




Abstract
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder caused by premutation expansions (55–200 CGG repeats) in the fragile X mental retardation 1 (FMR1) gene. The pathologic hallmark of FXTAS is the ubiquitin-positive intranuclear inclusion found in neurons and astrocytes in broad distribution throughout the brain. The pathogenesis of FXTAS is likely to involve an RNA toxic gain-of-function mechanism, and the FMR1 mRNA has recently been identified within the inclusions. However, little is known about the proteins that mediate the abnormal cellular response to the expanded CGG repeat allele. As one approach to identify the protein mediators, we have endeavoured to define the protein complement of the inclusion itself. Fluorescence-activated flow-based methods have been developed for the efficient purification of inclusions from the post-mortem brain tissue of FXTAS patients. Mass spectrometric analysis of the entire protein complement of the isolated inclusions, combined with immunohistochemical analysis of both isolated nuclei and tissue sections, has been used to identify inclusion-associated proteins. More than 20 inclusion-associated proteins have been identified on the basis of combined immunohistochemical and mass spectrometric analysis, including a number of neurofilaments and lamin A/C. There is no dominant protein species in the inclusions, and ubiquitinated proteins represent only a minor component; thus, inclusion formation is not likely to reflect a breakdown in proteasomal degradation of nuclear proteins. The list of proteins includes at least two RNA binding proteins, heterogeneous nuclear ribonucleoprotein A2 and muscle blind-like protein 1, which are possible mediators of the RNA gain-of-function in FXTAS.
Introduction
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a recently identified neurodegenerative disorder that affects many older adults (predominantly males) who carry premutation expansions (55–200 CGG repeats) of the fragile X mental retardation 1 (FMR1) gene (Hagerman et al., 2001; Berry-Kravis et al., 2003; Jacquemont et al., 2003, 2004; Leehey et al., 2003; Hagerman and Hagerman, 2004). Common features of the syndrome include progressive intention tremor, gait ataxia, parkinsonism, autonomic dysfunction and cognitive decline. Based on a premutation carrier frequency of ∼1 in 800 males (Dombrowski et al., 2002), nearly 1 in 3000 males in the general population has a lifetime risk of developing FXTAS (Jacquemont et al., 2004), which would make FXTAS one of the most common single-gene causes of tremor, ataxia and cognitive decline among older adults.
The pathogenic mechanism of FXTAS is not known. However, two observations have led us to propose an RNA toxic gain-of-function model for disease pathogenesis in FXTAS (Hagerman et al., 2001; Greco et al., 2002; Jacquemont et al., 2003; Hagerman and Hagerman, 2004). First, FMR1 mRNA levels are elevated by up to 8-fold in carriers, despite the FMR1 protein (FMRP) levels that appear to remain at, or slightly below, normal levels (Tassone et al., 2000a, b; Kenneson et al., 2001). Second, FXTAS has not been observed among older adults with full mutation alleles (>200 CGG repeats), in whom the FMR1 gene is silenced (i.e. absence of FMR1 mRNA and FMRP). Supporting evidence for this RNA toxicity model comes from the work of Jin et al. (2003), who demonstrated that the expanded CGG repeat, as RNA, could induce neuropathologic changes in a Drosophila melanogaster model.
The neuropathologic hallmark of FXTAS is the presence of ubiquitin-positive intranuclear inclusions, in both neurons and astrocytes, and in broad distribution throughout the cerebrum and brainstem (Greco et al., 2002). The inclusions are immunohistochemically negative for tau isoforms, α-synuclein or polyglutamine peptides, and appear to reflect a new class of inclusion disorder (reviewed in Hagerman and Hagerman, 2004).
One recently verified prediction of the RNA toxicity model is that the FMR1 mRNA itself is present within the FXTAS inclusions (Tassone et al., 2004). A second prediction of the model is that one or more RNA binding proteins is/are acting to mediate the effect of the abnormal mRNA to produce the clinical phenotype. In myotonic dystrophy, another (non-coding) repeat-expansion disorder also thought to result from RNA toxicity (Finsterer, 2002; Mankodi and Thornton, 2002), one protein mediator is believed to be muscle blind-like protein 1 (MBNL1), the human homologue of Drosophila muscleblind (Miller et al., 2000; Fardaei et al., 2001; Ranum and Day, 2004). Both muscular dystrophy mRNA and MBNL1 are found within intranuclear foci in muscular dystrophy, the sequestration of MBNL1 in turn leading to dysregulation of the splicing of several other mRNAs.
In the current instance, the FMR1-mRNA-containing intranuclear inclusions represent a singular clue to the pathogenesis of FXTAS. In particular, the inclusions are likely to harbour one or more proteins that mediate the toxic effects of the expanded-repeat FMR1 mRNA, either directly or indirectly. To investigate this proposal, we have undertaken a systematic analysis of the protein complement of the purified inclusions. Through the application of gel-based methods, as well as tandem mass spectrometry (MS) and Fourier transform ion cyclotron resonance (FT-ICR) MS approaches, more than 20 protein species have been identified thus far. At least one of the identified proteins, hnRNP A2, is an RNA binding protein and thus could serve as a mediator of the expanded-repeat FMR1 mRNA in FXTAS.
Materials and methods
Preparation of crude inclusions
Frontal cortex was obtained at autopsy of FXTAS patients, in accordance with University of California, Davis, IRB approved human subjects protocols. Isolates were derived principally from case 1 of Greco et al. (2002) or from a second FXTAS case (75 years old at death; 85 CGG repeats) (Hagerman et al., 2003). Frontal cortex from a 67-year-old male who died of arteriosclerotic cardiovascular disease was used for a set of control experiments; the latter tissue was obtained from the University of Maryland Brain and Tissue Bank. Approximately 1 g of tissue was cut into 1 mm2 pieces and suspended in 4 volumes of homogenizing buffer (HB) (0.32 M sucrose, 50 mM Tris, 5 mM Na EDTA, 17 μg/ml phenylmethanesulphonyl fluoride) supplemented with Complete (Roche) protease inhibitors tablet (HB-Complete). The tissue was homogenized in a Dounce homogenizer and filtered successively through 500 and 105 μm nylon mesh (Small Parts Inc., Miami Lakes, FL). Filtered homogenates were spun at 1500 g for 10 min at 5°C. Pelleted nuclei were washed/pelleted three additional times (1500 g, 10 min, 5°C) in HB-Complete, resuspended in BC-Complete [20 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulphonic acid, 400 mM NaCl, 1 mM dithiothreitol, 1 mM Na EDTA, 1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid supplemented with Complete (Roche) tablet] containing 0.25% Nonidet P-40, and incubated with rotation at 5°C for 15 min. Nuclei were homogenized with a Dounce homogenizer fitted with a tight pestle and centrifuged at 2000 g for 5 min at 5°C. Pelleted crude inclusions were washed and resuspended in BD-Complete [40 mM Tris–HCl, 10 mM NaCl, 10 mM CaCl2, 5 mM MgCl2, pH 7.9, supplemented with Complete (Roche)], incubated for 30 min at room temperature with 500 U/ml DNase I and centrifuged at 2000 g for 5 min at 5°C. The pellet was resuspended and washed three times in BC-Complete. The crude inclusion pellet was resuspended in BC-Complete (1.0 ml per gram of tissue), aliquoted and stored at −80°C.
Immunofluorescence staining
Nuclei obtained just prior to detergent lysis were spread on SuperFrost Plus slides (Fisher Scientific), fixed in 70% methanol and washed in phosphate-buffered saline (PBS)-T [10 mM sodium phosphate, 150 mM sodium chloride, pH 7.4, 0.1% polyethylene (20) sorbitan monolaurate]. Frozen sections of frontal cortex were formalin fixed and paraffin embedded for in situ staining. Sections of 5 μm were cut, de-paraffinized and washed in PBS-T. Following blocking with 5% goat serum in PBS-T for 2 h, slides were incubated overnight at 4°C with primary antibodies diluted 1:500 in blocking solution: rabbit polyclonal anti-ubiquitin (Novus), mouse monoclonal anti-ubiquitin (AbCam), rabbit polyclonal anti-tau (DAKO), mouse monoclonal anti-synuclein (Zymed), mouse monoclonal anti-αB-crystallin (Stressgen), rabbit polyclonal anti-Hsp 70 (Stressgen), mouse monoclonal anti-Hsp 27 (Stressgen), rabbit polyclonal anti-11s regulator to 20s proteasome (Affinity Research Products), mouse monoclonal anti-GFAP (Calbiochem), mouse monoclonal anti-lamin A/C (BD Transduction Laboratories), mouse monoclonal anti-hnRNP A2/B1 (ImmuQuest), mouse monoclonal anti-myelin basic protein (anti-MBP) (Upstate), rabbit polyclonal anti-golli-MBP (kindly provided by C. Campagnoni) and mouse anti-muscleblind (MBNL1) 3A4 and 2D9 (kindly provided by M. Swanson). Slides were washed extensively and incubated with 1:500 (v/v) Alexa 488 labelled goat anti-rabbit IgG and/or Alexa 594 labelled goat anti-mouse IgG (Molecular Probes) for 2 h at 20°C. Nuclei were stained with 2 μM DAPI (4′, 6-diamidino-2-phenyindole di-lactate). Slides were coverslipped in the presence of ProLong mounting media (Molecular Probes).
Preparative flow sorting of inclusions
Crude inclusions were blocked with 5% goat serum in PBS-T for 2 h and then incubated overnight at 4°C on a rotator with rabbit polyclonal anti-ubiquitin (1/100 dilution) and mouse monoclonal anti-αB-crystallin (1/100 dilution). Following extensive washing in PBS-T, inclusions were incubated for 2 h at 4°C with Alexa 488-conjugated goat anti-rabbit (1/200 dilution) and Alexa 594-conjugated goat anti-mouse (1/200 dilution) antibodies (Molecular Probes, Eugene, OR, USA). Labelled inclusions were washed extensively, resuspended in PBS-T with Complete and passed through a 40 μm cell sieve (BD Falcon, Bedford, MA, USA).
Fluorescently labelled inclusions were purified by preparative flow-cytometric sorting on a MoFlo cell sorter (DakoCytomation, Fort Collins, CO, USA) fitted with a 70 μm nozzle tip. The Alexa 488 fluorescent probe (ubiquitin target) was excited with an argon laser (488 nm line) and detected with a 530/50 nm bandpass filter. The Alexa 594 fluorescent probe (αB-crystallin target) was excited with a 568 nm laser line and detected with a 605/655 nm bandpass filter. To determine approximate inclusion size and complexity, unlabelled inclusions were characterized on a scattergram using forward scatter (FSC) and side scatter (SSC) on a linear scale. The size distribution of particles present in this region was estimated based on a comparison with 2.5 μm diameter beads, labelled with Alexa 488 (Molecular Probes, Eugene, OR, USA). From this information, an initial gate was set, based on scatter, to exclude very small particles (<1 μm) and large debris (>10 μm).
Following gating for size, a set of control experiments was performed to determine proper gating for the sorting of inclusions that were positive for both ubiquitin and αB-crystallin. First, inclusions incubated with Alexa-conjugated secondary antibodies only were examined on scattergrams to determine the distributions of unstained inclusions on each axis. Next, inclusions incubated with anti-ubiquitin primary plus Alexa 488-conjugated secondary, or anti-αB-crystallin primary plus Alexa 594-conjugated secondary, were examined to determine the distributions of singly (ubiquitin or αB-crystallin) positive inclusions, respectively, on the scattergram representing size-gated particles. Single-positive inclusions were also used to set compensation for Alexa 488 and Alexa 594 signals. Finally, inclusions labelled with both anti-ubiquitin and anti-αB-crystallin antibodies were gated and subjected to purification by flow sorting. As an additional control, particles falling within gates defining the ubiquitin and αB-crystallin ‘double-positive’ population were examined on scattergrams for Alexa 488 and Alexa 594 signal intensity versus forward scatter to verify positive staining.
Event rates for sorting averaged 19 000 events per second, with a coincidence rate at or below 9%. After initial sorting, a fraction of the ubiquitin and αB-crystallin-positive inclusions were re-analysed by flow-cytometry to assess purity of the sorted inclusions. Sorted inclusions were collected into PBS-T buffer at 4°C with Complete mini protease inhibitors (Roche).
2D gel electrophoresis
Purified inclusions were pelleted by centrifugation for 1 h at 164 000 g and resuspended in Trizol (Invitrogen). The inclusions were completely soluble in Trizol, with no residue following centrifugation. Proteins were precipitated from the phenol/chloroform phase with isopropanol, washed several times with 0.3 M guanidine hydrochloride in 95% ethanol, followed by several washes with 100% ethanol. The protein pellet was resuspended in ReadyPrep 2D Rehydration/Sample buffer (163-2106, BioRad, Hercules, CA, USA) and spun at 16 000 g. The supernatant (containing 2–10 μg of protein) was applied to a pH 3–10 (or pH 3–11) non-linear, immobilized gradient (7 or 11 cm) IPG strip (Amersham). Following rehydration for 12 h at 30 V, the sample was focused. For the pH 3–10 strip, focusing was carried out in three 1.5 h steps: 500, 1000 and 5000 V (total 9750 V h), with a current limit of 50 μA per strip. For the pH 3–11 strip, focusing was accomplished in four steps: 500 V for 500 V h, gradient to 1000 V for 800 V h, gradient to 6000 V for 7000 V h and 6000 V for 1500 V h. The focused strips were equilibrated, first in 64 mM dithiothreitol, then in 135 mM iodoacetamide in equilibration buffer (50 mM Tris, pH 8.8, 6 M urea, 30% glycerol, 2% SDS, 0.0002% bromophenol blue). Second-dimension electrophoresis was performed on a 10–20% linear gradient Criterion Tris–HCl gel (BioRad) at 20 mA per gel in running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3). The gel was fixed in 10% methanol, 7% acetic acid for 1 h, followed by incubation overnight in SyproRuby (BioRad).
2D western analysis
Proteins separated by 2D gel electrophoresis were transferred onto nitrocellulose (0.2 μm pore size) at 4°C in 25 mM Tris/192 mM glycine/20% methanol. Initial transfer was at 100 V in a Criterion Blotter (BioRad, Hercules, CA, USA), followed by overnight transfer at 30 V. The blots were incubated for 1 h in blocking buffer (5% BSA, 0.1% polyethylene (20) sorbitan monolaurate in PBS), followed by incubation overnight at 5°C with 1:1000 (v/v) anti-αB-crystallin, 1:500 (v/v) anti-ubiquitin, or 1:1000 (v/v) anti-hnRNPA2 antibodies. Following extensive washing with washing buffer (PBS with 0.1% polyethylene (20) sorbitan monolaurate), the blots were incubated with 1/5000 (v/v) peroxidase-conjugated goat anti-mouse IgG (BioRad, Hercules, CA, USA). Detection was accomplished with SuperSignal West Dura Extended Duration Substrate (Pierce, Rockford, IL, USA).
In-solution digestion of purified inclusions with trypsin
Purified inclusions (4 μg) were completely dissolved in 60 μl of 8 M urea (0.1 M Tris–HCl buffer, pH 7.8) and incubated with 3 μl of 450 mM dithiothreitol for 45 min at 55°C. The reduced sample was allowed to cool to room temperature, 6 μl of 500 mM iodoacetamide was added and incubated at room temperature for 20 min in the dark. The reaction was terminated by adding water to give a final concentration of 2 M urea. The reduced and alkylated proteins were digested with 5 μg of modified trypsin (modified by reductive methylation to reduce autolysis or pseudotrypsin activity: Promega) at 37°C overnight. The digest was placed at −20°C to terminate the reaction.
Protein identification by peptide mass fingerprinting
Trypsin-digested inclusions were desalted and purified using a C18 Ziptip (Millipore). The peptide solution was loaded onto the Ziptip, prewashed with 50% (v/v) acetonitrile (ACN)/H2O followed by 0.1% (v/v) trifluoroacetic acid (TFA) in H2O, and washed with 0.1% (v/v) TFA in H2O to remove salts and buffer. The peptides were eluted with 50% ACN, 0.1% TFA in H2O. Mass spectra were recorded on an external source HiResMALDI (IonSpec Corporation, Irvine, CA, USA) with a 7.0 Tesla magnet. FT-ICR MS, known for its accurate mass (∼10 p.p.m.) and high-resolution capabilities (Marshall et al., 1998) was coupled with MALDI for peptide mass determinations. The HiResMALDI instrument was equipped with a pulsed Nd:YAG laser (266 nm). 2,5-Dihydroxybenzoic acid was used as a matrix (5 mg/100 μl in 50% ACN in H2O) for both positive and negative modes. The tryptic peptide solution (1 μl) was applied to the MALDI probe, followed by matrix solution (1 μl). The sample was dried under a stream of air prior to mass spectrometric analysis. Peptide mass fingerprinting was performed by Mascot search (www.matrixscience.com) of the subdatabase comprising ‘Homo sapiens’ in the NCBI protein database.
Protein analysis by liquid chromatography MS/MS (LC-MS/MS)
An HPLC system (Paradigm MG4, Michrom Bio Resources, Auburn, CA, USA), directly coupled with an ion trap mass spectrometer (LCQ Deca XP plus, Finnigan, San Jose, CA, USA), was used for microcapillary 1D LC-MS/MS data acquisition. A lab-fabricated fritless reverse phase (RP) microcapillary column (0.1 mm × 180 mm) was packed with C18 (Phenomenex, Aqua: 5 μm 300Å) (Gatlin et al., 1998). Tryptic peptide mixtures were desalted and concentrated using the RP trap column (0.15 mm × 30 mm), followed by chromatographic separation on the microcapillary RP column at a flow rate of 300 nl/min. Eluted peptides were then directly sprayed into the mass spectrometer. MS/MS spectra were acquired for the most intense peptide ion from the previous MS spectra, with dynamic exclusion for 3 min. Two buffers, 5% ACN/0.1% formic acid (buffer A) and 80% ACN/0.1% FA (buffer B), were used for the RP chromatography. A 2.5 h gradient (0–10% B for 10 min, 10–45% B for 110 min, 45–100% B for 20 min, 100% B for 10 min) was used for optimal separation of the peptides.
Protein identification by LC-MS/MS
SEQUEST searches (Eng et al., 1994) were performed for each MS/MS dataset using the DTASelect software (Tabb et al., 2002) using the subdatabase comprising ‘Homo sapiens’ in the NCBI protein database. SEQUEST search parameters suggested by Washburn et al. (2001) were applied; namely, a minimum cross-correlation value (XCorr) of 1.9 was required for fully tryptic, singly charged peptide ions. XCorr values of 2.2 and 3.75 were used for the doubly and triply charged precursor ions, respectively, for either fully or partially cleaved tryptic peptide ions. In addition, the difference in cross-correlation score, ΔCn, between the potential sequences yielding the highest and second highest XCorr values, was required to exceed a value of 0.08 for a potential sequence match to be accepted. Proteins were identified on the basis of at least three peptides satisfying the criteria above; although, in most instances at least five peptide sequences were used for protein identification. At least one of the MS/MS spectra was manually reviewed for those proteins that were implicated on the basis of only three peptides. A probabilistic criterion was applied at the level of protein identification; specifically, each peptide cross-correlation value was normalized versus the auto-correlation value for the input spectrum. The normalized XCorr values for each peptide were used to determine the absolute probability of a correct protein match (MacCoss et al., 2002; Sadygov and Yates, 2003). Protein identification was restricted to the 99% confidence level or greater.
Results
Isolation of inclusion-bearing nuclei from post-mortem cortical tissue of FXTAS patients
To reduce the effect of autolysis of post-mortem brain tissue on our preparations of purified inclusions, we have developed a two-phased protocol for the purification of inclusions from frontal cortex of FXTAS brains. The first phase, isolation of intact nuclei from cerebral cortex, is based on the rationale that an intact nuclear membrane is associated with better preservation of the inclusion contained within that nucleus. The nuclear isolation step would also protect the inclusions from mechanical degradation through early purification steps (e.g. cell disruption). The second phase of the protocol involves the purification of the inclusions from the isolated nuclei. A separate rationale for the preparation of isolated nuclei is that background autofluorescence is much lower for the isolated nuclei than for whole cells, a distinct advantage for immunohistochemical studies of the localization of candidate proteins within the inclusion.
Preparations of isolated nuclei were subsequently used to screen for a number of additional proteins that are known to be present in the inclusions of several other neurodegenerative disorders (Fig. 1; Table 1). The results of these initial staining experiments confirm that the intranuclear inclusions found in cases of FXTAS represent a unique class of inclusion disorder. In particular, they do not belong to the classes of inclusions found in the tauopathies (e.g. frontotemporal dementia, Alzheimer disease, progressive supranuclear palsy, Galvin et al., 2001; Lee et al., 2001), the synucleinopathies (e.g. Lewy body disorders, multiple system atrophy, Jellinger, 2003) or the polyQ disorders (e.g. Huntington's disease, Zoghbi and Orr, 2000; Wood et al., 2003).
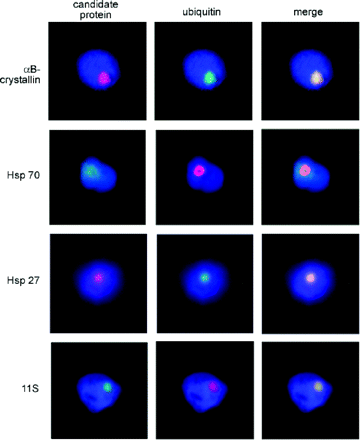
Double immunofluorescence staining demonstrates co-localization of several proteins with ubiquitin-positive inclusions. Secondary antibodies, Alexa 488-conjugated anti-rabbit (green) or Alexa 594-conjugated anti-mouse (red), were used to visualize the primary antibodies. Nuclei were counterstained with DAPI (blue).
Double immunofluorescence staining for candidate proteins in isolated nuclei of FXTAS frontal cortex
| Protein | Antibody* | Clone | Presence in FXTAS nuclei |
|---|---|---|---|
| Ubiquitin | Rabbit polyclonal | R5 | Yes |
| Ubiquitin | Mouse monoclonal | 1B4-UB | Yes |
| tau | Rabbit polyclonal | No | |
| αSynuclein | Mouse monoclonal | LB-509 | No |
| αB-crystallin | Mouse monoclonal | 1B6.1-3G4 | Yes |
| hsp 70 | Rabbit polyclonal | Yes | |
| hsp 27 | Mouse monoclonal | G3.1 | Yes |
| 11S regulator to 20S proteasome | Rabbit polyclonal | Yes |
| Protein | Antibody* | Clone | Presence in FXTAS nuclei |
|---|---|---|---|
| Ubiquitin | Rabbit polyclonal | R5 | Yes |
| Ubiquitin | Mouse monoclonal | 1B4-UB | Yes |
| tau | Rabbit polyclonal | No | |
| αSynuclein | Mouse monoclonal | LB-509 | No |
| αB-crystallin | Mouse monoclonal | 1B6.1-3G4 | Yes |
| hsp 70 | Rabbit polyclonal | Yes | |
| hsp 27 | Mouse monoclonal | G3.1 | Yes |
| 11S regulator to 20S proteasome | Rabbit polyclonal | Yes |
Candidate proteins co-localizing with ubiquitin were considered to be inclusion-related proteins.
See Materials and methods for sources and dilutions.
Double immunofluorescence staining for candidate proteins in isolated nuclei of FXTAS frontal cortex
| Protein | Antibody* | Clone | Presence in FXTAS nuclei |
|---|---|---|---|
| Ubiquitin | Rabbit polyclonal | R5 | Yes |
| Ubiquitin | Mouse monoclonal | 1B4-UB | Yes |
| tau | Rabbit polyclonal | No | |
| αSynuclein | Mouse monoclonal | LB-509 | No |
| αB-crystallin | Mouse monoclonal | 1B6.1-3G4 | Yes |
| hsp 70 | Rabbit polyclonal | Yes | |
| hsp 27 | Mouse monoclonal | G3.1 | Yes |
| 11S regulator to 20S proteasome | Rabbit polyclonal | Yes |
| Protein | Antibody* | Clone | Presence in FXTAS nuclei |
|---|---|---|---|
| Ubiquitin | Rabbit polyclonal | R5 | Yes |
| Ubiquitin | Mouse monoclonal | 1B4-UB | Yes |
| tau | Rabbit polyclonal | No | |
| αSynuclein | Mouse monoclonal | LB-509 | No |
| αB-crystallin | Mouse monoclonal | 1B6.1-3G4 | Yes |
| hsp 70 | Rabbit polyclonal | Yes | |
| hsp 27 | Mouse monoclonal | G3.1 | Yes |
| 11S regulator to 20S proteasome | Rabbit polyclonal | Yes |
Candidate proteins co-localizing with ubiquitin were considered to be inclusion-related proteins.
See Materials and methods for sources and dilutions.
Subsequent disruption of nuclei was accomplished with gentle homogenization in the presence of a mild detergent. DAPI staining of the crude inclusion isolates revealed significant amounts of DNA surrounding the inclusions. Treatment with DNase I successfully released the DNA and any proteins that may have been non-specifically associated with the inclusions through interactions with DNA (e.g. histones), and reduced the tendency of the crude inclusions to aggregate. It is, of course, possible that the DNase I treatment causes the release of some proteins that are specifically related to the inclusions. The crude, DNase I-treated, inclusions constitute the starting material for further purification based on flow methods.
Preparative flow cytometry provides substantial purification of intact inclusions from the nuclear isolates
In the second phase of the purification process, a fluorescence-activated, flow-cytometric particle sorting procedure was utilized that was based on specific properties of the inclusions; namely, particle size and known protein constituents, the latter identified through tagging with fluor-labelled antibodies. This general strategy is commonly used to separate cells based on fluorescent surface marker staining, and has been utilized for the purification of Lewy bodies (Iwatsubo et al., 1996a, b) and polyglutamine inclusions (Hazeki et al., 2002). In our initial approach, the crude inclusion isolates were singly labelled with anti-ubiquitin (primary) and fluor-labelled (secondary) antibodies. Size-gated particles (<1 to ∼10 μm dia; region R1) were then sorted on the basis of fluorescence intensity (Fig. 2A–D). At a flow rate of 19 000 events/s, ∼3.4 × 106 inclusions/h (∼2 μg protein) could be isolated with high levels of ubiquitin-targeted fluorescence (Fig. 2B) from an estimated 6 × 107 size-gated particles comprising the crude inclusion preparations (FXTAS; frontal cortex).
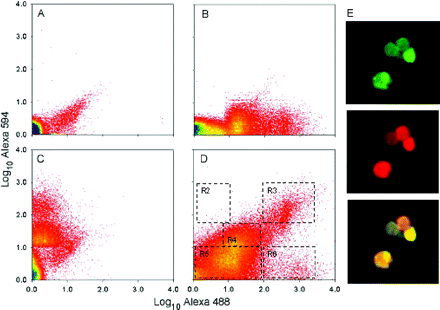
Purification of FXTAS intranuclear inclusions by flow cytometric sorting. (A) Secondary antibodies only. (B) Rabbit anti-ubiquitin (primary) and Alexa 488 anti-rabbit (secondary). (C) Mouse anti-αB-crystallin (primary) and Alexa 594 anti-mouse (secondary). (D) Both anti-ubiquitin primary (Alexa 488 secondary) and anti-αB-crystallin primary (Alexa 594 secondary) antibodies. (E) Fluorescence microscopy of inclusions purified using the dual-label selection criterion for flow sorting. (Upper panel) Ubiquitin immunostaining (green); (middle panel) αB-crystallin staining (red); (lower panel) merged images.
Repeat-analysis of the highly-ubiquitinated particles by analytical fluorescence-activated cell sorting (FACS) demonstrated that at least 80% of the ubiquitin-positive particles remained stable to a second round of flow analysis (data not shown). The diminished fluorescence is probably due to photobleaching of the fluorophore by the sorting process itself. Subsequent microscopic examination of the singly (ubiquitin) labelled inclusions demonstrated substantial purification; however, many of the sorted particles were clearly not inclusions (data not shown), reflecting the widespread ubiquitination of other subcellular structures within the nuclear preparations.
To improve the selectivity of the sorting protocol, we utilized a second fluorescent probe, targeted to αB-crystallin, which consistently co-localizes with ubiquitin in the inclusions (Fig. 1). The crude inclusions were labelled with both anti-ubiquitin (rabbit, polyclonal) and anti-αB-crystallin (mouse monoclonal) primary antibodies, and with Alexa 488-conjugated (goat anti-rabbit) and Alexa 594-conjugated (goat anti-mouse) secondary antibodies (see Materials and methods). The dual-labelled preparations were then subjected to preparative flow sorting. Four discrete populations of particles were observed in each experiment (Fig. 2D): (i) particles that had high levels of both ubiquitin- and αB-crystallin-targeted fluorescence (region R3), representing 1–3% of the R1-gated particles; (ii) particles with a high level of ubiquitin-targeted fluorescence, but with no αB-crystallin-targeted fluorescence (region R6), representing <1% of the sorted particles; (iii) particles with intermediate levels of both targets, representing 40–60% of the gated particles (region R4); and (iv) particles with little intensity for either target (region R5). Essentially none of the gated particles demonstrated αB-crystallin labelling in the absence of ubiquitin labelling (region R2). Particles collected within region R3 demonstrate a morphology (Fig. 2E) similar to the spherical inclusions present in both the whole cell mounts and the nuclear isolates. Moreover, they appear to be substantially free of non-inclusion materials, suggesting that the additional selection criterion renders the flow-sorting protocol highly selective for the inclusions. Finally, the size distribution of the purified inclusions was not reduced relative to the size distribution of the inclusions observed within whole nuclei. Thus, the flow-sorting procedure does not appear to eliminate a major component of the inclusions (also, see below).
One important (and fortunate) feature of the inclusions is that they appear to remain substantially intact throughout the flow-sorting procedure, thus affording the degree of purification necessary for subsequent analysis of the composition of the inclusions. Another important feature of the inclusions is that they are completely soluble in both 8 M urea and 2% SDS detergent solutions; this latter feature is of particular importance for subsequent gel and mass spectrometric analyses.
Application of the flow-based isolation protocol to an age-matched control brain does not yield particles with morphology similar to the FXTAS inclusions
Although purification of inclusions by flow cytometry appeared to be specific to tissue derived from FXTAS brain tissue, it is formally possible that the methods co-purify more generic (i.e. non-disease-specific) nuclear material. To assess this issue, human cortical tissue from a control male was subjected to the same isolation procedure. Our protocol yielded <10% (mass) of the ‘crude inclusion’ fraction (relative to the yield for FXTAS tissue) per gram of brain tissue. Furthermore, ubiquitin staining revealed no inclusion-like particles. Nevertheless, the crude material was subjected to dual-label (crystallin/ubiquitin) preparative sorting. No ubiquitin/αB-crystallin-positive particles were detected under the gating conditions used to isolate inclusions (<2% of the number of particles detected upon sorting of FXTAS brain tissue; data not shown). Since the preparative sorting procedure utilized an equivalent amount of the crude isolate for both FXTAS and control brains, with the latter representing >10-fold more nuclei than for FXTAS tissue, generic nuclear material is likely to represent not more than ∼0.1–0.2% of the total mass of the purified inclusions.
Initial characterization of the purified inclusions by 2D gel electrophoresis reveals a relatively complex pattern of protein spots with no single dominant species
To assess the complexity of the protein complement of the purified inclusions, isolates (∼2–10 μg) were subjected to 2D gel electrophoresis (see Materials and methods) followed by staining with SyproRuby (Fig. 3A). No precipitate remained following centrifugation at 16 000 g after resuspension in urea-based sample buffer, and no insoluble material was retained in the sample holder or wells following either first or second dimension separation. Resolution in the first dimension used an immobilized pH 3–10 (or pH 3–11) non-linear gradient; second dimension resolution utilized a 10–20% Tris–HCl gradient gel (0.1% SDS). Approximate positions and relative intensities of the principal spots are presented in Table 2.

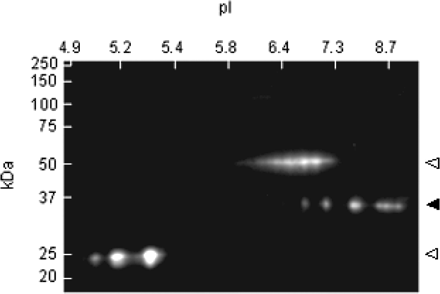
Electrophoretic (2D) separation of inclusions. Inclusions were solubilized in urea/CHAPS buffer. First dimension (horizontal), 7 cm pH 3–10 (non-linear gradient) IPG strip; second dimension (vertical), 10–20% Tris/glycine SDS–PAGE. (A) 2D gel of sorted inclusions stained with SyproRuby. Electrophoretic (2D) Western analysis of purified inclusions using antibodies directed against (B) αB-crystallin (closed triangles) or (C) ubiquitin. Open triangle, primary antibody.
List of the 10 most prominent protein species represented in the 2D gel (Fig. 6) of purified inclusion proteins
| Spot number* | Estimated mol wt† | Estimated pI† | Spot intensity‡ | Percentage of total§ |
|---|---|---|---|---|
| 1 | 35 612 | 9.3 | 8346 | 7.3 |
| 2 | 22 720 | 7.8 | 7566 | 6.6 |
| 3 | 52 876 | 5.3 | 5632 | 4.9 |
| 4 | 32 675 | 5.8 | 5032 | 4.4 |
| 5 | 52 876 | 5.2 | 4312 | 3.8 |
| 6 | 65 671 | 4.8 | 4080 | 3.6 |
| 7 | 75 541 | 3.5 | 3780 | 3.3 |
| 8 | 21 659 | 6.4 | 3240 | 2.8 |
| 9 | 44 228 | 9.2 | 2624 | 2.3 |
| 10 | 20 321 | 3.9 | 2520 | 2.2 |
| Spot number* | Estimated mol wt† | Estimated pI† | Spot intensity‡ | Percentage of total§ |
|---|---|---|---|---|
| 1 | 35 612 | 9.3 | 8346 | 7.3 |
| 2 | 22 720 | 7.8 | 7566 | 6.6 |
| 3 | 52 876 | 5.3 | 5632 | 4.9 |
| 4 | 32 675 | 5.8 | 5032 | 4.4 |
| 5 | 52 876 | 5.2 | 4312 | 3.8 |
| 6 | 65 671 | 4.8 | 4080 | 3.6 |
| 7 | 75 541 | 3.5 | 3780 | 3.3 |
| 8 | 21 659 | 6.4 | 3240 | 2.8 |
| 9 | 44 228 | 9.2 | 2624 | 2.3 |
| 10 | 20 321 | 3.9 | 2520 | 2.2 |
Numbers refer to labelled spots on Fig. 6.
Estimates for molecular weight and pI are based on molecular weight and ampholyte standards, and are approximate.
SyproRuby stained gels were analysed using the FluorChem 8000 software. Spot intensities were calculated based on pixel values, with black = 0 and white = 255; backgrounds were determined as the average of the 10 lowest pixel values surrounding the spot. Integrated intensities are the sums of all pixel values after background correction.
Spot intensities converted to percentages of total protein on the gel (∼100 spots).
List of the 10 most prominent protein species represented in the 2D gel (Fig. 6) of purified inclusion proteins
| Spot number* | Estimated mol wt† | Estimated pI† | Spot intensity‡ | Percentage of total§ |
|---|---|---|---|---|
| 1 | 35 612 | 9.3 | 8346 | 7.3 |
| 2 | 22 720 | 7.8 | 7566 | 6.6 |
| 3 | 52 876 | 5.3 | 5632 | 4.9 |
| 4 | 32 675 | 5.8 | 5032 | 4.4 |
| 5 | 52 876 | 5.2 | 4312 | 3.8 |
| 6 | 65 671 | 4.8 | 4080 | 3.6 |
| 7 | 75 541 | 3.5 | 3780 | 3.3 |
| 8 | 21 659 | 6.4 | 3240 | 2.8 |
| 9 | 44 228 | 9.2 | 2624 | 2.3 |
| 10 | 20 321 | 3.9 | 2520 | 2.2 |
| Spot number* | Estimated mol wt† | Estimated pI† | Spot intensity‡ | Percentage of total§ |
|---|---|---|---|---|
| 1 | 35 612 | 9.3 | 8346 | 7.3 |
| 2 | 22 720 | 7.8 | 7566 | 6.6 |
| 3 | 52 876 | 5.3 | 5632 | 4.9 |
| 4 | 32 675 | 5.8 | 5032 | 4.4 |
| 5 | 52 876 | 5.2 | 4312 | 3.8 |
| 6 | 65 671 | 4.8 | 4080 | 3.6 |
| 7 | 75 541 | 3.5 | 3780 | 3.3 |
| 8 | 21 659 | 6.4 | 3240 | 2.8 |
| 9 | 44 228 | 9.2 | 2624 | 2.3 |
| 10 | 20 321 | 3.9 | 2520 | 2.2 |
Numbers refer to labelled spots on Fig. 6.
Estimates for molecular weight and pI are based on molecular weight and ampholyte standards, and are approximate.
SyproRuby stained gels were analysed using the FluorChem 8000 software. Spot intensities were calculated based on pixel values, with black = 0 and white = 255; backgrounds were determined as the average of the 10 lowest pixel values surrounding the spot. Integrated intensities are the sums of all pixel values after background correction.
Spot intensities converted to percentages of total protein on the gel (∼100 spots).
A striking feature of the 2D gel pattern is that no single protein is predominant; the most abundant single species (Fig. 3A; spot 1; mw ∼37 kDa, pI ∼9) constitutes only ∼7% of the total protein mass (Table 2). A 2D gel analysis of the crude inclusions (prior to sorting) revealed additional protein species; however, no single protein species accounted for more than ∼10% of the protein mass. This observation is consistent with the lack of any size reduction of the inclusions upon flow-sorting. Most of the individual gel spots have not yet been associated with specific proteins, since most are present in fM quantities on individual gels. However, proteins present within the inclusions are being identified through direct sequencing of the entire peptide pool from the purified inclusions (see below). Certain relationships among the spots (e.g. spots 3 and 5) suggest that some proteins may exist in multiple forms, presumably due to various forms of post-translational modification; however, the nature of most post-translational modifications remains to be determined.
The 2D gel results can be used to make several additional observations, particularly with respect to the absence of specific proteins within the inclusions. For example, based on the estimated molecular weights (∼67–71 kDa) and pIs (∼6.8–8.6) of the principal isoforms of FMRP, the protein product of the FMR1 gene does not appear to be a significant component of the inclusions (Fig. 3A), although its message is present within the inclusions (Tassone et al., 2004). This observation is consistent with the absence of detectable FMRP by 1D western analysis (data not shown). Interestingly, αB-crystallin does not appear to be a major constituent of the inclusions, although it is clearly present as shown both by immunostaining of the intact inclusions and by 2D western analysis (see below).
2D western analysis indicates that multiple forms of αB-crystallin are present within the purified inclusions
To further characterize the nature of αB-crystallin in the inclusions, 2D western analysis was performed on blot-transferred proteins, using αB-crystallin monoclonal antibody (see Materials and methods). The purified inclusions clearly contain two variants of αB-crystallin (Fig. 3B), created by either variant splicing or post-translational modification; the nature of these isoforms is currently being investigated. However, it is clear that the protein detected by immunofluorescence is not a major species within the inclusions.
2D western analysis for the presence of ubiquitin reveals that only a small subset of the proteins is ubiquitinated within the inclusions
One of the principal working hypotheses of ‘aggresome’ models of inclusion formation is that inclusions arise in part through impeded proteasomal degradation, with consequent accumulation of multiple polyubiquitinated protein species (Keller et al., 2002; Ardley et al., 2005). To evaluate this possibility for the nuclear inclusions found in FXTAS, 2D western analysis was performed (Fig. 3C) using an antibody (see Materials and methods) that recognizes both mono- and polyubiquitinated proteins.
Two features of the blot are readily apparent. First, only a small number of proteins, perhaps as few as five, appear to be ubiquitinated. Second, there is little evidence that any of the proteins is polyubiquitinated. This suggests that the targets of ubiquitination, which have not yet been identified due to their low abundance within the inclusions, may have been ubiquitinated for reasons other than as targets for degradation. Furthermore, it is evident from the lack of accumulation of polyubiquitinated proteins that impeded proteasomal degradation of polyubiquitinated proteins is not the basis for inclusion formation.
Peptide sequence analysis of the purified inclusions by tandem mass spectrometry reveals the presence of heterogeneous nuclear ribonucleoprotein A2 (hnRNP A2) by LC-MS/MS
As an independent approach for characterizing the protein composition of the inclusions, peptide sequencing of the total protein complement of the isolated inclusions was carried out using tandem MS. This approach has yielded ∼20 protein candidates that meet the dual criteria of XCorr and ΔCn (Table 3).
List of the 19 candidate proteins identified by tandem MS of the entire protein complement of the inclusions
| Protein | Mol wt | pI | Category | % Coverage* |
|---|---|---|---|---|
| Similar to H2A histone family, member Z | 13 449 | 10.4 | Histone family | 18 |
| H2B histone family | 13 615 | 10.1 | Histone family | 20 |
| HIST1H4D protein | 11 339 | 11.4 | Histone family | 29 |
| H2A histone family A (L) | 14 135 | 11.1 | Histone family | 30 |
| H2A histone family, member Q (O) | 13 988 | 10.9 | Histone family | 52 |
| Neurofilament 3 | 102 448 | 4.9 | Intermediate filament | 15 |
| Lamin A/C isoform 1 | 74 139 | 6.6 | Intermediate filament | 16 |
| Vimentin | 53 652 | 5.1 | Intermediate filament | 26 |
| Internexin neuronal intermediate filament protein | 55 391 | 5.3 | Intermediate filament | 29 |
| NEFL protein | 61 517 | 4.6 | Intermediate filament | 32 |
| Glial fibrillary acidic protein (GFAP) | 49 808 | 5.5 | Intermediate filament | 35 |
| β5 tubulin | 49 672 | 4.8 | Microtubule | 17 |
| Tubulin, alpha 6 | 50 136 | 4.9 | Microtubule | 28 |
| Tubulin, alpha, ubiquitous | 50 152 | 4.9 | Microtubule | 28 |
| Myelin/oligodendrocyte glycoprotein beta 3 | 11 284 | 6.1 | Myelin associated protein | 16 |
| 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) | 47 579 | 9.2 | Myelin associated protein | 24 |
| Myelin basic protein | 21 493 | 11.4 | Myelin associated protein | 26 |
| Heterogeneous nuclear ribonucleoprotein A2/B1 | 37 430 | 9.0 | RNA binding protein | 18 |
| αB-crystallin | 22 449 | 7.2 | Stress related protein | 12 |
| Protein | Mol wt | pI | Category | % Coverage* |
|---|---|---|---|---|
| Similar to H2A histone family, member Z | 13 449 | 10.4 | Histone family | 18 |
| H2B histone family | 13 615 | 10.1 | Histone family | 20 |
| HIST1H4D protein | 11 339 | 11.4 | Histone family | 29 |
| H2A histone family A (L) | 14 135 | 11.1 | Histone family | 30 |
| H2A histone family, member Q (O) | 13 988 | 10.9 | Histone family | 52 |
| Neurofilament 3 | 102 448 | 4.9 | Intermediate filament | 15 |
| Lamin A/C isoform 1 | 74 139 | 6.6 | Intermediate filament | 16 |
| Vimentin | 53 652 | 5.1 | Intermediate filament | 26 |
| Internexin neuronal intermediate filament protein | 55 391 | 5.3 | Intermediate filament | 29 |
| NEFL protein | 61 517 | 4.6 | Intermediate filament | 32 |
| Glial fibrillary acidic protein (GFAP) | 49 808 | 5.5 | Intermediate filament | 35 |
| β5 tubulin | 49 672 | 4.8 | Microtubule | 17 |
| Tubulin, alpha 6 | 50 136 | 4.9 | Microtubule | 28 |
| Tubulin, alpha, ubiquitous | 50 152 | 4.9 | Microtubule | 28 |
| Myelin/oligodendrocyte glycoprotein beta 3 | 11 284 | 6.1 | Myelin associated protein | 16 |
| 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) | 47 579 | 9.2 | Myelin associated protein | 24 |
| Myelin basic protein | 21 493 | 11.4 | Myelin associated protein | 26 |
| Heterogeneous nuclear ribonucleoprotein A2/B1 | 37 430 | 9.0 | RNA binding protein | 18 |
| αB-crystallin | 22 449 | 7.2 | Stress related protein | 12 |
Percent amino acid sequence coverage.
List of the 19 candidate proteins identified by tandem MS of the entire protein complement of the inclusions
| Protein | Mol wt | pI | Category | % Coverage* |
|---|---|---|---|---|
| Similar to H2A histone family, member Z | 13 449 | 10.4 | Histone family | 18 |
| H2B histone family | 13 615 | 10.1 | Histone family | 20 |
| HIST1H4D protein | 11 339 | 11.4 | Histone family | 29 |
| H2A histone family A (L) | 14 135 | 11.1 | Histone family | 30 |
| H2A histone family, member Q (O) | 13 988 | 10.9 | Histone family | 52 |
| Neurofilament 3 | 102 448 | 4.9 | Intermediate filament | 15 |
| Lamin A/C isoform 1 | 74 139 | 6.6 | Intermediate filament | 16 |
| Vimentin | 53 652 | 5.1 | Intermediate filament | 26 |
| Internexin neuronal intermediate filament protein | 55 391 | 5.3 | Intermediate filament | 29 |
| NEFL protein | 61 517 | 4.6 | Intermediate filament | 32 |
| Glial fibrillary acidic protein (GFAP) | 49 808 | 5.5 | Intermediate filament | 35 |
| β5 tubulin | 49 672 | 4.8 | Microtubule | 17 |
| Tubulin, alpha 6 | 50 136 | 4.9 | Microtubule | 28 |
| Tubulin, alpha, ubiquitous | 50 152 | 4.9 | Microtubule | 28 |
| Myelin/oligodendrocyte glycoprotein beta 3 | 11 284 | 6.1 | Myelin associated protein | 16 |
| 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) | 47 579 | 9.2 | Myelin associated protein | 24 |
| Myelin basic protein | 21 493 | 11.4 | Myelin associated protein | 26 |
| Heterogeneous nuclear ribonucleoprotein A2/B1 | 37 430 | 9.0 | RNA binding protein | 18 |
| αB-crystallin | 22 449 | 7.2 | Stress related protein | 12 |
| Protein | Mol wt | pI | Category | % Coverage* |
|---|---|---|---|---|
| Similar to H2A histone family, member Z | 13 449 | 10.4 | Histone family | 18 |
| H2B histone family | 13 615 | 10.1 | Histone family | 20 |
| HIST1H4D protein | 11 339 | 11.4 | Histone family | 29 |
| H2A histone family A (L) | 14 135 | 11.1 | Histone family | 30 |
| H2A histone family, member Q (O) | 13 988 | 10.9 | Histone family | 52 |
| Neurofilament 3 | 102 448 | 4.9 | Intermediate filament | 15 |
| Lamin A/C isoform 1 | 74 139 | 6.6 | Intermediate filament | 16 |
| Vimentin | 53 652 | 5.1 | Intermediate filament | 26 |
| Internexin neuronal intermediate filament protein | 55 391 | 5.3 | Intermediate filament | 29 |
| NEFL protein | 61 517 | 4.6 | Intermediate filament | 32 |
| Glial fibrillary acidic protein (GFAP) | 49 808 | 5.5 | Intermediate filament | 35 |
| β5 tubulin | 49 672 | 4.8 | Microtubule | 17 |
| Tubulin, alpha 6 | 50 136 | 4.9 | Microtubule | 28 |
| Tubulin, alpha, ubiquitous | 50 152 | 4.9 | Microtubule | 28 |
| Myelin/oligodendrocyte glycoprotein beta 3 | 11 284 | 6.1 | Myelin associated protein | 16 |
| 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) | 47 579 | 9.2 | Myelin associated protein | 24 |
| Myelin basic protein | 21 493 | 11.4 | Myelin associated protein | 26 |
| Heterogeneous nuclear ribonucleoprotein A2/B1 | 37 430 | 9.0 | RNA binding protein | 18 |
| αB-crystallin | 22 449 | 7.2 | Stress related protein | 12 |
Percent amino acid sequence coverage.
One interesting component of the inclusions is hnRNP A2. As a well-known RNA binding protein (Dreyfuss et al., 2002), hnRNP A2 is a candidate as a mediator of the toxic gain-of-function of the FMR1 mRNA, the latter now known to be present within the FXTAS inclusions (Tassone et al., 2004). hnRNP A2 was identified using the tandem MS spectra of five tryptic peptides, representing 19.1% sequence coverage. Representative tandem MS data for hnRNP A2 are presented in Fig. 4. In Fig. 4A, tandem MS data are displayed for a 15-amino acid peptide (m/z, 1378.58) with a relatively high glycine composition. The tandem MS pattern shows cleavages of 9 of the 13 peptide bonds, with most of the cleavages represented by both b and y ions. Peptide sequence coverage for hnRNP A2 is displayed in Fig. 4B.
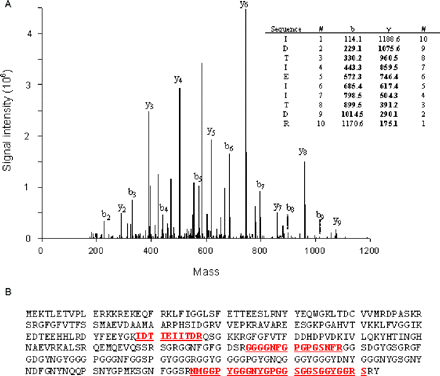
(A) Tandem MS spectrum of a doubly charged, tryptic peptide (m/z 1378.58) and sequence data interpretation for a peptide associated with hnRNP A2. Fragmentation of N-terminal and C-terminal peptide fragments correspond to b-type and y-type ions, respectively. (B) Peptides identified and matched with hnRNP A2 (underlined).
The presence of hnRNP A2 was further confirmed by 2D western analysis (Fig. 5), which additionally suggests that the RNA binding protein is post-translationally modified (multiple pIs). We have not yet identified the nature of such modifications, although their presence raises the possibility of regulation of hnRNP protein function at the post-translational level.
Western blot analysis of sorted inclusion proteins reveals several isoforms of hnRNP A2 (closed triangle). Open arrows represent anti-hnRNP A2 antibody components from FACS purification.
Identification of MBP by peptide mass mapping within the whole inclusion protein complement
One puzzling entry in Table 3 is the myelin basic protein, an important, high-abundance component of myelin and, hence, of oligodendroglial cells. Thirteen peptides have been identified for MBP by peptide mass mapping of the trypsinized inclusion complexes, followed by accurate mass analyses with FT-ICR mass spectrometry. All peptides were matched within 50 p.p.m. mass tolerance, with a total of 38% sequence coverage for the 21.5 kDa isoform (Fig. 6). A second approach, ion trap with tandem MS, also identified MBP as a significant component of the inclusions. With this latter method, nine peptides were identified, representing 26% sequence coverage of the protein.

Sequence coverage of MBP and Golli-MBP isoforms. MBP and golli-MBP peptide sequences identified by MS/MS analysis of inclusion proteins are indicated in boldface.
In addition, we have identified two peptides that map uniquely to the golli form of MBP (Pribyl et al., 1993) (Fig. 6). The identification of the golli-MBP protein is interesting in that it is known to be expressed in non-oligodendroglial cells, and is found in the nuclear compartment, suggesting a possible regulatory role for this protein.
Identification of MBNL1 by peptide mass fingerprinting within whole inclusion protein complement
Tryptic peptides of another RNA binding protein, MBNL1, (Kino et al., 2004), were found in the inclusion digests. Peptide fingerprinting yielded five peptides (mass accuracy within 20 p.p.m.) matching MBNL1, representing sequence coverage of ∼19%. While further analysis by tandem MS was not performed, immunostaining results support the suggestion that the trinucleotide binding protein is associated with the inclusions (see below).
Immunofluorescence staining of inclusions within intact, isolated nuclei provides an efficient method for identifying inclusion-associated proteins in a low-autofluorescence (DAPI-counterstained) background
One of the main caveats associated with the 2D western and MS analyses, as with the purification protocol itself, is that abundant cellular proteins may become non-specifically associated with the inclusions during the course of the purification. To address this issue, we utilized Immunofluorescence staining of isolated nuclei to assess the localization of various candidate proteins that were identified through western and/or MS analysis of isolated inclusions (Fig. 7). As a secondary confirmation of the nuclear staining results, we also performed in situ staining of fixed tissues (Fig. 8) for four of the proteins represented in Fig. 7, with essentially identical results. The use of nuclear isolates avoids the problem of autofluorescence that occurs with histological sections. Double Immunofluorescence staining confirmed co-localization of several proteins, including hnRNP A2, golli-MBP, MBP and MBNL1, within ubiquitin-positive intranuclear inclusions. Negative controls, non-immune rabbit and mouse immunoglobulins, at concentrations comparable to primary antibodies, did not stain inclusions. Additionally, fluorescent secondary antibodies failed to localize to inclusions in the absence of primary antibodies. While the MS analysis of purified inclusions detected peptides for 2′3′ CNPase, immunostaining failed to detect it (data not shown). Thus, some 2′3′ CNPase may be non-specifically trapped during the purification process. We note that immunostaining of >20 000 cells in a series of normal control brains has failed to detect a single intranuclear inclusion (Greco et al., unpublished data).
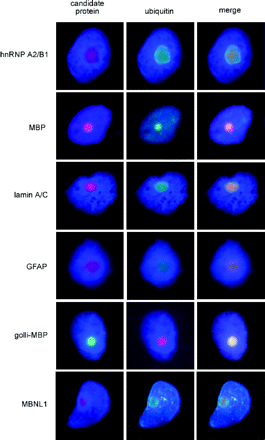
Double immunofluorescence staining of protein candidates identified by tandem MS analysis of purified inclusions, to assess co-localization of proteins with ubiquitin-positive inclusions. Immunostaining for various candidate proteins (left column); anti-ubiquitin immunostaining (centre column); and merged images demonstrating co-localization of candidate proteins with ubiquitin-positive inclusions (right column).
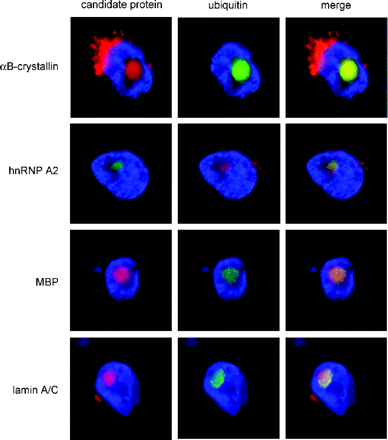
Double immunofluorescence staining of frontal cortex sections from FXTAS brains for four of the proteins represented in Fig. 7.
Discussion
Using a combination of nuclear isolation and preparative particle sorting protocols, the latter employing dual immunofluorescence labelling, we have successfully isolated highly purified intranuclear inclusions from post-mortem brain tissue of two FXTAS patients. To our knowledge, this study represents the most detailed examination to date of any inclusion associated with a neurodegenerative disorder. Determination of the identities of the proteins comprising the inclusions has been greatly facilitated by the use of modern proteomics approaches. In particular, we have been able to make multiple protein assignments from tryptic digests of whole, purified inclusions, without the need for further fractionation. This approach provides a rapid and general method for studying similar, complex systems. Further analysis of the purified inclusions by 2D gel electrophoresis and additional immunochemical probing enabled us to broadly characterize the protein complement of the inclusions.
The absence of a predominant protein species in the inclusions, with the most prominent protein spots on the 2D gels accounting for not more than ∼7% of the total protein mass, argues against pathways of inclusion formation that involve accretion of specific, abnormal proteins (Wood et al., 2003). Such mechanisms are thought to occur with many of the expanded CAG (polyglutamine) repeat disorders (Paulson, 1999; Zoghbi and Orr, 2000; Tarlac and Storey, 2003), Alzheimer's disease (β-amyloid), the tauopathies (tau protein isoforms) and Parkinson's disease (α-synuclein) (Taylor et al., 2002; Ross and Poirier, 2004). The lack of a principal protein species in the inclusions is not surprising, since there is no known abnormal protein product associated with FXTAS. In particular, the protein product of the FMR1 gene, FMRP, is not structurally abnormal, as the expanded CGG repeat is located in a non-coding portion (5′-UTR, 5′-untranslated region) of the gene. Moreover, FMRP levels are not elevated among premutation carriers, nor do we detect significant quantities of FMRP within the inclusions. However, the current results do not preclude models for aggregation in which a minor protein species acts as a trigger for the aggregation of numerous other proteins (see below).
The current observations also argue against more specific aggregation models in which the generation of misfolded and/or polyubiquitinated proteins exceeds the capacity of the proteasomal degradation pathway (Johnston et al., 1998; Kopito, 2000; Bence et al., 2001; Chung et al., 2001; Waelter et al., 2001; Klimaschewski, 2003; Miller and Wilson, 2003). We observe that as few as five or six proteins appear to be ubiquitinated in the purified inclusions, contrary to the expectation for a general failure of proteasomal degradation of ubiquitinated proteins. Indeed, such proteins may not be polyubiquitinated, but rather mono-ubiquitinated, and would thus be less likely to be substrates for proteasomal degradation (Ciechanover, 1998; Hagerman et al., 2003; Li et al., 2003). We have not yet identified the proteins that are ubiquitinated, due to their very low abundance; however, their identification is of interest due to the potential regulatory importance of mono-ubiquitination events (Hicke, 2001; Bach and Ostendorff, 2003; Muratani and Tansey, 2003).
Our finding of hnRNP A2 within the inclusions raises the possibility that this well-known RNA binding protein (Dreyfuss et al., 2002) is involved in the pathogenesis of FXTAS. A central prediction of the RNA toxic gain-of-function model for FXTAS (Hagerman et al., 2001; Greco et al., 2002; Jin et al., 2003; Hagerman and Hagerman, 2004) is that one or more RNA binding proteins is necessary to ‘transduce’ the effect of the abnormal FMR1 mRNA, which has now been identified within the inclusions of FXTAS (Tassone et al., 2004). In myotonic dystrophy, pathogenesis is also thought to involve a toxic gain-of-function of the non-coding CUG repeat within the 3′-UTR of the myotonic dystrophy protein kinase gene (DMPK) mRNA (muscular dystrophy type 1, DM1), or the non-coding (intronic) CCUG repeat of the zinc finger protein 9 gene (ZNF9) mRNA (muscular dystrophy type 2, DM2) (Finsterer, 2002; Mankodi and Thornton, 2002). The transducing function appears to be mediated, at least in part, by MBNL1, a homologue of Drosophila muscleblind (Miller et al., 2000; Fardaei et al., 2001; Ranum and Day, 2004). Interestingly, MBNL1 was identified by FT-ICR as an inclusion-associated protein. Since MBNL1 is a CUG/CCUG binding protein, it could interact with the 5′-UTR of the FMR1 mRNA; this possibility is currently being investigated.
Although any postulated role of hnRNP A2 in FXTAS would be speculative at this point, both the co-localization of FMR1 mRNA and hnRNP A2 within the inclusions and preliminary evidence (C. K. Iwahashi and P. J. Hagerman, unpublished data) for the direct binding of hnRNP A2 to the 5′-UTR of FMR1 mRNA are consistent with some form of involvement of hnRNP A2 in FXTAS. For example, were hnRNP A2 to be partially sequestered through its interaction with expanded-repeat FMR1 mRNA, transport of other mRNAs that are also mediated by hnRNP A2 (including MBP mRNA within oligodendroglial cells) may be adversely affected (Ainger et al., 1993, 1997; Munro et al., 1999; Barbarese et al., 1999; Carson and Barbarese, 2005). Partial sequestration of hnRNP A2 could also interfere with its function as a translational activator (Barbarese et al., 1999; Kwon et al., 1999). It is noteworthy that white matter disease, with apparent loss of axons and myelin, is a central feature of the neuropathology of FXTAS (Brunberg et al., 2002; Greco et al., 2002; Jacquemont et al., 2003). This raises the possibility that reduced efficiency of transport of MBP mRNA, secondary to partial sequestration of hnRNP A2, could contribute to the loss of myelinated axons, or to the inability to adequately maintain myelinated cells in FXTAS. These possibilities are currently being investigated.
The inclusions also appear to contain several intermediate filament (IF) proteins, including lamins A/C, α-internexin and other neurofilament (NF) proteins. Although the role(s) of the A-type nuclear lamins is only beginning to emerge, they may be involved with the regulation of RNA synthesis and processing (Hutchison and Worman, 2004; Zastrow et al., 2004). Thus, the lamins could also be involved in mediating the effects of the expanded-repeat FMR1 mRNA. α-internexin has also been found within the (cytoplasmic) inclusions of neuronal IF inclusion disease (Cairns et al., 2004), and accumulations of NF proteins have been described in a growing number of neurological disorders (Omary et al., 2004). Such accumulations could be due to failure of axonal transport (Cairns et al., 2004), which could also contribute to the axonal loss observed in FXTAS (Greco et al., 2002). In any case, the presence of IF proteins and the RNA binding proteins, hnRNP A2 and MBNL1, within the inclusions signals a clear avenue for further research on the pathogenesis of FXTAS.
Although the inclusions associated with FXTAS contain both ubiquitin and αB-crystallin, neither protein is unique to this disorder. Indeed, ubiquitin is present within the intracellular aggregates of a wide range of neurological disorders. In the current instance, the presence of ubiquitin, reported earlier in Greco et al. (2002), has been exploited as a marker for isolation. Similarly, αB-crystallin, a small heat shock protein (HSP) used as our second marker, is a component of the Lewy bodies of Parkinson's disease (Gai et al., 1999) and a component of Rosenthal fibres, a cytoplasmic inclusion found in the astrocytes of Alexander's disease patients (Gordon, 2003). In addition, αB-crystallin is associated with the astroglial inclusions that are found in ‘tauopathies’ such as progressive supranuclear palsy (Dabir et al., 2004). Furthermore, although both proteins are clearly present in the inclusions of FXTAS, neither is abundant. It is interesting that αB-crystallin acts as a protein chaperone, facilitating the correct folding of misfolded proteins (den Engelsman et al., 2004). Small HSPs, including αB-crystallin, oligomerize to form a complex quaternary structure that is essential for its chaperone function (MacRae, 2000). Phosphorylation of serine residues (ser-19, ser-45 and/or ser-59) reduces the size of the oligomer, and alters its activity and intracellular location (Ito et al., 2001; Aquilina et al., 2004). Our 2D Western analysis of purified inclusions revealed the presence of several αB-crystallin isoforms. However, it is unknown at present what role(s) (if any) these various isoforms play in disease formation in FXTAS. Finally, we have found no evidence for the presence of either tau isoforms or α-synuclein in the inclusions.
MBP, an oligodendrocyte related protein, is a major component of the myelin sheath. The 18.5 kDa isoform, ‘classic’ MBP, is one of the four isoforms that arises as a result of differential splicing of a single transcript in humans (Kamholz et al., 1986, 1988). The largest isoforms, 21.5 and 20.2 kDa, contain a 26 amino acid sequence encoded by exon II. These larger forms do not appear to be membrane-associated, but have been localized in the nucleus (Pedraza et al., 1997). Based on MS/MS sequences, we were able to eliminate the 20.2 kDa isoform, but could not further discriminate among the remaining three isoforms. The seven exons of the MBP gene are part of a larger complex, golli-MBP, which contains three upstream exons. Expression of golli-MBP is not restricted to the nervous system and is found in tissues and cells of the immune systems (Pribyl et al., 1993). Golli-MBP is localized primarily in axonal and dendritic processes of neurons but is also found transiently in neuronal nuclei and in oligodendrocytes (Landry et al., 1996). Two golli-MBP peptides were identified in the inclusion preparations. As one of the golli-MBP isoforms contains the 18.5 kDa MBP, it is possible that the inclusions contain either golli-MBP alone or both golli-MBP and MBP.
Potential clinical significance of the current studies
Clinical involvement of FXTAS has recently been described in women carriers of premutation alleles (Hagerman and Hagerman, 2004), who frequently also experience focal sensory deficits and/or muscle weakness that can have a clinical presentation similar to multiple sclerosis (Hagerman and Hagerman, 2004; Zhang et al., 2005). In fact, peripheral neuropathy, common in both male and female carriers with FXTAS, may be the presenting clinical feature, occurring even before the onset of tremor or ataxia. In particular, we have recently seen two males with FXTAS whose initial clinical diagnosis was Charcot-Marie-Tooth (CMT), but who were negative for known genetic forms of CMT. These clinical observations may, in fact, be mechanistically linked to FXTAS, since abnormalities of the αB-crystallin domain of HSP 27 have been linked to CMT (Evgrafov et al., 2004). Thus, as the pathogenic mechanism of FXTAS is further elucidated, we may be better able to understand the basis for the more variable features of the disorder in addition to the core features of the disease.
This work was supported by NIH grants HD 40661 (P.J.H.) and HD36071 (R.J.H.), by the Boory Family Fund, and by the UC Davis M.I.N.D. Institute for general laboratory support. The authors wish to thank Drs C. Campagnoni and M. Swanson for generously providing antibodies to golli-MBP and MBNL1, respectively. The authors have no financial conflicts of interest.
References
Ainger K, Avossa D, Morgan F, Hill SJ, Barry C, Barbarese E, et al. Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes.
Ainger K, Avossa D, Diana AS, Barry C, Barbarese E, Carson JH. Transport and localization elements in myelin basic protein mRNA.
Aquilina JA, Benesch JL, Ding LL, Yaron O, Horwitz J, Robinson CV. Phosphorylation of alphaB-crystallin alters chaperone function through loss of dimeric substructure.
Ardley HC, Hung CC, Robinson PA. The aggravating role of the ubiquitin-proteasome system in neurodegeneration.
Bach I, Ostendorff HP. Orchestrating nuclear functions: ubiquitin sets the rhythm.
Barbarese E, Brumwell C, Kwon S, Cui H, Carson JH. RNA on the road to myelin.
Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation.
Berry-Kravis E, Lewin F, Wuu J, Leehey M, Hagerman R, Hagerman P, et al. Tremor and ataxia in fragile X premutation carriers: blinded videotape study.
Brunberg JA, Jacquemont S, Hagerman RJ, Berry-Kravis EM, Grigsby J, Leehey MA, et al. Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction.
Cairns NJ, Zhukareva V, Uryu K, Zhang B, Bigio E, Mackenzie IR, et al. alpha-internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease.
Carson JH, Barbarese E. Systems analysis of RNA trafficking in neural cells.
Chung KK, Dawson VL, Dawson TM. The role of the ubiquitin-proteasomal pathway in Parkinson's disease and other neurodegenerative disorders.
Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life.
Dabir DV, Trojanowski JQ, Richter-Landsberg C, Lee VM, Forman MS. Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology.
den Engelsman J, Bennink EJ, Doerwald L, Onnekink C, Wunderink L, Andley UP, et al. Mimicking phosphorylation of the small heat-shock protein alphaB-crystallin recruits the F-box protein FBX4 to nuclear SC35 speckles.
Dombrowski C, Levesque S, Morel ML, Rouillard P, Morgan K, Rousseau F. Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles.
Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry.
Eng JK, McCormack AL, Yates I, John R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database.
Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy.
Fardaei M, Larkin K, Brook JD, Hamshere MG. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts.
Gai WP, Power JH, Blumbergs PC, Culvenor JG, Jensen PH. Alpha-synuclein immunoisolation of glial inclusions from multiple system atrophy brain tissue reveals multiprotein components.
Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications.
Gatlin CL, Kleemann GR, Hays LG, Link AJ, Yates JR, 3rd. Protein identification at the low femtomole level from silver-stained gels using a new fritless electrospray interface for liquid chromatography-microspray and nanospray mass spectrometry.
Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers.
Greco C, Berman R, Martin R, Tassone F, Schwartz P, Brunberg J, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain (In press).
Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective.
Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X.
Hagerman P, Iwahashi C, Babineau B, Yasui D, Greco CM, Duncan M, et al. Fragile X-associated tremor/ataxia syndrome (FXTAS): a common heritable neuronal inclusion disorder.
Hazeki N, Tsukamoto T, Yazawa I, Koyama M, Hattori S, Someki I, et al. Ultrastructure of nuclear aggregates formed by expressing an expanded polyglutamine.
Ito H, Kamei K, Iwamoto I, Inaguma Y, Nohara D, Kato K. Phosphorylation-induced change of the oligomerization state of alpha B-crystallin.
Iwatsubo T, Yamaguchi H, Fujimuro M, Yokosawa H, Ihara Y, Trojanowski JQ, et al. Purification and characterization of Lewy bodies from the brains of patients with diffuse Lewy body disease.
Iwatsubo T, Yamaguchi H, Fujimuro M, Yokosawa H, Ihara Y, Trojanowski JQ, et al. Lewy bodies: purification from diffuse Lewy body disease brains.
Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates.
Jacquemont S, Farzin F, Hall D, Leehey M, Tassone F, Gane L, et al. Aging in individuals with the FMR1 mutation.
Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, Moses K, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila.
Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins.
Kamholz J, de Ferra F, Puckett C, Lazzarini R. Identification of three forms of human myelin basic protein by cDNA cloning.
Kamholz J, Toffenetti J, Lazzarini RA. Organization and expression of the human myelin basic protein gene.
Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers.
Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats.
Kwon S, Barbarese E, Carson JH. The cis-acting RNA trafficking signal from myelin basic protein mRNA and its cognate trans-acting ligand hnRNP A2 enhance cap-dependent translation.
Landry C, Ellison J, Pribyl T, Campagnoni C, Kampf K, Campagnoni A. Myelin basic protein gene expression in neurons: developmental and regional changes in protein targeting within neuronal nuclei, cell bodies, and processes.
Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies.
Leehey MA, Munhoz RP, Lang AE, Brunberg JA, Grigsby J, Greco C, et al. The fragile X premutation presenting as essential tremor.
Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2.
MacCoss MJ, Wu CC, Yates JR, 3rd. Probability-based validation of protein identifications using a modified SEQUEST algorithm.
MacRae TH. Structure and function of small heat shock/alpha-crystallin proteins: established concepts and emerging ideas.
Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: a primer.
Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy.
Miller RJ, Wilson SM. Neurological disease: UPS stops delivering!
Munro TP, Magee RJ, Kidd GJ, Carson JH, Barbarese E, Smith LM, et al. Mutational analysis of a heterogeneous nuclear ribonucleoprotein A2 response element for RNA trafficking.
Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription.
Omary MB, Coulombe PA, McLean WH. Intermediate filament proteins and their associated diseases.
Paulson HL. Protein fate in neurodegenerative proteinopathies: polyglutamine diseases join the (mis)fold.
Pedraza L, Fidler L, Staugaitis SM, Colman DR. The active transport of myelin basic protein into the nucleus suggests a regulatory role in myelination.
Pribyl TM, Campagnoni CW, Kampf K, Kashima T, Handley VW, McMahon J, et al. The human myelin basic protein gene is included within a 179-kilobase transcription unit: expression in the immune and central nervous systems.
Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus.
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease.
Sadygov RG, Yates JR, 3rd. A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases.
Tabb DL, McDonald WH, Yates JR, 3rd. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics.
Tarlac V, Storey E. Role of proteolysis in polyglutamine disorders.
Tassone F, Hagerman RJ, Chamberlain WD, Hagerman PJ. Transcription of the FMR1 gene in individuals with fragile X syndrome.
Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in fragile X syndrome.
Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS).
Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease.
Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, et al. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation.
Washburn MP, Wolters D, Yates JR, 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology.
Wood JD, Beaujeux TP, Shaw PJ. Protein aggregation in motor neurone disorders.
Zastrow MS, Vlcek S, Wilson KL. Proteins that bind A-type lamins: integrating isolated clues.
Zhang L, Apperson M, Coffey S, Agius M, Nowicki S, Tartaglia NR, et al. Clinical and MR abnormalities in females carrying the FMR1 premutation. 57th Annual Meeting of the American Academy of Neurology. Miami Beach, FL: Neurology,
Author notes
1Department of Biochemistry and Molecular Medicine, 2Department of Pathology and 3M.I.N.D. Institute, School of Medicine and 4Department of Chemistry, University of California, Davis, CA, USA


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}