
Abstract
Abnormally hyperphosphorylated tau aggregated as intraneuronal neurofibrillary tangles is one of the two neuropathological hallmarks of Alzheimer’s disease (AD). The majority of AD cases are sporadic with numerous environmental, biological and genetic risks factors. Interestingly, insulin dysfunction and hyperglycaemia are both risk factors for sporadic AD. However, how hyperglycaemia and insulin dysfunction affect tau pathology, is not well understood. In this study, we examined the effects of insulin deficiency on tau pathology in transgenic hTau mice by injecting different doses of streptozotocin (STZ), a toxin that destroys insulin-producing cells in the pancreas. One high dose of STZ resulted in marked diabetes, and five low doses led to a milder diabetes. Both groups exhibited brain tau hyperphosphorylation but no increased aggregation. Tau hyperphosphorylation correlated with inhibition of Protein Phosphatase 2A (PP2A), the main tau phosphatase. Interestingly, insulin injection 30 minutes before sacrifice partially restored tau phosphorylation to control levels in both STZ-injected groups. Our results confirm a link between insulin homeostasis and tau phosphorylation, which could explain, at least in part, a higher incidence of AD in diabetic patients.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most widespread neurodegenerative disease affecting mainly elderly. Sporadic, late onset AD is the most common (99%), while genetic forms occurring earlier in life represent less than 1% of AD patients. The exact causes for sporadic AD are not fully understood but epidemiological studies have identified numerous environmental, biological and genetic risks factors, with aging being the most important1. Interestingly, there is increasing evidence suggesting that insulin dysfunction is an important risk factor for sporadic AD2,3. Studies have reported altered glucose metabolism and abnormalities in insulin and insulin receptors levels in brains of AD patients4,5,6,7,8, and a higher incidence of AD has been detected in patients with diabetes mellitus (DM)9,10,11,12. Moreover, the 2014 Alzheimer’s Association report13 reported 29% of co-morbidity between DM and AD.
Abnormally hyperphosphorylated tau aggregated as intraneuronal neurofibrillary tangles is one of the two neuropathological hallmarks of AD14 along with extracellular accumulation of senile plaques composed of amyloid-β peptide (Aβ)15. Numerous studies have demonstrated that insulin is able to modulate tau phosphorylation both in vitro and in vivo (see ref. 16 for review). This impact of insulin on tau could explain, at least in part, co-morbidity between DM and AD since tau hyperphosphorylation has been shown to induce tau pathology17, which correlates with the degree of cognitive impairment in Alzheimer’s disease18,19,20,21.
Previous studies have evaluated tau phosphorylation in wild type rodents with insulin-dependent DM induced by intra-peritoneal injection of streptozotocin toxin (STZ)9,22,23,24,25. However, the mechanisms underlying this hyperphosphorylation are controversial; some studies have implicated Glycogen Synthase Kinase-3β (GSK-3β activation24, while others Protein Phosphatase 2A (PP2A) inhibition22,23 or both25. These differences in mechanisms might come from the doses of STZ used. Interestingly, whatever the dose of STZ used, insulin injection restores physiological tau phosphorylation in WT mice22,23, but it is unknown whether it could rescue tau pathology.
The goals of our study were: (i) to determine whether a single high dose or multiple low doses of STZ induce tau hyperphosphorylation by different mechanisms; and (ii) to determine whether insulin can rescue both tau hyperphosphorylation and pathology in a mouse model of tauopathy.
Therefore, we examined the effects of insulin deficiency on tau pathology in transgenic hTau mice, expressing human tau protein without mutations (tau mutations cause FTDP-17, not AD). We compared two protocol of Type 1 DM (T1DM) induction: one high dose of STZ resulting in marked diabetes (STZ HD group) or 5 low doses leading to a milder T1DM (STZ LD group). Both groups exhibited tau hyperphosphorylation without aggregation in the brain. Tau hyperphosphorylation correlated with inhibition of PP2A, the main tau phosphatase. Some mice exhibited more widespread tau hyperphosphorylation due to diabetes-induced hypothermia. Interestingly, insulin injection 30 minutes before sacrifice partially restored physiological tau phosphorylation to control levels in both STZ-injected groups. Our results confirmed a link between insulin homeostasis and tau phosphorylation, which could, at least, contribute to co-morbidity between diabetes and AD.
Results
Reminders (Fig. 1)

Experimental design of the study.
STZ LD group: mice injected with 5 low doses of STZ
STZ HD group: mice injected with 1 high dose of STZ
STZ LD Ins group: mice injected with 5 low doses of STZ + 1 injection of insulin 30 min before sacrifice
STZ HD Ins group: mice injected with 1 high dose of STZ + 1 injection of insulin 30 min before sacrifice
STZ HD Hypo group: mice injected with 1 high dose of STZ that developed hypothermia.
Physiological parameters of STZ injected hTau mice
We first measured key physiological parameters, including weight, glycaemia, insulinemia and body temperature (Table 1). One high dose of STZ induced significant weight loss of mice (groups: STZ HD, STZ HD Insulin, and STZ HD Hypothermic) while five low dose injections did not. As expected, STZ injected mice (high dose and low dose injections groups) exhibited hyperglycaemia along with obvious hypoinsulinemia in comparison with control mice. Physiological glycaemia was restored in mice injected with insulin 30 minutes before sacrifice. Interestingly, 7 mice injected with a high dose of STZ exhibited a significant drop in body temperature (34.8 ± 0.6 vs. 36.9 ± 0.5 °C) and were analyzed as a distinct group of STZ HD-injected mice (STZ HD Hypo) as hypothermia can impact on tau phosphorylation26.
Tau hyperphosphorylation in STZ injected hTau mice
We tested whether type 1 diabetes induced tau phosphorylation in the hippocampus, using several anti-phospho-tau antibodies (Table S1). The specificity of the tau antibodies used in this study has been extensively characterized in our laboratory27. We first evaluated whether STZ low doses injections, inducing mild T1DM, led to tau hyperphosphorylation (STZ LD group). We observed significant tau hyperphosphorylation at AT8 (pSer202/pThr205), PHF-1 (pSer396/Ser404) and pThr205 epitopes (Fig. 2, panels 2, 3, 5). We also noticed a significant decrease of Tau-1 signal, which recognize non-phosphorylated tau at Ser195/Ser198/Ser199/Ser202 (Fig. 2, panel 6). A decrease of Tau-1 signal reflects tau hyperphosphorylation. We next examined tau phosphorylation in a group with marked T1DM, due to a single high dose injection of STZ (STZ HD group). This group also exhibited tau hyperphosphorylation according to Tau-1, AT8 and PHF1 signals but not to pT205 (Fig. 2, panels 5 & 6). Seven mice of the STZ HD group developed hypothermia following STZ injection (STZ HD Hypo group). As mentioned before, we separated these mice in a distinct group since hypothermia induces tau hyperphosphorylation26. Tau hyperphosphorylation on all tau phospho-epitopes tested (CP13, AT8, pT205, AT180, PHF-1, and, Tau-1) was observed in this group in comparison with controls (Fig. 2, panels 1 to 6) by western blot. We confirmed tau hyperphosphorylation in high dose groups by immunohistochemistry (Fig. 3). Total tau and β-actin levels remained unchanged (Fig. 2, panels 7 & 8).

Hippocampal proteins from 7–9 months-old mice were probed with the following antibodies: 1. CP13, 2. AT8, 3. pT205, 4. AT180, 5. PHF1, 6. Tau-1, 7. Total tau and 8. β-actin (loading control). Two lanes from representative immunoblots are displayed for each condition. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Quantifications of tau phosphoepitopes were normalized to total tau levels. Total tau quantification was normalized to β-actin. Results are expressed as percentage of control group (CTL). Data are mean ± SD. Asterisks indicate significant differences, with *p < 0.05, **p < 0.01 and ***p < 0.001. Data from AT8, pT205, AT180, PHF1, Tau-1 and total tau following a normal distribution were analyzed with one-way ANOVA of variance followed by a Bonferroni’s post hoc test, while Kruskal-Wallis test followed by a Dunn’s post hoc test was used for CP13 and β-actin analysis since data deviated from a normal distribution.

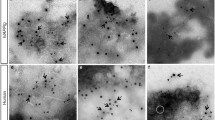
Regional anatomical localization of phosphorylated tau. Immunolabelling of hippocampal sagittal sections (CA1 region) are shown with AT8 for the following conditions: Control group, STZ HD group, STZ HD + Insulin group and hypothermic STZ-injected mice. Red scale bar: 20μm.
Impact of insulin injection on tau phosphorylation in STZ injected hTau mice
Since STZ injection lead to hypoinsulinaemia resulting in hyperglycaemia, we next tested whether insulin injection, restoring physiological insulinaemia and glycaemia, impacts on tau hyperphosphorylation in STZ injected mice (STZ LD Ins and STZ HD Ins groups). Interestingly, 30 minutes after insulin injection, physiological tau phosphorylation comparable to control mice was partially restored in both STZ LD Ins and STZ HD Ins groups on most phospho-epitopes exhibiting hyperphosphorylation (Fig. 2, panels 1 to 6). However, statistical significance between STZ-injected mice treated with or without insulin injection was observed only at Tau-1 phosphoepitope (STZ HD group vs. STZ HD Ins group) (Fig. 2.6). These results suggest that tau hyperphosphorylation in STZ-injected mice follows, at least in part, insulin deficiency in these mice. Total tau and β-actin levels remained unchanged (Fig. 2 panels 7 & 8). We confirmed the effect of insulin on tau hyperphosphorylation in STZ high dose groups by immunohistochemistry (Fig. 3). Insulin injection in control mice did not change tau phosphorylation (Fig. S1).
Tau solubility in STZ injected hTau mice
As tau hyperphosphorylation has been shown to induce insoluble tau aggregation and tangle formation in vitro17, we analyzed insoluble and soluble fraction of tau in our different groups (Fig. 4). However, neither STZ nor insulin injections altered tau solubility (Fig. 4A,B).

A. Soluble tau: Cortical soluble tau were probed with the following antibodies: 1. Total tau and 2. Human tau. B. Insoluble tau: Cortical aggregated of tau protein were probed with the following antibodies: 1. Total tau and 2. Human tau. Two lanes from representative immunoblots are displayed for each condition. There was no significant difference between STZ-injected mice and their controls (quantification data not shown). Data from insoluble total and human tau and soluble human tau following a normal distribution were analyzed with one-way ANOVA of variance followed by a Bonferroni’s post hoc test, while Kruskal-Wallis test followed by a Dunn’s post hoc test was used for soluble total tau analysis since data deviated from a normal distribution.
Tau kinases in STZ injected hTau mice
We next evaluated the mechanisms underlying tau hyperphosphorylation in STZ-injected mice. Tau hyperphosphorylation can be due to either activation of tau kinases, or inhibition of tau phosphatases. To test these two possibilities, we first examined a panel of major tau kinases activation/inhibition and expression (Table S1), including Ca2 + calmodulin-dependent protein kinase II (CamKII), c-Jun N-terminal kinase (JNK), P38 mitogen-activated protein kinase (P38), glycogen synthase kinase-3β (GSK-3β), mitogen activated protein kinase/extracellular signal-regulated kinase (ERK) and cyclin-dependent kinase 5 (cdk5) (Fig. 5). No change in tau kinases was observed in STZ-injected mice independently of insulin injection or hypothermia (Fig. 5, panels 2, 6, 8, 10, 11, 12). However, there was an increase of GSK-3β phosphorylation at Ser9 (indicating inhibition) in STZ HD and STZ HD hypothermic groups when compared to control mice (Fig. 5 panel 5), confirming previous reports22,23. Physiological Ser9 phosphorylation of GSK-3β comparable to control mice was restored after insulin injection (Fig. 5, panel 5). GSK-3β inhibition was, however, inconsistent with the elevation of tau phosphorylation in STZ-injected mice.

Hippocampal proteins from 7–9 months-old mice were probed with the following antibodies: 1. pCamKII, 2. CamKII, 3. pJNK, 4. JNK, 5. pS9-GSK3β, 6. GSK3β, pP38, 8. P38, 9. pERK, 10. ERK, 11. cdk5, 12. P35. Two lanes from representative immunoblots are displayed for each condition. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Quantifications of phosphoepitopes were normalized to total form of respective protein. Total protein expression was quantified vs. β-actin. Except for pS9-GSK3β, there was no difference between STZ-injected mice and their controls (quantification data not shown). Results of pS9-GSK3β quantification are expressed as percentage of control group (CTL). Data are mean ± SD. Asterisks indicate significant differences from controls, with *p < 0.05 and **p < 0.01. Data from CamKII, pJNK, JNK, pERK, ERK, cdk5 and p35 following a normal distribution were analyzed with one-way ANOVA of variance followed by a Bonferroni’s post hoc test, while Kruskal-Wallis test followed by a Dunn’s post hoc test was used for pCaMKII, pP38, P38, pS9-GSK3β GSK3β analysis since data deviated from a normal distribution.
Tau phosphatases in STZ injected hTau mice
As our prior results failed to explain tau hyperphosphorylation, we next turned to tau Ser/Thr protein phosphatases (PP) in the hippocampus, including PP1, PP2A, PP2B (calcineurin), and PP528. All of them are highly expressed in the mammalian brain29. Biochemical studies have previously demonstrated that all these PP can dephosphorylate tau in vitro30. Therefore, we examined the profiles of these four PP using specific antibodies (Table S1). We observed no change in PP1, PP2B and PP5 expression in STZ injected hTau mice (Fig. 6, panels 1, 7, 8). We explored further the PP2A components since STZ injection has previously been show to reduce PP2A activity in the brain22,23. We thus investigated the levels of PP2A subunits: PP2A-A, PP2A-B (α and β, PP2A-C expression levels, as well as the demethylated PP2A-C (inactivated PP2A) in STZ injected hTau mice. No change was observed on PP2A subunits expression as well as its demethylation (Fig. 6, panels 2 to 6). We next analyzed PP2A activity with a commercial assay kit. After immunoprecipitation of PP2A from the samples, this assay measures the phosphates released from a synthetic phosphopeptide, which corresponds to the activity of PP2A in each sample. This assay revealed a decrease of PP2A activity in all STZ-injected groups (Fig. 6, panel 11) because fewer phosphates were released in these groups, suggesting that tau hyperphosphorylation reported in STZ-injected mice could be a consequence of PP2A inhibition. On the other hand, insulin injection did not restore physiological PP2A activity in STZ-injected mice. These data suggest that the beneficial effect of insulin injection on tau hyperphosphorylation might go through a different pathway than restoring PP2A activity.

Hippocampal proteins from 7–9 months-old mice were probed with the following antibodies: 1. PP1, 2. PP2A-A, 3. PP2A-Bα, 4. PP2A-Bβ, 5. demPP2A-C, 6. PP2A-C, 7. PP2B, 8. PP5, 9. pPTEN, 10. PTEN. Two lanes from representative immunoblots are displayed for each condition. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Quantification of demethylated or phosphorylated phosphatase was normalized to total phosphatase. Total protein expression was quantified vs. β-actin. There was no significant difference between STZ-injected mice and their controls (quantification data not shown). 11. PP2A activity: Results are expressed as percentage of control group (CTL). Data are mean ± SD. Asterisks indicate significant differences from controls, with **p < 0.01 and ***p < 0.001. Data from PP1, PP2A-Bβ, demPP2A-C, PP2A-C, pPTEN and PP2A activity following a normal distribution were analyzed with one-way ANOVA of variance followed by a Bonferroni’s post hoc test, while Kruskal-Wallis test followed by a Dunn’s post hoc test was used for PP2A-A, PP2A-Bα, PP2B, PP5 and PTEN analysis since data deviated from a normal distribution.
Finally, we assessed PTEN expression and phosphorylation since PTEN phosphatase as been previously shown to impact on insulin signaling and resistance31 and affect tau phosphorylation, aggregation, and binding to microtubules through PIP3 signalling pathway32. We observed no change on both PTEN expression and phosphorylation at serine 380 in STZ-injected mice compared to controls (Fig. 6, panels 9 & 10).
Brain insulin signaling in STZ injected hTau mice
We also evaluated brain insulin signalling since impaired central insulin signaling can induce tau hyperphosphorylation16,33. We investigated the activation state of three keys protein of insulin signaling: insulin receptor (IR), Akt and PI3K kinases, as well as IGF1 receptor, since IGF1-signaling pathway can be activated by insulin. Interestingly, Akt is also a tau kinase34. No change was observed for these four proteins. However, AKT and insulin receptors had increased phosphorylation in STZ HD and STZ HD Hypothermic groups compared to controls, reflecting their activation (Fig. 7, panels 1 & 7). Increased phosphorylation was also found on PI3K in STZ HD hypothermic group (Fig. 7, panel 5). Surprisingly, this hippocampal insulin pathway activation in STZ HD groups disappeared after i.p. insulin injection (Fig. 7.1,7.7). These results suggest a deregulation of central insulin signaling pathway in peripheral hypoinsulinemic conditions.

Hippocampal proteins from 7–9 months-old mice were probed with the following antibodies: 1. pIR, 2. IR, 3. pIGF1 Rc, 4. IGF1 Rc, 5. pPI3K, 6. PI3K, 7. pAKT, 8. AKT. Two lanes from representative immunoblots are displayed for each condition. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Quantifications of phosphoepitopes were normalized to total form of respective protein. Total protein expression was quantified vs. β-actin. Results are expressed as percentage of control group (CTL). Data are mean ± SD. Asterisks indicate significant differences from controls, with *p < 0.05 and **p < 0.01. Data from pIGF-1 Rc, IGF-1 Rc, pPI3K, PI3K and AKT following a normal distribution were analyzed with one-way ANOVA of variance followed by a Bonferroni’s post hoc test, while Kruskal-Wallis test followed by a Dunn’s post hoc test was used for pIR, IR and pAKT analysis since data deviated from a normal distribution.
Synaptic markers in STZ-injected hTau mice
We also tested some synaptic markers to assess synaptic integrity in STZ-injected mice, since synapse loss is an early event in AD35. As tau hyperphosphorylation is thought to trigger dendritic atrophy and/or synaptic alterations36, we evaluated pre- and post- synaptic connections by measuring synaptic proteins such as drebrin, synaptophysin, SNAP25 and PSD95 (Fig. 8). Interestingly, STZ injections impacted on both pre- and post-synaptic markers as there was a decrease in drebrin, and a tendency to decrease in SNAP25 (both STZ LD and STZ HD groups vs. CTL group) (Fig. 8, panels 1 & 3). Insulin injection restored drebrin reduction compared to control (Fig. 8, panel 1), while it increased SNAP25 when compared to STZ-injected groups (Fig. 8, panel 3). However, synaptophysin and PSD95 remained however unchanged regardless of mice treatment (Fig. 8, panel 2 and 4). These results suggest that T1DM could promote synaptic impairments, which might be restored by insulin injection.

Hippocampal proteins from 7–9 months-old mice were probed with the following antibodies: 1. Drebrin, 2. Synaptophysin, 3. SNAP25, 4. PSD95. Two lanes from representative immunoblots are displayed for each condition. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Total protein expression was quantified vs. β-actin. Results are expressed as percentage of control group (CTL). Data are mean ± SD. Asterisks indicate significant differences, with *p < 0.05 and **p < 0.01. Data from this figure following a normal distribution were analyzed with one-way ANOVA of variance followed by a Bonferroni’s post hoc test.
Discussion
In this study, we addressed whether insulin deprivation would induce tau hyperphosphorylation and aggregation by administration of different doses of STZ in hTau mice, a humanized tau transgenic mouse. We report that hypoinsulinemia induced tau hyperphosphorylation without affecting tau aggregation. Our data indicated that tau hyperphosphorylation following STZ injection is likely a consequence of PP2A inhibition. Interestingly, insulin injection restoring physiological glycaemia partially restored normal tau phosphorylation to control levels in STZ injected mice.
Previous studies have examined the impact of STZ injection resulting in hypoinsulinemia on tau phosphorylation in wild-type rodents to explore the link between AD and insulin dysfunction9,22,23,24,25. Most used a single high dose of STZ injection to quickly induce a marked T1DM in wild-type rodents, and reported tau hyperphosphorylation at several phospho-epitopes such as AT8, PHF1 and Tau-19,22,23,25,37. Similarly, we observed tau hyperphosphorylation in STZ HD group at these epitopes. AT8 (Ser202/Thr205) is an antibody routinely used for the staging of AD-associated neurofibrillary pathology38, and the phosphorylation sites detected by PHF-1 (Ser396/Ser404) are associated with neurofibrillary tangle pathology39. Unfortunately, few of these studies monitored mice body temperature after STZ injection, which is important since STZ injection can induce hypothermia resulting in massive tau hyperphosphorylation22. Therefore, it is difficult to conclude if the tau hyperphosphorylation observed in studies without temperature monitoring is the result of T1DM per se or consequent hypothermia or both. To avoid this artefact, mice developing hypothermia after STZ injection formed a distinct group from the other mice. As expected, these mice displayed extensive tau hyperphosphorylation on all six epitopes tested here, while tau was hyperphosphorylated at only three epitopes in normothermic mice, underlying the importance of recording mice body temperature in diabetic models.
It is important to examine tau aggregation after diabetes induction, since hyperphosphorylation has been shown to induce the formation of insoluble tau aggregates and neurofibrillary tangle in vitro17. Transgenic mice overexpressing human tau are the tool of choice since murine tau does not readily aggregate; surprisingly, only one study has used such mice models. Ke et al. observed increased tau aggregation in pR5 mice overexpressing P301L mutant human tau after T1DM induction with 1 high dose of STZ (200 mg/kg)37. Interestingly, in hTau mice (overexpressing non-mutant human tau), we did not detect any change in tau solubility after STZ injection despite marked hyperphosphorylation. The differences between the two lines could be due to the fact that P301L tau mutant is more prone to aggregation than non-mutant tau. Indeed, the P301L mutation has been shown to accelerate the formation of paired helical filaments and promote β-sheet formation40,41,42. While hTau mice do develop tau aggregates, they do it slower than pR5 mice. Therefore, the lack of mutation on tau in our model could account for the lack of exacerbation observed here. However, this does not mean absence of toxic tau species. Indeed, some forms of hyperphosphorylated tau have been shown to have cytotoxic proprieties43, and the synaptic deficits present in many models of tauopathies are often observed before tau aggregation and tangle formation44,45,46. Here, tau hyperphosphorylation might have contributed to synaptic deficits, along with the effects of diabetes per se47,48.
Other studies used multiple lower doses of STZ to induce a milder T1DM9,24,49,50. The results from these multiple doses of STZ are more variable. For example, Kim et al. did not report change in tau phosphorylation after 5 low doses (5 × 50 mg/kg) of STZ in WT mice brains9, while Jolivalt et al. observed tau hyperphosphorylation in brain of mice after 2 injections of low dose (2 × 90 mg/kg) of STZ24. In our study, we observed a roughly equivalent hyperphosphorylation of tau in mice injected with one high dose or five low doses of STZ, suggesting that the important factor is the diabetic state.
At the mechanistic level, the results diverge in the literature to explain tau hyperphosphorylation after STZ injection. Some studies have linked tau hyperphosphorylation to an increased activity of GSK-3β, a major tau kinase24,25, while others have suggested inhibition of PP2A as the main mechanism22,23,25. In STZ-injected hTau mice, we confirmed PP2A inhibition in both STZ LD and STZ HD groups, suggesting that it underlies tau hyperphosphorylation in these groups. Moreover, tau hyperphosphorylation along with inhibition of PP2A was observed in NOD mice, a spontaneous mouse model of T1DM51. At the kinase level, the only change observed was an increase of GSK-3β inhibitory phosphorylation at Ser9 HD, a result opposite to what was observed by some studies24,25, but in line with other observations (for review ref. 16). This result might be explained by the decrease in PP2A activity, since the inhibition of PP2A increases GSK-3β Ser9 phosphorylation and inhibits its activity52,53. These results are in line with others demonstrating that when PP2A is inhibited, it is the dominant factor inducing tau hyperphosphorylation, overriding the inhibition of key tau kinases54,55. While it is difficult to give a definitive mechanistic explanation to tau hyperphosphorylation in STZ-injected mice because of discrepancies in previous studies, our data support the PP2A inhibition hypothesis.
Surprisingly, only two studies assessed central insulin signaling in STZ-injected rodents24,25, despite the fact that impaired central insulin signalling can induce tau hyperphosphorylation16,33. Both reported a decrease of AKT phosphorylation, a key protein of insulin signaling in STZ injected rodent24,25. Jolivalt et al. also observed a reduction of insulin receptor phosphorylation at Ser972. In our study, we observed opposite results: increased insulin receptor and AKT phosphorylation in STZ-injected mice. These results are in line with the inhibitory phosphorylation of GSK-3β at Ser9 observed in STZ-injected hTau mice. Interestingly, a similar AKT activation was observed in NOD mice, a spontaneous mouse model of T1DM51. These results suggest a deregulation of brain insulin signalling in T1DM mice models.
Finally, we assessed whether insulin injection can restore physiological tau phosphorylation in STZ-injected mice. Previous studies reported that insulin treatment is sufficient to abolish tau hyperphosphorylation in STZ-injected rodents22,23,25. Tau hyperphosphorylation was attenuated in STZ-injected mice after peripheral insulin injection 30 minutes before sacrifice, confirming a link between insulin homeostasis and tau phosphorylation. However, insulin injection significantly decreased tau hyperhosphorylation only at one phosphoepitope compared to STZ-injected mice without insulin injection 30 minutes before sacrifice. We could hypothesized that one acute insulin supplementation is not enough to totally restore physiological tau phosphorylation in mice deprived of insulin for 4 weeks. Indeed, previous studies observed a rescue of tau phosphorylation after daily insulin administration in STZ-injected rodents22,25. Interestingly, insulin did not restore physiological PP2A activity, suggesting the beneficial effect of insulin on tau hyperphosphorylation goes through different pathway. Interestingly, GSK-3β Ser9 levels in STZ-injected mice were restored to normal levels after insulin injection, alleviating its inhibition. Although it could not directly explain decrease of tau hyperphosphorylation after insulin injection, restoring GSK-3β indirectly contribute to the reduction of tau hyperphosphorylation by activating PP1. Indeed, GSK-3β is able to promote PP1 activity by phosphorylation of PP1 Inhibitor-256,57. Further studies are needed to confirm this hypothesis. Insulin administration also increased synaptic markers reduced by STZ injection. This effect of insulin on synaptic markers as previously reported58,59,60 and insulin is known to regulate synapse number and dendritic plasticity60,61. Our data support that brain insulin dysregulation impacts on synaptic alterations and that tau hyperphosphorylation could contribute to this phenomenon.
To conclude, our study provides important data on the impact of insulin deprivation on tau phosphorylation and implicates the deregulation of PP2A activity (Fig. 9). Although the link between T1DM and Alzheimer’s disease is not as clearly established in humans as it is for T2DM10,11,12, these data provide valuable information by evaluating the effects of hyperglycemia and hypoinsulinemia seen in advanced stages of T2DM on AD pathology. Indeed, the patients in advanced stage of T2DM have higher risk of AD11. Therefore, Insulin deregulation resulting in decrease of PP2A activity and tau hyperphosphorylation might explain the higher tau phosphorylation and pathology found in T2DM patients and could underlie, at least in part, the high risk od AD in DM patients. These observations highlight the importance of glycaemia and insulinemia control in diabetic patients.

STZ injection induces insulin deprivation by destroying insulin-producing cells in pancreas. The resulting T1DM lead to PP2A inhibition in the brain, which induce tau hyperphosphorylation. Insulin injection 30 minutes before sacrifice partially restores physiological phosphorylation of tau comparable to non-T1DM mice without rescue of PP2A activity, suggesting involvement of different pathway.
Materials and Methods
Animals
The founders of our hTau mice colony were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) (B6.Cg-Mapttm1 (EGFP) Klt Tg(MAPT)8cPdav/J) on C57BL/6J background. They were initially generated by the group of Dr Peter Davies62 by crossing tau knockout mice that have a targeted disruption of exon one of murine tau63, with 8c mice that express a tau transgene containing the coding intronic and regulatory regions of the human gene64. These mice develop tau pathology in a time course and distribution comparable to that occurring in the early stages on human AD62.
7-9 months-old mice of either sex were maintained in a temperature-controlled room (~23 °C) with a light/dark cycle of 12/12 h. Experiments were performed during the light period. All animals had access to food and water ad libitum. Animals were handled according to procedures approved by the Comité de Protection des Animaux du CHUL under the guidelines of the Canadian Council on Animal Care.
Streptozotocin and insulin injections
STZ
Nonfasted animals were injected following two different protocols from the Animal Models for Diabetic Complications Consortium (www.diacomp.org). Two groups of mice received one intraperitoneal injection of 50 mg/kg of STZ (2-deoxy-2-(3-(methyl-3-nitrosoureido)-D-glucopyranose); Sigma, St. Louis, MO) per day for 5 consecutive days according with Multiple Low-Dose STZ induction Protocol (STZ LD groups). This protocol induces a mild and progressive T1DM that can last for many months without overt toxicity for the mice. Two other groups received only one intraperitoneal injection of 150 mg/kg of STZ (Sigma, St. Louis, MO) according with the High-Dose STZ induction Protocol (STZ HD groups). This protocol quickly induces a marked T1DM. STZ was freshly dissolved in 0.05 M citrate buffer, pH 4.5. Vehicle-injected mice were used as (Fig. 1) controls.
Insulin
Thirty minutes before mice sacrifice, half of each STZ group (STZ-HD and STZ-LD) received insulin injection (4 IU/kg) to reverse the hypoinsulinemic effects of STZ. The second half of each group and control group received injection of 0.9% saline as vehicle. For control purpose, the same insulin and vehicle injections was done in non-diabetic hTau mice (n = 8 for each group) to evaluate the specific effect of insulin on tau phosphorylation.
Physiological parameters
All mice were weighed at the time of sacrifice, and body temperature was monitored using a rectal probe (Thermalert TH-5, Physitemp, Clifton, NJ). Fasting blood glucose and insulin were measured using a glucometer with reagent strips (ACCU-CHEK ® Aviva Nano; Roche Diagnostics GmbH, Mannheim, Germany) and a sandwich enzyme immunoassay (Ultrasensitive Mouse Insulin ELISA, Mercodia, Sweden) respectively. Weight, temperature fasting blood glucose and insulin of mice group are reported in Table 1.
Protein extraction
Four weeks after STZ injections, the mice were killed by decapitation without anaesthesia, as anaesthesia can lead to hypothermia-induced tau hyperphosphorylation65. Brains were immediately removed and the tissues dissected on ice, frozen on dry ice, and kept at −80 °C until they were processed as described66. Briefly, dissected brain structures (hippocampus and cortex) were homogenized, without thawing, in 5 times volume/weight of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 1% NP-40, 150 mM NaCl, 0.25% Na-deoxycholate, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 10 μl/ml of Proteases Inhibitors Cocktail (P8340, Sigma-Aldrich, St. Louis, MO)), using a mechanical homogenizer (TH, Omni International, Marietta, GA). Samples were then centrifuged for 20 min at 20,000 g at 4 °C. The supernatant was recovered, diluted in sample buffer (NuPAGE LDS; Invitrogen, Carlsbad, CA) containing 5% of 2-β-mercapto-ethanol, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 10 μl/ml of Proteases Inhibitors Cocktail (P8340; Sigma-Aldrich), boiled for 5 min. and kept at −20 °C.
Purification of aggregates
Tau aggregates were extracted according to our protocol previously used to isolate tau aggregates in mouse models of tauopathies67. This procedure uses 1% sarkosyl and is derived from one used to isolate tau aggregates from the brains of AD68.
Briefly, the RIPA supernatant was adjusted to 1% sarkosyl (N-lauroylsarcosine), incubated for 30 min at room temperature with constant shaking, and centrifuged at 100,000 × g for 1 hour at 20 °C. The pellet containing sarkosyl-insoluble aggregated (SP fraction) was resuspended and diluted in Sample buffer (NuPAGE LDS) containing 5% of 2-β-mercapto-ethanol, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 10 μl/ml of Proteases Inhibitors Cocktail (P8340, Sigma-Aldrich), boiled for 5 min, and kept at −20 °C.
For heat stable soluble tau, the RIPA supernatant was boiled for 5 min and centrifuged at 20,000 × g for 20 min. The supernatant was recovered, diluted in sample buffer (NuPAGE LDS; Invitrogen, Carlsbad, CA) containing 5% of 2-β-mercapto-ethanol, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 10 μl/ml of Proteases Inhibitors Cocktail (P8340; Sigma-Aldrich) and boiled for 5 min.
Western-blot analysis
SDS-PAGE and Western blot analysis were done as previously described27. All antibodies used in this study are listed in Table 1. Depending on the antibody used, 5–15 μg of brain protein were analyzed. Brain homogenates were separated on a SDS-10% polyacrylamide gel and then transferred onto nitrocellulose membranes (Amersham Biosciences, Pittsburgh, PA). Non-specific binding sites were blocked with 5% nonfat dry milk in Phosphate-buffered saline containing 0.1% Tween 20 (PBS-T) for one hour at room temperature and were afterwards incubated overnight at 4 °C with primary antibodies directed against the specific antibody. The following day, membranes were washed 3 times in PBS-T and then incubated for 1 hour at room temperature with the corresponding secondary antibody in 5% nonfat dry milk in PBS-T, and the immunoreactive signal intensity was visualized by enhanced chemiluminescence (ECL Plus, GE Healthcare Biosciences, Piscataway, NJ). Some anti-tau monoclonal antibodies were revealed with anti-LightChain secondary antibody according to our previous study27. Immunoreactive bands were visualized using ImageQuant LAS 4000 imaging system (Fujifilm USA, Valhalla, NY) and densitometric analyses were performed with Image Gauge analysis software (Fujifilm USA, Valhalla, NY).
Immunohistochemistry
Tissue fixation was done according to the “Cold Bouin’s method”69. Briefly, mice were sacrificed by decapitation, the brain was quickly removed and immersed in ice-cold Bouin’s solution (saturated picric acid, formalin, acetic acid at 15:5:1) for 24 h and then embedded in paraffin blocks. Five μm thick sections were processed for immunostaining analyses. Prior to immunostaining, slides were deparaffinized with Citrisolv for 2 × 10 minutes. The sections were subsequently rehydrated by immersing them in graded ethanol solutions (100%, 95%, 70% and 50%) during 5 minutes for each condition. After further rinses in water, deparaffinized and hydrated sections were incubated for 1 hour in citrate buffer previously warmed at 80°C for enhancement of the immunoreactivity. The slides were next washed in PBS 0, 1 M (pH 7.4) for 3 × 10 minutes, then incubated in a solution of PBS containing 3% of peroxide for 30 minutes, and washed again in PBS for 3 × 10 minutes. The specimens were blocked for 30 minutes with a solution of PBS containing 5% NGS and 1% of Triton X-100 10%. The sections were next incubated overnight in primary antibodies diluted in 5% NGS in PBS 0.1 M (pH 7.4) containing 1% of Triton X-100 10% at 4°C. Bound antibodies were visualized with biotinylated anti-rabbit or anti-mouse IgG (Vector Laboratories Inc., Burlingame, CA). Immunolabeled tissues were observed under a Carl Zeiss AxioCamIC (Carl Zeiss, Jena, Germany) microscope.
PP2A activity assay
Brain PP2A activity was evaluated using a kit from R&D Systems according to the manufacturer’s instructions (Human/Mouse/Rat Active PP2A DuoSet IC, R&D Systems, Minneapolis, MN, USA). Briefly, after immunoprecipitation of PP2A for each sample, PP2A activity was determined by measuring the released phosphate from a chemically synthetized phosphopeptide after 30 minutes of incubation at 37°C. The level of phosphate release corresponds to PP2A activity and was determined by the absorbance of malachite green-phosphate at 620 nm.
Statistical analysis
Statistical analyses were performed with GraphPad Prism software 4.0 (Graphpad Software, La Jolla, CA). We used one-way ANOVA of variance followed by a Bonferroni’s post hoc test, if the effects were significant (p < 0.05) as assessed by ANOVA under normal distribution. If samples deviated from a normal distribution, the statistical analysis was performed using Kruskal-Wallis test followed by a Dunn’s post hoc test, when indicated (p < 0.05). For figure S1, statistical analyses were performed using unpaired t-tests. *, **, and *** symbols indicate significant differences vs. control with p < 0.05, p < 0.01, p < 0.001, respectively. Quantitative data were presented as mean ± SD.
Additional Information
How to cite this article: Gratuze, M. et al. Insulin deprivation induces PP2A inhibition and Tau hyperphosphorylation in hTau mice, a model of Alzheimer’s disease-like tau pathology. Sci. Rep. 7, 46359; doi: 10.1038/srep46359 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Ritchie, K. & Lovestone, S. The dementias. Lancet 360, 1759–1766 (2002).
De Felice, F. G. Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. The Journal of clinical investigation 123, 531–539, doi: 10.1172/JCI64595 (2013).
Craft, S. & Watson, G. S. Insulin and neurodegenerative disease: shared and specific mechanisms. The Lancet. Neurology 3, 169–178, doi: 10.1016/S1474-4422(04)00681-7 (2004).
Rivera, E. J . et al. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. Journal of Alzheimer’s disease: JAD 8, 247–268 (2005).
Craft, S. et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology 50, 164–168 (1998).
Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. European journal of nuclear medicine and molecular imaging 32, 486–510, doi: 10.1007/s00259-005-1762-7 (2005).
Steen, E. et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? Journal of Alzheimer’s disease: JAD 7, 63–80 (2005).
Talbot, K. et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. The Journal of clinical investigation 122, 1316–1338, doi: 10.1172/JCI59903 (2012).
Kim, B., Backus, C., Oh, S., Hayes, J. M. & Feldman, E. L. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 150, 5294–5301, doi: 10.1210/en.2009-0695 (2009).
Leibson, C. L. et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol 145, 301–308 (1997).
Ott, A. et al. Diabetes mellitus and the risk of dementia: The Rotterdam Study [see comments]. Neurology 53, 1937–1942 (1999).
Arvanitakis, Z., Wilson, R. S., Bienias, J. L., Evans, D. A. & Bennett, D. A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol 61, 661–666 (2004).
Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 10, e47–92 (2014).
Grundke-Iqbal, I. et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83, 4913–4917 (1986).
Glenner, G. G. & Wong, C. W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120, 885–890 (1984).
El Khoury, N. B., Gratuze, M., Papon, M. A., Bretteville, A. & Planel, E. Insulin dysfunction and Tau pathology. Frontiers in cellular neuroscience 8, 22, doi: 10.3389/fncel.2014.00022 (2014).
Alonso, A. C., Zaidi, T., Novak, M., Grundke-Iqbal, I. & Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments / straight filaments. Proc. Natl. Acad. Sci. USA 98, 6923–6928 (2001).
Bretteville, A. & Planel, E. Tau aggregates: toxic, inert, or protective species? J Alzheimers Dis 14, 431–436 (2008).
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T. & Hyman, B. T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639 (1992).
Duff, K. & Planel, E. Untangling memory deficits. Nat Med 11, 826–827 (2005).
Tremblay, C. et al. Biochemical characterization of Abeta and tau pathologies in mild cognitive impairment and Alzheimer’s disease. Journal of Alzheimer’s disease: JAD 12, 377–390 (2007).
Planel, E. et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 13635–13648, doi: 10.1523/JNEUROSCI.3949-07.2007 (2007).
Clodfelder-Miller, B. J., Zmijewska, A. A., Johnson, G. V. & Jope, R. S. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 55, 3320–3325, doi: 10.2337/db06-0485 (2006).
Jolivalt, C. G. et al. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. Journal of neuroscience research 86, 3265–3274, doi: 10.1002/jnr.21787 (2008).
Qu, Z. et al. Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain research 1383, 300–306, doi: 10.1016/j.brainres.2011.01.084 (2011).
Planel, E. et al. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J Neurosci 24, 2401–2411, doi: 10.1523/JNEUROSCI.5561-03.2004 (2004).
Petry, F. R. et al. Specifity of anti-tau antibodies when analyzing mice models of Alzheimer’s disease: problems and solutions. PLoS ONE 9, e94251 (2014).
Liu, F., Grundke-Iqbal, I., Iqbal, K. & Gong, C. X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22, 1942–1950, doi: EJN4391 [pii]10.1111/j.1460-9568.2005.04391.x (2005).
Cohen, P. The structure and regulation of protein phosphatases. Annual Review of Biochemistry 58, 453–508, doi: 10.1146/annurev.bi.58.070189.002321 (1989).
Tian, Q. & Wang, J. Role of serine/threonine protein phosphatase in Alzheimer’s disease. Neuro-signals 11, 262–269, doi: 67425 (2002).
Gupta, A. & Dey, C. S. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Molecular biology of the cell 23, 3882–3898, doi: 10.1091/mbc.E12-05-0337 (2012).
Zhang, X. et al. Tumor-suppressor PTEN affects tau phosphorylation, aggregation, and binding to microtubules. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 20, 1272–1274, doi: 10.1096/fj.06-5721fje (2006).
Deng, Y. et al. Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: Implication for Alzheimer’s disease. Am J Pathol 175, 2089–2098, doi: ajpath.2009.090157 [pii]10.2353/ajpath.2009.090157 (2009).
Ksiezak-Reding, H., Pyo, H. K., Feinstein, B. & Pasinetti, G. M. Akt/PKB kinase phosphorylates separately Thr212 and Ser214 of tau protein in vitro . Biochimica et biophysica acta 1639, 159–168 (2003).
Terry, R. D. et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30, 572–580, doi: 10.1002/ana.410300410 (1991).
Kimura, T. et al. Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J 26, 5143–5152, doi: 10.1038/sj.emboj.7601917 (2007).
Ke, Y. D., Delerue, F., Gladbach, A., Gotz, J. & Ittner, L. M. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PloS one 4, e7917, doi: 10.1371/journal.pone.0007917 (2009).
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H. & Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112, 389–404 (2006).
Kimura, T. et al. Sequential changes of tau-site-specific phosphorylation during development of paired helical filaments. Dementia 7, 177–181 (1996).
Barghorn, S. et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 39, 11714–11721 (2000).
Fischer, D. et al. Structural and microtubule binding properties of tau mutants of frontotemporal dementias. Biochemistry 46, 2574–2582, doi: 10.1021/bi061318s (2007).
von Bergen, M. et al. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. The Journal of biological chemistry 276, 48165–48174, doi: 10.1074/jbc.M105196200 (2001).
Fath, T., Eidenmuller, J. & Brandt, R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. J Neurosci 22, 9733–9741 (2002).
Le Corre, S. et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci U S A 103, 9673–9678, doi: 10.1073/pnas.0602913103 (2006).
Oddo, S. et al. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem 281, 39413–39423, doi: 10.1074/jbc.M608485200 (2006).
Yoshiyama, Y. et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351, doi: 10.1016/j.neuron.2007.01.010 (2007).
Duarte, J. M., Carvalho, R. A., Cunha, R. A. & Gruetter, R. Caffeine consumption attenuates neurochemical modifications in the hippocampus of streptozotocin-induced diabetic rats. J Neurochem 111, 368–379, doi: 10.1111/j.1471-4159.2009.06349.x (2009).
Wang, X. et al. Contribution of single-minded 2 to hyperglycaemia-induced neurotoxicity. Neurotoxicology 35, 106–112, doi: 10.1016/j.neuro.2013.01.003 (2013).
Li, Y . et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. Journal of Alzheimer’s disease: JAD 19, 1205–1219, doi: 10.3233/JAD-2010-1314 (2010).
Currais, A. et al. Diabetes exacerbates amyloid and neurovascular pathology in aging-accelerated mice. Aging cell 11, 1017–1026, doi: 10.1111/acel.12002 (2012).
Papon, M. A. et al. Deregulation of protein phosphatase 2A and hyperphosphorylation of tau protein following onset of diabetes in NOD mice. Diabetes 62, 609–617, doi: 10.2337/db12-0187 (2013).
Qian, W. et al. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J Alzheimers Dis 19, 1221–1229, doi: 10.3233/JAD-2010-1317 (2010).
Sutherland, C., Leighton, I. A. & Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 296 (Pt 1), 15–19 (1993).
Planel, E., Yasutake, K., Fujita, S. C. & Ishiguro, K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem 276, 34298–34306, doi: 10.1074/jbc.M102780200 (2001).
Bretteville, A. et al. Hypothermia-induced hyperphosphorylation: a new model to study tau kinase inhibitors. Sci Rep 2, 480, doi: 10.1038/srep00480 (2012).
Aitken, A. et al. Amino acid sequence at the site on protein phosphatase inhibitor-2, phosphorylated by glycogen synthase kinase-3. Biochimica et biophysica acta 790, 288–291 (1984).
DePaoli-Roach, A. A. Synergistic phosphorylation and activation of ATP-Mg-dependent phosphoprotein phosphatase by F A/GSK-3 and casein kinase II (PC0.7). The Journal of biological chemistry 259, 12144–12152 (1984).
Lee, C. C., Huang, C. C., Wu, M. Y. & Hsu, K. S. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem 280, 18543–18550, doi: 10.1074/jbc.M414112200 (2005).
Chen, Y. et al. Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces Abeta level and microglia activation in the brains of 3xTg-AD mice. Exp Neurol 261, 610–619, doi: 10.1016/j.expneurol.2014.06.004 (2014).
Moult, P. R. & Harvey, J. Hormonal regulation of hippocampal dendritic morphology and synaptic plasticity. Cell Adh Migr 2, 269–275 (2008).
Huang, C. C., Lee, C. C. & Hsu, K. S. The role of insulin receptor signaling in synaptic plasticity and cognitive function. Chang Gung Med J 33, 115–125 (2010).
Andorfer, C. et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. Journal of neurochemistry 86, 582–590 (2003).
Tucker, K. L., Meyer, M. & Barde, Y. A. Neurotrophins are required for nerve growth during development. Nat Neurosci 4, 29–37 (2001).
Duff, K. et al. Characterization of Pathology in Transgenic Mice Over-Expressing Human Genomic and cDNA Tau Transgenes. Neurobiol Dis 7, 87–98 (2000).
Planel, E. et al. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. Journal of Neuroscience 27, 3090–3097 (2007).
Julien, C. et al. Dimethyl sulfoxide induces both direct and indirect tau hyperphosphorylation. PLoS One 7, e40020, doi: 10.1371/journal.pone.0040020 (2012).
Julien, C., Bretteville, A. & Planel, E. Biochemical isolation of insoluble tau in transgenic mouse models of tauopathies. Methods in Molecular Biology 849, 473–491, doi: 10.1007/978-1-61779-551-0_32 (2012).
Greenberg, S. G. & Davies, P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proceedings of the National Academy of Sciences of the United States of America 87, 5827–5831 (1990).
Planel, E. et al. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. Journal of Neuroscience 24, 2401–2411 (2004).
Acknowledgements
We would like to thank Dr. Peter Davies (Hofstra North Shore-LIJ School of Medicine, Manhasset, NY, USA) for the generous gift of multiple anti-tau antibodies. This work was supported by Biomedical Doctoral Awards from the Alzheimer Society of Canada (to M.G. and F.R.P.) and grants to E.P. from the Canadian Institute of Health Research (MOP-106423, PCN-102993), Fonds de Recherche en Santé du Québec (16205, 20048) and the Natural Sciences and Engineering Research Council of Canada (354722). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. E.P. is the guarantor of the manuscript presented here and takes full responsibility for the work as a whole, including the study design, access to data, and the decision to submit and publish the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: M.G., E.P. Performed the experiments: M.G., J.J., F.R.P., F.M. Analyzed the data: M.G. Wrote the manuscript: M.G. and E.P. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gratuze, M., Julien, J., Petry, F. et al. Insulin deprivation induces PP2A inhibition and tau hyperphosphorylation in hTau mice, a model of Alzheimer’s disease-like tau pathology. Sci Rep 7, 46359 (2017). https://doi.org/10.1038/srep46359
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46359
This article is cited by
-
Pathogenesis of miR-155 on nonmodifiable and modifiable risk factors in Alzheimer’s disease
Alzheimer's Research & Therapy (2023)
-
Crosstalk between Alzheimer’s disease and diabetes: a focus on anti-diabetic drugs
Metabolic Brain Disease (2023)
-
A Potential Link Between Visceral Obesity and Risk of Alzheimer’s Disease
Neurochemical Research (2023)
-
Ser/Thr phosphatases: One of the key regulators of insulin signaling
Reviews in Endocrine and Metabolic Disorders (2022)
-
Repurposing beta-3 adrenergic receptor agonists for Alzheimer’s disease: beneficial effects in a mouse model
Alzheimer's Research & Therapy (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
