






Abstract
The worldwide increase in antibiotic resistance is a concern for public health. The fact that the choice of dose and treatment duration can affect the selection of antibiotic-resistant mutants is becoming more evident, and an increased number of studies have used pharmacodynamic models to describe the drug exposure and pharmacodynamic breakpoints needed to minimize and predict the development of resistance. However, there remains a lack of sufficient data, and future work is needed to fully characterize these target drug concentrations. More knowledge is also needed of drug pharmacodynamics versus bacteria with different resistance mutations and susceptibility levels. The dosing regimens should exhibit high efficacy not only against susceptible wild-type bacteria but, preferably, also against mutated bacteria that may exist in low numbers in “susceptible” populations. Thus, to prolong the life span of existing and new antibiotics, it is important that dosing regimens be carefully selected on the basis of pharmacokinetic and pharmacodynamic properties that prevent emergence of preexisting and newly formed mutants.
Antibiotic Resistance
Antibiotic resistance has become a serious global problem and affects almost every bacterial species for which treatment with antibiotics is available. Resistance to multiple antibiotics has developed among many common pathogens, such as staphylococci, pneumococci, Pseudomonas organisms, and extended-spectrum β-lactamase (ESBL)-producing strains of Enterobacteriaceae, and the resistance problem is steadily increasing worldwide. Consequently, the therapeutic options for the treatment of some infections are limited, especially in developing countries, where second- and third-line antibiotics are unavailable or unaffordable. Infectious diseases cause >11 million deaths annually [1], and treatment failure caused by antibiotic-resistant bacteria is a contributing factor in these deaths, although the quantitative burden of antibiotic resistance is not certain.
The development and spread of antibiotic-resistant bacteria are affected by several factors. Some factors are bacteria specific, such as the mutation rate, the transmission rate, the biological fitness costs, and the ability to compensate for such costs. Other factors of major importance in the emergence of resistance are the volume and quality of antibiotic use, including prescription when there is no clinical indication, over-the-counter sales or sales by drug vendors, inappropriate drug choice, and suboptimal dosing [2, 3]. Dissemination of antibiotic resistance is also influenced by environmental factors in the community and hospitals. Direct or indirect person-to-person transmission is affected by population density and hospital structure and significantly increases in association with poor hygiene.
Mutation Rate
In a bacterial cell, spontaneous mutations normally occur at a rate of 10-9–10-10 mutations/bp/generation during DNA replication [4, 5]. In contrast to the mutation rate, the mutation frequency measures all of the mutants present in a given population, irrespective of when the mutations occurred. Because one antibiotic-resistance phenotype usually can be caused by several mutations, both in different base pairs and in different genes, the mutation frequency is generally higher than the general mutation rate. Bacteria with highly increased mutation rates are called “mutators” and can have mutation rates that are 10-fold to nearly 1000-fold higher than those of wild-type strains [6]. The mutator phenotype is often associated with inactivation of the methyl-directed mismatch repair, a repair system that recognizes mismatches on newly replicated DNA and restores the sequence with the correct base [7].
Because resistance development is very much dependent on mutations, the mutation frequency may play a significant role in the development of resistance. Studies have shown that mutators accelerate resistance development in vitro [8] and in animals [9] and that strains that are clinically fluoroquinolone resistant are often associated with a mutator phenotype [10]. In addition, Gustafsson et al. [11] presented evidence that antibiotic treatment selected for commensal bacteria with increased ciprofloxacin resistance and that there was a correlation between increased mutation frequency and resistance development.
Biological Cost of Antibiotic Resistance
Antibiotic resistance often confers a biological fitness cost, because the resistance mutations typically occur in genes of target molecules with essential functions in the cell. For example, mutations in the rpoB gene that encodes the β subunit of RNA polymerase cause rifampicin resistance in Escherichia coli and may also affect the rate of transcription [12]. Fusidic acid resistance caused by mutations in the ribosomal elongation factor G of Staphylococcus aureus [13] and Salmonella enterica serotype Typhimurium [14, 15] is often associated with a decreased rate of protein synthesis. These cost-associated mutations confer a reduction in bacterial fitness that can be measured as relative growth rates within and outside the host, transmission rates between hosts, and clearance from the hosts [16]. In normal settings, consequently, resistant bacteria usually are inferior to normal bacteria and can easily be outcompeted when no antibiotic pressure is present. For example, fosfomycin resistance, which can be conferred by mutations in several genes and by any loss-of-function mutations in these genes, is easily selected for E. coli under experimental conditions. However, fosfomycin resistance is rarely found in clinical isolates, in spite of the relatively high mutation frequency in vitro. This can be explained by the fact that fosfomycin resistance is often associated with a reduced bacterial growth rate. A mathematical model showed that such fitness costs rapidly reduced the probability of development of fosfomycin resistance, because they prevented the establishment of resistant bacteria in the bladder [17]. In a human competition model, it has also been shown that biological fitness is a significant determinant of bacterial survival [18].
Compensatory Evolution
Bacteria may reduce the biological cost associated with resistance through compensatory evolution. Such compensation can involve true reversion of the resistance mutation or acquisition of second-site compensatory mutations (intra- or extragenic suppressor mutations). Amelioration of the costs of resistance by compensatory mutations has been noted for several antibiotics and bacterial species in vitro, in animal experiments, and in clinical settings [19]. For example, a number of compensatory intragenic mutations have partly or fully restored the biological fitness in fusidic acid-resistant mutants of S. aureus and S. Typhimurium [13, 15], rifampin-resistant mutants of E. coli [12], and clinical isolates of isoniazid-resistant Mycobacterium tuberculosis [20]. A study of the epidemiology of M. tuberculosis has shown that, among clinical isolates with streptomycin resistance, strains with a low biological fitness cost are more frequent [21]. It has been shown that resistant bacteria can persist in humans for a long time without any antibiotic pressure [22, 23]. In one study, Staphylococcus epidermidis isolates with high-level resistance to clarithromycin were recovered from patients up to 4 years after antibiotic treatment [23]. Antibiotic resistance can, thus, be stabilized with fitness-restoring compensatory mutations and may allow fully resistant strains to compete successfully with susceptible strains in an antibiotic-free environment [24]. Consequently, restrictions in antibacterial use might not lead to reversibility of antibiotic resistance in the community.
Dosage-Related Selection
The fact that resistance development can be affected by antibiotic dosing regimens is evident from the results of a number of in vitro and animal experiments. A few selected examples will be given here. In a study by Blaser et al. [25], Klebsiella pneumoniae was challenged with the quinolone enoxacin in different dosing regimens in an in vitro kinetic model. When enoxacin was administered in a twice-daily dosage, bacteria with increased MICs were selected. When, instead, the total daily dose was given as 1 dose daily, the bacteria were eradicated. The need for high peak fluoroquinolone levels for the suppression of resistant bacteria was also noted by Marchbanks et al. [26], who exposed Pseudomonas aeruginosa to ciprofloxacin in an in vitro model. A single dose prevented growth of the ciprofloxacin-resistant bacteria that were selected during exposure to smaller fractionated doses [26]. The impact of dose fractionation on survival was also investigated in a neutropenic rat model of P. aeruginosa sepsis. Once-daily administration of the fluoroquinolone lomefloxacin gave a higher survival rate for the rats, compared with the use of smaller doses administered after shorter intervals [27]. In an in vivo abscess model, dose-dependent selection of mutants of Enterobacter cloacae was demonstrated after treatment with ceftizoxime [28]. When healthy pigs were infected with Salmonella organisms and were treated orally with the recommended dose of the fluoroquinolone enrofloxacin (2.5 mg/kg of body weight), resistant Salmonella organisms were rapidly selected. By instead using the intramuscular route of administration and by escalating the dose to 3 or 6 times the recommended dose, resistance selection was reduced [29]. The association between β-lactam use and penicillin-resistant Streptococcus pneumoniae was analyzed in an observational study of schoolchildren. Low daily doses and a long duration of treatment with a β-lactam antibiotic were associated with an increased risk of pharyngeal carriage of penicillin-resistant S. pneumoniae [30].
Clinical and experimental studies thus indicate that the choice of dose and treatment duration can influence the selection of antibiotic-resistant mutants. For example, the initially recommended dose of levofloxacin has been questioned on the basis of clinical experience indicating that selection of resistance can take place during treatment of infection with S. pneumoniae [31]. Still, there is poor knowledge of the optimal dosing strategies to treat bacterial infections while simultaneously preventing the selection and emergence of resistance.
Selective Concentrations
During antibiotic therapy, gradients of antibiotic concentrations are formed in the human body. These gradients will depend on the rates of drug diffusion into various tissues and the variation in the rate of elimination from different compartments (figure 1). In the body, diverse environmental antibiotic selective pressures will, thus, be created, and the selection process can be expected to differ accordingly.
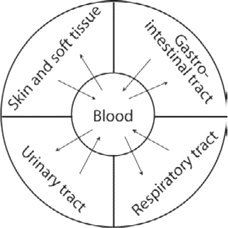
Multicompartment pharmacokinetics. Antibiotic concentrations in the blood will be transported to the interstitial fluid of different body sites, such as those in the figure. The differences in pharmacokinetics within the different sites will depend on the rate of diffusion, potential diffusion barriers, and the volume of compartments.
A subpopulation of resistant bacteria often exists and will be selected by certain drug concentrations, leading to a regrowth during treatment [32] (figure 2). Low antibiotic concentrations can select for low-level resistance, which could have a major effect on the emergence of high-level antibiotic resistance [33, 34]. During antibiotic treatment, selection of resistance may take place at several sites in the human body. In the evaluation of optimal drug concentrations, it is important to focus not only on the infective pathogens and the infectious sites of the body but, also, on the commensal flora (e.g., in the intestinal tract or the skin), in which much higher numbers of bacteria exist and which perhaps is even more important in the selection of resistance. When the fluoroquinolone clinafloxacin was administered to healthy humans, ecological disturbances in the intestinal microflora were created. The treatment effectively eradicated aerobic bacteria in most subjects, whereas the effect on anaerobic bacteria was somewhat less effective and resistance was noted to develop in Bacteroides species [35].
![Emergence of resistance. The susceptible (wild-type) population is readily killed after drug exposure, whereas the resistant (mutant) population may be amplified. The total population is explained by the changes in the 2 different populations. Adapted from Drusano [32], with permission from the Nature Publishing Group.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cid/45/Supplement_2/10.1086_519256/1/m_45-Supplement_2-S129-fig002.gif?Expires=1716320182&Signature=aH1PG-SCj8wNm66kNodT7SEg378wVZIA-T7A3P7MkHhIrDo3z5V0LQXoa2toIiK67ZoI6oNI0pnJeXyVInSyMhneXBv2fPjdsAwMpENcEHzmqgAlpSDFu9~GdMQkSIaWV9jsuXGpN2Att2rYgu4B4WZQSQskjhqQ2cZVmN8n22dHIUlmblDD3Ji1i5DagGuL9x22C8nIaBrfafzDBno9OJZVaiOClB5owhat4l8TkVESsiottniG5N8HLP0b3UXjAQh5s8r8TMo2F1L0yuJpwFTB5sLnz68m1kGOlfvGk0iE1W~DbvuvfH0pm2XD~3DpPzo17PR0mENE8UF4-bx4nQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Emergence of resistance. The susceptible (wild-type) population is readily killed after drug exposure, whereas the resistant (mutant) population may be amplified. The total population is explained by the changes in the 2 different populations. Adapted from Drusano [32], with permission from the Nature Publishing Group.
However, because of these large fluctuations in drug concentrations and because of the difficulties associated with accurately measuring the concentrations at some sites, the actual drug concentrations are often uncertain. To simplify this complex system, in vitro models in which serum concentrations can be simulated are increasingly being used. It has been shown that the free (nonprotein-bound) concentrations in interstitial tissue fluid, where most bacterial infections take place, are similar to those in serum [36–38].
Pharmacokinetics (Pk) and Pharmacodynamics (Pd)
During the past decades, the knowledge of the interaction between the PK and PD of antibiotics has significantly increased, and the International Society of Anti-Infective Pharmacology has proposed standardization of PK/PD terminology [39, 40]. However, studies of antibiotic dosing regimens that consider the development of resistance have not been of high priority, and most PK/PD indices thus focus on clinical or microbiological efficacy. Not until recently has some research included resistance development as an end point, and several studies, especially studies of fluoroquinolones, have used PD models in the search for PK/PD principles and breakpoints that predict the emergence of resistance. Another important issue of concern is that most PK/PD studies include susceptible, wild-type bacteria as target organisms. More knowledge is needed of drug pharmacodynamics versus bacteria with different resistance mutations and susceptibility levels.
Resistance Prevention Studies
End points. To investigate the optimal dosing regimens and to determine the breakpoints needed for prevention of resistance, different end points can be used. A simple method to measure resistance and detect changes in susceptibility is MIC determination. The disadvantage of MIC determination is that often only 1 or 2 colonies are tested, and minor resistant subpopulations may, thus, not be detected. A more sensitive method, cultivation of bacterial samples on agar plates containing antibiotics, makes it possible to measure the changes in the number of resistant bacteria during experiments [25, 26, 41–43]. Another method of studying resistance selection is through the use of competition assays that use strains with different drug susceptibilities. The different populations can be measured using a discriminative marker; the presence of a mutation in the arabinose gene results in a different colony color on a certain agar and allows for the simple identification of each strain in a mixed culture [34, 44]. Measurement of resistance development in the clinical setting is, as for efficacy studies, limited by discontinuous end points. Ideally, successive samples for cultures should be obtained during treatment and the results related to the individual drug exposure, but this is most often not possible. Therefore, studies of resistance development during treatment of patients with antibiotics have used single measurements determined before treatment and either at the end of treatment or at clinical failure.
Resistance prevention predictors. An increased number of studies have attempted to describe the drug exposure needed to minimize resistance development. Thomas et al. [45] retrospectively evaluated 107 patients treated in 4 clinical trials of nosocomial lower respiratory tract infection, and they found that the selection of antibiotic resistance among the pathogens was significantly decreased when the area under the concentration-time curve (AUC)/MIC was ⩾100. These results have been criticized because of the great time differential between the studies and because of combination in the analysis of drugs that have different pharmacodynamic indices for efficacy [46]. Using mixed cultures of susceptible, intermediately susceptible, and resistant pneumococci in an in vitro kinetic model, Odenholt et al. [47] studied resistance selection after exposure to different concentrations of benzylpenicillin. Although the study design did not allow evaluation of the exact time above the MIC needed to suppress mutant growth, no regrowth was noted when the time above the MIC was 48%, 38%, and 46% for the penicillin-susceptible, -intermediately susceptible, and -resistant subpopulations, respectively.
The hypothesis that the selection of resistant bacteria is driven by particular selective antibiotic concentrations was first suggested by Baquero et al. [33, 34, 48]. They predicted that there would be a range of concentrations for which selection of strains with increased levels of resistance to a drug would be the most intense, and they called this range a “selective window” [34]. Using the same bacterial strains, we have confirmed, in an in vitro kinetic model, that the duration of antibiotic concentrations within the selective window affected the selection of resistant bacteria [44].
The mutant prevention concentration (MPC) is a relatively new pharmacodynamic parameter that has been introduced as a measure of a drug's resistance prevention potency. MPC is defined as the lowest antibiotic concentration that prevents growth of the least susceptible single-step mutants. Resistant subpopulations are proposed to be selectively enriched in the mutant selection window (MSW)—that is, the drug concentration range between the MIC and the MPC [49, 50], as described in figure 3. The methodology of MPC determination has not been standardized; however, in most experimental designs, a bacterial population of 1010 cells is applied to agar plates containing antibiotics, and the MPC is the minimum concentration that allows no colony recovery. For bacteria to grow in the presence of antibiotic concentrations above the MPC, ⩾2 resistance mutations have to arise. If the mutation frequency is ⩽10-7 mutations, then a bacterial population of >1014 cells is needed for 2 mutations to arise, a population size that is unlikely to be achieved in most clinical infections [51].
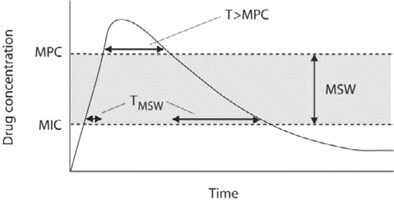
Illustration of the drug concentration over time and the concept of the mutant selection window (MSW). The hypothesis is that selection of resistant mutants occurs when drug concentrations are within the MSW (shaded area)—that is, between the MIC and the mutant prevention concentration (MPC). T > MPC, time above the MPC; TMSW, time within the MSW.
The MPC corresponds to the MIC of the least drug-susceptible single-step mutant in a population. It has been questioned whether the MPC can be predicted from the MIC. In a study by Marcusson et al. [52], the MICs and MPCs of ciprofloxacin for 22 clinical urinary tract infection E. coli isolates were measured. Comparing the MICs and MPCs showed only weak correlation (e.g., there were several isolates with low MICs and high MPCs) (figure 4). This poor correlation between the MIC and the MPC has been reported for a variety of fluoroquinolones and different bacteria [53]. Mutations that can lead to resistance may arise in many different combinations, and the correlation (or lack of correlation) between the MIC and the MPC is probably dependent on where the mutation is likely to arise. In S. pneumoniae, it has been shown that the presence of a parC mutation increases the MPC for fluoroquinolones [54]. A consequence of the variation in the MPC relative to the MIC is that the MPC cannot be accurately predicted from the MIC on an individual basis but, rather, has to be measured separately. This implies that, at present, MPC determination might be more applicable during the process of antibiotic development than in clinical practice. To be used in the latter context, MPC methodology needs to be further developed and simplified.
![MICs and mutant prevention concentrations (MPCs) of ciprofloxacin for 22 clinical urinary tract infection Escherichia coli isolates [52].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cid/45/Supplement_2/10.1086_519256/1/m_45-Supplement_2-S129-fig004.gif?Expires=1716320182&Signature=QzQnH0qXw~jGSn~jgitc7gjpDMmFexdufg5LK92xBr5CuQgxqg3m-TMC7J4C3UzBTJWyLk2eeqfkq8B2it5m47edRbfKAg2NKUJG5~delvskmKjXZlciX6uJGq1VKILJAsEev1EUV1962~Blnp3ADXYkAiOQ5M9ASsAP6-CGu4LEkIzX7ZKJaDi7dF2ydlVk0-INANyEtYw~EWb2UmguKbandAEhVfPz53X3RwX0OA67KuV258MaQ0o30VEgziODppcMk3s~3GBTN1crNJtFdxoWqw8OCeEhe~eyp8XTIcMcDG97amB-8IGzEvSBV8wQlJ1zwDXFGfzTOwfyy3hk-A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MICs and mutant prevention concentrations (MPCs) of ciprofloxacin for 22 clinical urinary tract infection Escherichia coli isolates [52].
The concept of the MSW is not equally applicable to all antibiotic-organism combinations. For MPC determination to be useful in the approach to restricting the selection of antibacterial resistance, the resistance mechanisms observed in the in vitro MPC studies must be the same as the resistance mechanisms observed in vivo. The MPC has mostly been studied for antibiotics for which resistance primarily develops by stepwise chromosomal point mutations, especially the fluoroquinolones. The application of MPC determination to other drugs has raised questions regarding the relevance of mutational events for such drugs as the β-lactams and the aminoglycosides and whether MPC measurements can be performed for drugs with other resistance mechanisms (e.g., efflux and β-lactamases) [55–57]. The significance of these concerns is based on whether the MSW hypothesis addresses the first step in resistance development (i.e., spontaneous mutations or horizontal DNA acquisition) or the second step (i.e., the selective enrichment of mutant subpopulations) [56]. If the second step is the most important, then the individual resistance mechanisms are of less relevance for the MSW concept.
In theory, drug dosing that yields concentrations above the MPC during the whole dosing interval would prevent resistance selection. This could possibly be achieved by using higher doses and/or shorter dosing intervals than would generally be needed to cure an infection. Furthermore, the time within the MSW can be narrowed using compounds with small differences between the MIC and the MPC or compounds that quickly pass through the MSW. However, for some currently available drugs, concentrations above the MPC cannot be reached in patients because of reasons related to PK or toxicity limits. Like the MIC, the MPC is measured with static concentrations. When evaluating dosing regimens from a resistance prevention perspective, it is important to consider the PK properties of a drug. Thus, different PK/PD indices, such as the AUC/MPC, the maximum drug concentration/MPC, and the time within the MSW, have been investigated for their propensity to correlate with antibiotic resistance in vitro and in vivo [41, 42, 58–64]. Zinner et al. [41] tested the theory of the MSW by use of moxifloxacin and S. pneumoniae, and they showed that an AUC/MIC <10 h and >100 h, which represented a time within the MSW of <20%, prevented resistance. Also, when S. aureus was exposed to 4 fluoroquinolones, no resistant bacteria were observed with an AUC/MIC >200 h and a time within the MSW of <20% [62]. Thus, the drug concentrations may not have to exceed the MPC for the entire dosing interval to prevent resistance development.
A study showed that the time above the MPC needed to prevent ciprofloxacin resistance in E. coli was shorter when the maximum drug concentration was increased, and the PK/PD index that was best associated with resistance development was the AUC/MPC [64]. The potential use of the AUC/MPC as a predictor of resistance development has also been suggested in a study exposing S. aureus to different fluoroquinolones [42]. of the MPC-related indices tested, the AUC/MPC was the only one found to correlate with the prevention of fluoroquinolone resistance. Thus, MPC determination and appropriate PK/PD indices could possibly be of use in making decisions about the dosing regimen with respect to a given dosing regimen's potential for the selection and enrichment of mutants. Among antibiotic-resistant mutants isolated from patients, there is a great variation in both the MIC and the MPC [10, 52], and the target breakpoint could vary significantly depending on these values. However, because of the lack of consistency in the ratio of the MIC to the MPC for individual bacterial strains, the use of the MPC as a denominator in these target PD indices would probably lead to less variability.
Implications For Clinical Practice and Drug Development
Antibiotic resistance is of concern for public health, and the rapid emergence of resistant bacteria has led to an increasing awareness of the problem. Still, strict regulations concerning antibiotic use are lacking, and the development of new antibiotics is becoming increasingly expensive and difficult. It is well known that antibiotic resistance can be selected during antibiotic treatment [31, 45, 65]; selection takes place both at the site of infection and in the commensal flora. However, the association between drug dosage and resistance development has been a neglected research area. To prolong the life span of existing and new antibiotics, it is of utmost importance that the dosing regimens are carefully selected on the basis of the PK/PD properties that prevent emergence of preexisting or newly formed mutants. In future drug development, other strategies to prevent resistance development should also be included. One such strategy should be to identify targets for which the resistance mechanism has the most negative effect on fitness. An ideal antibiotic would be a drug for which resistance mutations are rare, the fitness cost of resistance is high, and the rate of fitness-restoring compensatory mutations is low.
To prevent and/or minimize resistance, dosing regimens should exhibit high efficacy to susceptible wild-type bacteria but, preferably, also to mutated bacteria with different degrees of resistance that may exist in low numbers. To achieve useful results, the resistance mechanisms studied in vitro should be the same as the resistance that is expressed in vivo. When an optimal resistance-preventing dosage regimen is limited by the PK and toxicity of the drug, this may be circumvented using combination therapy. The concurrent administration of 2 antibiotics of different classes would require 2 concurrent mutations for bacterial growth to occur. For M. tuberculosis, concentrations above the MPC cannot be attained for most of the first-line agents [66]. In the case of tuberculosis, treatment duration is long, and, to minimize the risk of resistance, 2–4 drugs usually are prescribed.
Although the number of studies that have demonstrated a PD target to prevent the emergence of resistance have been increasing during the past few years, the knowledge gained is far from complete (e.g., only a few studies have addressed the fact that an antibiotic dosage that may prevent the emergence of resistance in the target pathogen may have a collateral effect in the commensal flora, where other pathogens may be selected). However, it is becoming obvious that dosage regimens, on the basis of studies of the drug exposure needed for eradication of the main (wild-type) bacterial population, seem to be clearly associated with a risk of selecting preexisting or newly formed mutants. Thus, higher dosages may be needed to slow down resistance development. Standardization of methods and future studies are needed to define such drug exposure targets for different classes of antibiotics versus different pathogens. These issues should be addressed by academic researchers, by the pharmaceutical industry during drug development, and by regulatory authorities.
Acknowledgments
Supplement sponsorship. This article was published as part of a supplement entitled “Annual Conference on Antimicrobial Resistance,” sponsored by the National Foundation for Infectious Diseases.
Potential conflicts of interest. S.K.O. and O.C.: no conflicts.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments