70, 5244–5251 (2004).
Weickert, M. J. & Chambliss, G. H. Site-directed mutagenesis of a catabolite repression operator sequence in Bacillus subtilis . Proc. Natl. Acad. Sci. USA 87, 6238–6242 (1990).
Stülke, J. & Hillen, W. Regulation of carbon catabolism in Bacillus species. Annu. Rev. Microbiol. 54, 849–80 (2000).
Leibig, M. et al. Pyruvate formate lyase acts as a formate supplier for metabolic processes during anaerobiosis in Staphylococcus aureus . J. Bacteriol. 193, 952–62 (2011).
Härtel, T. et al. Characterization of central carbon metabolism of Streptococcus pneumoniae by isotopologue profiling. J. Biol. Chem. 287, 4260–74 (2012).
Vey, J. L. et al. Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc. Natl. Acad. Sci. USA 105, 16137–41 (2008).
Peng, Y., Veneziano, S. E., Gillispie, G. D. & Broderick, J. B. Pyruvate formate-lyase, evidence for an open conformation favored in the presence of its activating enzyme. J. Biol. Chem. 285, 27224–27231 (2010).
Aung-Hilbrich, L. M., Seidel, G., Wagner, A. & Hillen, W. Quantification of the influence of HPrSer46P on CcpA-cre interaction. J. Mol. Biol. 319, 77–85 (2002).
Giammarinaro, P. & Paton, J. C. Role of RegM, a homologue of the catabolite repressor protein CcpA, in the virulence of Streptococcus pneumoniae . Infect. Immun. 70, 5454–61 (2002).
Leonhartsberger, S., Korsa, I. & Böck, A. The molecular biology of formate metabolism in enterobacteria. J. Mol. Microbiol. Biotechnol. 4, 269–76 (2002).
Huang, Y., Suyemoto, M., Garner, C. D., Cicconi, K. M. & Altier, C. Formate acts as a diffusible signal to induce Salmonella invasion. J. Bacteriol. 190, 4233–41 (2008).
Moreno, M. S., Schneider, B. L., Maile, R. R., Weyler, W. & Saier, M. H. Catabolite repression mediated by the CcpA protein in Bacillus subtilis: novel modes of regulation revealed by whole-genome analyses. Mol. Microbiol. 39, 1366–81 (2001).
Blencke, H.-M. et al. Transcriptional profiling of gene expression in response to glucose in Bacillus subtilis: regulation of the central metabolic pathways. Metab. Eng. 5, 133–49 (2003).
Ludwig, H., Rebhan, N., Blencke, H.-M., Merzbacher, M. & Stülke, J. Control of the glycolytic gapA operon by the catabolite control protein A in Bacillus subtilis: a novel mechanism of CcpA-mediated regulation. Mol. Microbiol. 45, 543–53 (2002).
Hendriksen, W. T. et al. Site-specific contributions of glutamine-dependent regulator GlnR and GlnR-regulated genes to virulence of Streptococcus pneumoniae . Infect. Immun. 76, 1230–8 (2008).
Gerlini, A. et al. The role of host and microbial factors in the pathogenesis of pneumococcal bacteraemia arising from a single bacterial cell bottleneck. PLoS Pathog. 10, e1004026 (2014).
Hillerich, B. & Westpheling, J. A new GntR family transcriptional regulator in Streptomyces coelicolor is required for morphogenesis and antibiotic production and controls transcription of an ABC transporter in response to carbon source. J. Bacteriol. 188, 7477–87 (2006).
Crain, A. V. & Broderick, J. B. Pyruvate formate-lyase and its activation by pyruvate formate-lyase activating enzyme. J. Biol. Chem. 289, 5723–5729 (2014).
Sambrook, J., Fritsch, E. F. & Maniatis, T. In (Cold Spring Harbor Laboratory Press, 1989).
Shafeeq, S., Afzal, M., Henriques-Normark, B. & Kuipers, O. P. Transcriptional profiling of UlaR-regulated genes in Streptococcus pneumoniae . Genomics Data 4, 57–59 (2015).
Yesilkaya, H. Studies on the role of superoxide dismutase (SOD) in the virulence of Streptococcus pneumoniae and the effects of interferon gamma on sensitivity of phagocytes to the toxin pneumolysin. PhD Thesis. Department of Infection, Immunity and Inflammation, (University of Leicester, 1999).
Guiral, S. et al. Construction and evaluation of a chromosomal expression platform (CEP) for ectopic, maltose-driven gene expression in Streptococcus pneumoniae . Microbiology 152, 343–9 (2006).
Bricker, A. & Camilli, A. Transformation of a type 4 encapsulated strain of Streptococcus pneumoniae . FEMS Microbiol. Lett. 172, 131–135 (1999).
Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
Solovyev, V. & Salamov, A. In Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies (ed. Li, R. ) 61–78 (Nova Science Publishers, 2011).
Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 (1994).
Jing, D., Agnew, J., Patton, W., Hendrickson, J. & Beechem, J. A sensitive two-color electrophoretic mobility shift assay for detecting both nucleic acids and protein in gels. Proteomics 3, 1172–1180 (2003).
Halfmann, A., Hakenbeck, R. & Brückner, R. A new integrative reporter plasmid for Streptococcus pneumoniae . FEMS Microbiol. Lett. 268, 217–24 (2007).
Miller, J. H. In 352–355 (Cold Spring Harbor Laboratory, 1972).
Stewart, G. et al. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology 148, 3129–3138 (2002).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Hajaj, B. et al. Thiol peroxidase is an important component of Streptococcus pneumoniae in oxygenated environments. Infect. Immun. 80, 4333–4343 (2012).
Buis, J. M. & Broderick, J. B. Pyruvate formate-lyase activating enzyme: elucidation of a novel mechanism for glycyl radical formation. Arch. Biochem. Biophys. 433, 288–96 (2005).
Acknowledgements
This work was supported by the Higher Committee for Education Development in Iraq (HCED) as part of Firas A.Y Al-Bayati’s scholarship. We thank Dr. Marc Prudhomme (Toulouse, France) and Dr. Reinhold Bruckner (Kaiserslautern, Germany) for providing the plasmids pCEP and pPP1.
Author information
Authors and Affiliations
Contributions
H.Y. and P.W.A. conceived the experiments, F.A.Y.A., H.F.H.K. A.D., and S.S. conducted the experiments, all authors analysed the results, H.Y. and F.A.Y.A. wrote the manuscript, and all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Al-Bayati, F., Kahya, H., Damianou, A. et al. Pneumococcal galactose catabolism is controlled by multiple regulators acting on pyruvate formate lyase. Sci Rep 7, 43587 (2017). https://doi.org/10.1038/srep43587
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43587
This article is cited by
-
Klebsiella pneumoniae peptide hijacks a Streptococcus pneumoniae permease to subvert pneumococcal growth and colonization
Communications Biology (2024)
-
Metagenomic sequencing reveals altered gut microbial compositions and gene functions in patients with non-segmental vitiligo
BMC Microbiology (2023)
-
Rgg-Shp regulators are important for pneumococcal colonization and invasion through their effect on mannose utilization and capsule synthesis
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
