





Abstract
Compaction of DNA into chromatin is a characteristic feature of eukaryotic organisms. The core (H2A, H2B, H3, H4) and linker (H1) histone proteins are responsible for this compaction through the formation of nucleosomes and higher order chromatin aggregates. Moreover, histones are intricately involved in chromatin functioning and provide a means for genome dynamic regulation through specific histone variants and histone post-translational modifications. ‘HistoneDB 2.0 – with variants’ is a comprehensive database of histone protein sequences, classified by histone types and variants. All entries in the database are supplemented by rich sequence and structural annotations with many interactive tools to explore and compare sequences of different variants from various organisms. The core of the database is a manually curated set of histone sequences grouped into 30 different variant subsets with variant-specific annotations. The curated set is supplemented by an automatically extracted set of histone sequences from the non-redundant protein database using algorithms trained on the curated set. The interactive web site supports various searching strategies in both datasets: browsing of phylogenetic trees; on-demand generation of multiple sequence alignments with feature annotations; classification of histone-like sequences and browsing of the taxonomic diversity for every histone variant. HistoneDB 2.0 is a resource for the interactive comparative analysis of histone protein sequences and their implications for chromatin function.
Database URL:http://www.ncbi.nlm.nih.gov/projects/HistoneDB2.0
Introduction
Nucleosomes constitute the elementary building blocks of chromatin and play important functional roles in epigenetic regulation of transcription, replication, cell development and reprogramming. Each nucleosome core particle consists of about 147 base pairs (bp) of DNA wrapped around an octamer of histone proteins—two copies of H3, H4, H2A and H2B (see Figure 1) (1, 2). Adjacent nucleosomes are separated by stretches of linker DNA of varying length up to about 100 bp. All four types of core histones share the same ‘histone fold’ while the sequence identity between them is rather low (3) (not exceeding 25% according to our estimates). The linker histone H1 usually binds to the nucleosome core and linker DNA to form ‘chromatosomes’ and promotes further chromatin compaction. A linker histone H1 has a different fold and makes a unique set of interactions with the linker DNA (4). The structure of the nucleosome core as revealed by X-ray crystallography is fairly conserved throughout all eukaryotes irrespective of histone sequence variants, mutations and post-translational modifications (5). However, it has been revealed that chromatin and nucleosomes may undergo substantial conformational changes and the balance between different conformations may be shifted even by subtle changes in histone sequences (6, 7) Most eukaryotes have histone variants for some or all of the histone families (H2A, H2B, H3, H4, H1) (see Figure 1). In addition, nucleosomes may employ different sets of histone variants and post-translational modifications, which may be essential for sustaining their diverse functions and responding to environmental stimuli (8–10).
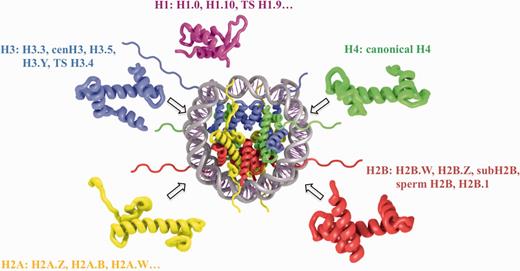
Schematic representation of nucleosome structure and its composition of different histone variants. The nucleosome core is formed by 147 bp of DNA and an octamer of H3, H4, H2A and H2B histones (depicted in blue, green, yellow and red, respectively). H1 linker histone (magenta) is associated with the nucleosome core near the DNA entry exit points. Selected histone variant names for each histone type are shown.
Classification of histones is a daunting task. They are usually subdivided into ‘canonical’ replication-dependent histones that are expressed during the S-phase of cell cycle and replication-independent histone ‘variants’, constitutively expressed during the cell cycle (11). This division is based on the history of their discovery and has its limitations. For example, ‘Canonical’ histones in plants and animals usually encompass a distinct set of replication-dependent H2A and H2B, while in many unicellular organisms, there is no special set of ‘canonical’ replication-coupled paralogs, and ‘variants’ fulfill their roles, rendering the distinction between these classes meaningless in these organisms. In animals, genes encoding canonical histones are typically clustered along the chromosome, lack introns and employ a specific type of regulation at the RNA level with a stem loop structure at the 3’ end instead of polyA tail. On the other hand, genes encoding histone variants are usually not clustered, have introns and their mRNAs are regulated with polyA tails similar to the mRNAs of most genes (12). In plants, canonical histone genes lack introns, but are not clustered and the mRNAs are polyadenylated (13). Remarkably, more complex multicellular organisms typically have a higher number of histone variants providing a variety of different functions. Recent data are accumulating about the roles of diverse histone variants highlighting the functional links between variants and the delicate regulation of organism development. Some of the most striking examples include the importance of the H2A.Z variant in memory consolidation (14) and the modulation of the olfactory neurons life span by histone variant H2B.E in mice (15).
Each histone variant has characteristic sequence and structural features that account for its specific function. The difference between canonical histones and variants can be minor with only very few amino acid changes (e.g. canonical H3 and H3.3) and overall conservation of most structural features. However, in some particular cases histone variants might vary from their canonical counterparts in sequence as much as different families of canonical histones differ from each other (up to 25% identity). Variant sequences may have shortened or extended N- and C-terminal tails and may have characteristic regions with physicochemical properties drastically dissimilar from the canonical histones. Many of these features and their functional implications are largely unknown and/or poorly annotated. The phylogenetic origin of histone variants has been addressed in several studies, which pointed to a monophyletic origin for some variants while others, including canonical histones, were found to originate repeatedly in evolution (16–19). Although some histone variants can have unique characteristic post-translational modification patterns, the majority of them remain to be characterized.
In this article, we present a database ‘HistoneDB 2.0 – with Variants’ that collects canonical histones and histone variants, their sequence, structural and functional features. This database is the successor to a previous ‘Histone Database’ which represented a curated collection of sequences and structures of histones and non-histone proteins containing histone folds (20–22). The HistoneDB 2.0 consists of two parts. First, we compiled a manually curated set of histone variants and their multiple sequence alignments with the expert annotated characteristic features and descriptions of their functions. Second, we constructed Hidden Markov Models (HMM) based on these alignments and used them together with the sequence motif identification algorithms to search a sequence of interest or all sequences from the non-redundant sequence (nr) database given that they pass rigorous criteria established in this study. As a result, automatic annotations of histone variants were produced. Furthermore, HistoneDB 2.0 allows for comparing variants and their features to each other within the same or different species. The phylogenetic tree of histone variants offers another important evolutionary aspect of the database so that it is feasible to browse through the lineage-specific or universally conserved variants and decipher their characteristic features. The database promotes the new nomenclature for histone variants proposed recently (16).
Database source and contents
The HistoneDB 2.0 database consists of two complimentary parts (i) a set of manually curated and annotated variants and (ii) a set of automatically extracted, classified and annotated histone sequences from non-redundant database of protein sequences maintained by NCBI (nr). Below we summarize each part of the database and its content, including feature descriptions and annotations. Data types, organization of the database and key widgets of the web site are highlighted in Figure 2.
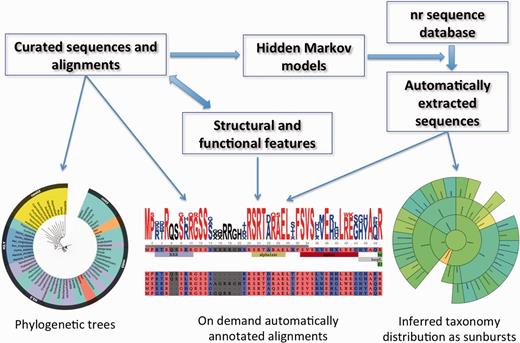
Information flow, main data types and widgets in HistoneDB 2.0 (see text for description).
Curated set of histone sequences and alignments
For each histone type, H2A, H2B, H3, H4 and H1, we collected histone variant sequences from the previous manual classification described in (16) (so called ‘curated sequences’ set) and appended it with a set of canonical histones for a wide set of species. These sequences were aligned using the MUSCLE alignment tool (23) and alignments were further checked manually to ensure they had a wide taxonomic span, did not contain insertions or deletions in the core histone fold regions and were in agreement with the structural alignment of the available PDB structures of histones and nucleosomes (‘curated alignments’ set).
The curated set contains histone sequences classified in total into 30 different groups representing major histone variants and canonical histones from core families H2A, H2B, H3, H4 and linker histone H1 family. The canonical sets of sequences for core histones and a generic set for H1 histone (see below) are provided as separate groups within the corresponding histone families. Note that for the majority of organisms no variants are available for H4 histone. Each histone type and variant has an annotation record in the database with a brief description, relevant references, structural and functional features.
We adhere to the naming convention of histone variants that was put forward in (16), while we provide alternative names on the summary page for every variant. The canonical histones are referenced using the name of histone type prefixed by ‘canonical’. The current version of the database focuses on indexing the major structurally distinct monophyletic clades of histone families, which according to new nomenclature are denoted with letter suffixes or prefixes (eg. H2A.Z, cenH3, etc.) (16). However, the database also includes certain variants that are denoted by number suffixes. According to (16) the variants with number suffixes should be assumed to be species-specific, but in related species, where unique orthologies are clear the variants with number suffixes should correspond to related proteins. In the current version of the database, we opted to include groups of number suffixed variants when the sequences within each group are known to be related within a certain taxonomic span.
Below we briefly describe different histone groups indexed in the database and their main features. For brevity we do not describe the general features and functions of histone type families, although they are also available in the database. The statistics of our curated and automatically annotated sets are given in Table 1.
A summary table of histone variants in HistoneDB 2.0
| Variant | # Curated sequences | # Automatically extracted sequences | # Features | Taxonomic span |
|---|---|---|---|---|
| Canonical H3 | 26 | 1606 | 11 | Eukaryotes |
| cenH3 | 14 | 276 | 15 | Eukaryotes |
| H3.3 | 17 | 541 | 12 | Eukaryotes |
| H3.5 | 2 | 1135 | 11 | Hominids |
| H3.Y | 8 | 89 | 12 | Primates |
| TS H3.4 | 2 | 2110 | 11 | Mammals |
| Canonical H4 | 14 | 7498 | 10 | Eukaryotes |
| Canonical H2A | 39 | 4096 | 17 | Eukaryotes |
| H2A.1 | 2 | 846 | 17 | Mammals |
| H2A.B | 15 | 139 | 21 | Mammals |
| H2A.L | 17 | 186 | 17 | Certain mammals |
| H2A.P | 11 | 95 | 17 | Placentalia |
| H2A.W | 9 | 870 | 19 | Plants |
| H2A.X | 22 | 1142 | 19 | Eukaryotes except nematode |
| H2A.Z | 25 | 2609 | 20 | Eukaryotes |
| macroH2A | 10 | 1215 | 19 | Vertebrates(?) |
| Canonical H2B | 27 | 6633 | 9 | Eukaryotes |
| H2B.1 | 4 | 443 | 9 | Mammals |
| H2B.W | 6 | 245 | 10 | Mammals |
| H2B.Z | 3 | 208 | 9 | Apicomplexa |
| Sperm H2B | 5 | 56 | 10 | Echinoidea(?) |
| subH2B | 11 | 86 | 11 | Primates, rodents, marsupials, and bovids |
| Generic H1 | 18 | 4340 | 7 | Eukaryotes |
| H1.0 | 15 | 681 | 7 | Metazoa |
| H1.10 | 6 | 146 | 7 | Vertebrates |
| OO H1.8 | 2 | 250 | 7 | Mammals |
| scH1 | 2 | 404 | 13 | Saccharomyces(?) |
| TS H1.6 | 8 | 474 | 7 | Mammals |
| TS H1.7 | 2 | 144 | 7 | Mammals |
| TS H1.9 | 4 | 101 | 7 | Mammals |
| Variant | # Curated sequences | # Automatically extracted sequences | # Features | Taxonomic span |
|---|---|---|---|---|
| Canonical H3 | 26 | 1606 | 11 | Eukaryotes |
| cenH3 | 14 | 276 | 15 | Eukaryotes |
| H3.3 | 17 | 541 | 12 | Eukaryotes |
| H3.5 | 2 | 1135 | 11 | Hominids |
| H3.Y | 8 | 89 | 12 | Primates |
| TS H3.4 | 2 | 2110 | 11 | Mammals |
| Canonical H4 | 14 | 7498 | 10 | Eukaryotes |
| Canonical H2A | 39 | 4096 | 17 | Eukaryotes |
| H2A.1 | 2 | 846 | 17 | Mammals |
| H2A.B | 15 | 139 | 21 | Mammals |
| H2A.L | 17 | 186 | 17 | Certain mammals |
| H2A.P | 11 | 95 | 17 | Placentalia |
| H2A.W | 9 | 870 | 19 | Plants |
| H2A.X | 22 | 1142 | 19 | Eukaryotes except nematode |
| H2A.Z | 25 | 2609 | 20 | Eukaryotes |
| macroH2A | 10 | 1215 | 19 | Vertebrates(?) |
| Canonical H2B | 27 | 6633 | 9 | Eukaryotes |
| H2B.1 | 4 | 443 | 9 | Mammals |
| H2B.W | 6 | 245 | 10 | Mammals |
| H2B.Z | 3 | 208 | 9 | Apicomplexa |
| Sperm H2B | 5 | 56 | 10 | Echinoidea(?) |
| subH2B | 11 | 86 | 11 | Primates, rodents, marsupials, and bovids |
| Generic H1 | 18 | 4340 | 7 | Eukaryotes |
| H1.0 | 15 | 681 | 7 | Metazoa |
| H1.10 | 6 | 146 | 7 | Vertebrates |
| OO H1.8 | 2 | 250 | 7 | Mammals |
| scH1 | 2 | 404 | 13 | Saccharomyces(?) |
| TS H1.6 | 8 | 474 | 7 | Mammals |
| TS H1.7 | 2 | 144 | 7 | Mammals |
| TS H1.9 | 4 | 101 | 7 | Mammals |
For each histone variant the numbers of sequences in curated and automatically extracted data sets, a number of annotated sequence features and inferred taxonomic span is given. Question marks denote ambiguous taxonomic spans.
A summary table of histone variants in HistoneDB 2.0
| Variant | # Curated sequences | # Automatically extracted sequences | # Features | Taxonomic span |
|---|---|---|---|---|
| Canonical H3 | 26 | 1606 | 11 | Eukaryotes |
| cenH3 | 14 | 276 | 15 | Eukaryotes |
| H3.3 | 17 | 541 | 12 | Eukaryotes |
| H3.5 | 2 | 1135 | 11 | Hominids |
| H3.Y | 8 | 89 | 12 | Primates |
| TS H3.4 | 2 | 2110 | 11 | Mammals |
| Canonical H4 | 14 | 7498 | 10 | Eukaryotes |
| Canonical H2A | 39 | 4096 | 17 | Eukaryotes |
| H2A.1 | 2 | 846 | 17 | Mammals |
| H2A.B | 15 | 139 | 21 | Mammals |
| H2A.L | 17 | 186 | 17 | Certain mammals |
| H2A.P | 11 | 95 | 17 | Placentalia |
| H2A.W | 9 | 870 | 19 | Plants |
| H2A.X | 22 | 1142 | 19 | Eukaryotes except nematode |
| H2A.Z | 25 | 2609 | 20 | Eukaryotes |
| macroH2A | 10 | 1215 | 19 | Vertebrates(?) |
| Canonical H2B | 27 | 6633 | 9 | Eukaryotes |
| H2B.1 | 4 | 443 | 9 | Mammals |
| H2B.W | 6 | 245 | 10 | Mammals |
| H2B.Z | 3 | 208 | 9 | Apicomplexa |
| Sperm H2B | 5 | 56 | 10 | Echinoidea(?) |
| subH2B | 11 | 86 | 11 | Primates, rodents, marsupials, and bovids |
| Generic H1 | 18 | 4340 | 7 | Eukaryotes |
| H1.0 | 15 | 681 | 7 | Metazoa |
| H1.10 | 6 | 146 | 7 | Vertebrates |
| OO H1.8 | 2 | 250 | 7 | Mammals |
| scH1 | 2 | 404 | 13 | Saccharomyces(?) |
| TS H1.6 | 8 | 474 | 7 | Mammals |
| TS H1.7 | 2 | 144 | 7 | Mammals |
| TS H1.9 | 4 | 101 | 7 | Mammals |
| Variant | # Curated sequences | # Automatically extracted sequences | # Features | Taxonomic span |
|---|---|---|---|---|
| Canonical H3 | 26 | 1606 | 11 | Eukaryotes |
| cenH3 | 14 | 276 | 15 | Eukaryotes |
| H3.3 | 17 | 541 | 12 | Eukaryotes |
| H3.5 | 2 | 1135 | 11 | Hominids |
| H3.Y | 8 | 89 | 12 | Primates |
| TS H3.4 | 2 | 2110 | 11 | Mammals |
| Canonical H4 | 14 | 7498 | 10 | Eukaryotes |
| Canonical H2A | 39 | 4096 | 17 | Eukaryotes |
| H2A.1 | 2 | 846 | 17 | Mammals |
| H2A.B | 15 | 139 | 21 | Mammals |
| H2A.L | 17 | 186 | 17 | Certain mammals |
| H2A.P | 11 | 95 | 17 | Placentalia |
| H2A.W | 9 | 870 | 19 | Plants |
| H2A.X | 22 | 1142 | 19 | Eukaryotes except nematode |
| H2A.Z | 25 | 2609 | 20 | Eukaryotes |
| macroH2A | 10 | 1215 | 19 | Vertebrates(?) |
| Canonical H2B | 27 | 6633 | 9 | Eukaryotes |
| H2B.1 | 4 | 443 | 9 | Mammals |
| H2B.W | 6 | 245 | 10 | Mammals |
| H2B.Z | 3 | 208 | 9 | Apicomplexa |
| Sperm H2B | 5 | 56 | 10 | Echinoidea(?) |
| subH2B | 11 | 86 | 11 | Primates, rodents, marsupials, and bovids |
| Generic H1 | 18 | 4340 | 7 | Eukaryotes |
| H1.0 | 15 | 681 | 7 | Metazoa |
| H1.10 | 6 | 146 | 7 | Vertebrates |
| OO H1.8 | 2 | 250 | 7 | Mammals |
| scH1 | 2 | 404 | 13 | Saccharomyces(?) |
| TS H1.6 | 8 | 474 | 7 | Mammals |
| TS H1.7 | 2 | 144 | 7 | Mammals |
| TS H1.9 | 4 | 101 | 7 | Mammals |
For each histone variant the numbers of sequences in curated and automatically extracted data sets, a number of annotated sequence features and inferred taxonomic span is given. Question marks denote ambiguous taxonomic spans.
Histone H2A family
Histone H2A has the highest number of known variants (nine sequence groups indexed in our database including canonical H2A), some of which are relatively well characterized:
H2A.X is the most common H2A variant, with the defining sequence motif ‘SQ(E/D)Φ’ (where Φ-represents a hydrophobic residue, usually Tyr in mammals). It becomes phosphorylated during the DNA damage response, chromatin remodeling, and X-chromosome inactivation in somatic cells. H2A.X and canonical H2A have diverged several times in phylogenetic history, but each H2A.X version is characterized by similar structure and function, suggesting it may represent the ancestral state.
H2A.Z regulates transcription, DNA repair, suppression of antisense RNA, and RNA Polymerase II recruitment. Notable features of H2A.Z include a sequence motif ‘DEELD,’ a one amino acid insertion in L1-loop, and a one amino acid deletion in the docking domain relative to canonical H2A. variant H2A.Z.2 was suggested to be driving the progression of malignant melanoma (24). Canonical H2A can be exchanged in nucleosomes for H2A.Z with special remodeling enzymes (25).
macroH2A contains a histone fold domain and an extra, long C-terminal macro domain which can bind poly-ADP-ribose. This histone variant is used in X-inactivation and transcriptional regulation. Structures of both domains are available, but the inter-domain linker is too flexible to be crystalized.
H2A.B (Barr body deficient variant) is a rapidly evolving mammal specific variant, known for its involvement in spermatogenesis. H2A.B has a shortened docking domain, which wraps a shorter DNA region.
H2A.L and H2A.P variants are closely related to H2A.B, but are less studied.
H2A.W is a plant specific variant with SPKK motifs at the N-terminus with a putative minor-groove-binding activity.
H2A.1 is a mammalian testis, oocyte and zygote specific variant (26). It can preferentially dimerize with H2B.1. So far characterized only in mouse, but a similar gene in human is available. The gene is located at the end of the largest histone gene cluster.
Currently other less extensively studied H2A variants are starting to emerge such as H2A.J (27), these may be included in our database at the next update.
Histone H2B family
H2B histone type is known to have a limited number of variants at least in mammals, apicomplexa and sea urchins.
H2B.1 is a testis, oocyte and zygote specific variant that forms subnucleosomal particles, at least, in spermatids. It can dimerize with H2A.L and H2A.1.
H2B.W is involved in spermatogenesis, telomere associated functions in sperm and is found in spermatogenic cells. It is characterized by the extension of the N-terminal tail.
subH2B participates in regulation of spermiogenesis and is found in non-nucleosomal particle in the subacrosome of spermatozoa. This variant has a bipartite nuclear localization signal.
H2B.Z is an apicomplexan specific variant that is known to interact with H2A.Z.
‘sperm H2B’ is a putative group in our database that contains sperm H2B histones from sea and sand urchins and potentially is common for Echinacea.
Recently discovered variant H2B.E is involved in the regulation of olfactory neuron function in mice and it might be included in the next update of the database.
Histone H3 family
Histone H3 participates in the formation of H3/H4 tetramer within a nucleosome and has a number of important variants present throughout all eukaryotes.
cenH3, or centromeric H3, replaces canonical H3 in centromeric nucleosomes and is essential for kinetochore formation in most eukaryotes. cenH3 typically has about 50–60% sequence identity to canonical H3 within the histone fold domain and a variable N-terminal region. It has an extended L1-loop and usually replaces F84 in canonical H3 with W, and T107 with A, C, or S (28). The structural studies of cenH3 nucleosomes have been controversial and cenH3 nucleosomes may differs between species in the number of component H4 molecules (29). For example, a hemisome structure of centromeric nucleosomes in budding yeast has been proposed, with one copy of cenH3, H4, H2A and H2B and DNA forming a hemisome instead of a full nucleosome, whereas fission yeast cenH3 nucleosomes have two H4 molecules, and may resemble conventional nucleosomes.
H3.3 refers to a replication-independent H3, which typically differs from canonical H3 by only a few amino acids that are necessary for replication-independent assembly. H3.3 and canonical H3 diverged independently in plants, animals and ciliates. Fungi typically have H3 that undergoes both replication-coupled and replication-independent assembly, similar to H3.3, which may represent the ancestral state.
Other H3 variants are lineage-specific, and often include germ cell or pollen variants. In our database we include TS H3.4 (TS—testis specific), H3.5 and H3.Y, specific for mammals, hominid and primates respectively.
Histone H1 family
Histone H1 (linker histone) variants lack histone fold and typically have a short basic N-terminal domain, a globular winged-helix domain and a lysine- alanine-rich C-terminal domain. Their binding to nucleosome may be variant-specific and also depends on DNA sequence (30, 31). They evolved faster than the core histone families and are abundant and diverse, which presents problems for their grouping and classification. Although certain H1 variants manifest replication independent or replication dependent behavior, it is difficult to name any group of sequences as canonical H1s because of the lack of conserved orthologs across even moderately distant classes or phyla. Hence, we opt to have in our database only several H1 histone sequence groups limited to certain taxonomic clades and a generic H1 group (generic H1), which collects various H1 sequences from a wide set of species. Currently, the following groups are present:
Generic H1 refers to a set of sequence variants across a broad span of taxa not specifically related to each other.
H1.0 is a replication independent linker histone found in animals expressed in terminally differentiated cells. Has a common monophyletic origin that can be traced Histones H1.0 have prior to the divergence of protostomes and deuterostomes, very early in metazoan evolution.
TS H1.6, TS H1.7 and TS H1.9 groups in our database currently encompass sequences that belong to the corresponding testis specific variants of H1 common in mammals.
OO H1.8 encompasses sequences that belong to an oocyte specific variant of H1 common in mammals.
H1.10 includes sequences of a vertebrate specific H1 variant.
scH1 denotes a special variant of H1 found in Saccharomyces cerevisiae and probably other yeast species that has two globular domains (32). Saccharomyces has only one gene that encodes H1 histone (HHO1).
Automatically extracted and annotated sets of histone sequences
Curated alignments of canonical histones and histone variants were used to train Hidden Markov Models (HMM), utilizing HMMER 3.1b2 (33), to create one HMM for each variant. These models were used as a part of automatic extraction and annotation pipeline. Namely, all sequences from the nr protein database have been classified by the HMM models (variants and canonical) and were added into the HistoneDB 2.0 as ‘automatically extracted sequences’. We assigned a model with a maximum HMMER score to a given sequence. The score was required to exceed a certain threshold identified as follows. For any given variant model, we searched the curated sequence set and calculated HMMER scores for all curated sequences. We then varied the HMMER score threshold in order to distinguish sequences of a specific variant from all other sequences with 90% specificity. Specificity of the retrieval was estimated based on the number of true negatives (TNs; is the number of true negatives (number of sequences correctly classified as not belonging to the variant)) and false positives (FPs; incorrectly predicted sequences) found above each HMMER score cutoff. The specificity was calculated as TN/(FP + TN). Then a desired score cutoff value was obtained from the interpolated inverse curve of the score cutoffs plotted versus specificity. The efficacy of classification based on HMM models depends significantly on whether the respective histone variant model is sufficiently divergent and forms a separate clade on the phylogenetic tree (see Supplementary Figure S1 and Supplementary Data for examples of different variants’ ROC curves). For certain variants, which emerged repeatedly in the course of evolution, the classification becomes difficult unless a characteristic signature is known. In the case of H2A.X, HMM search algorithm was supplemented by the pattern matching, since this variant is characterized by the presence of ‘SQ(E/D)Φ’ motif as described above. Any sequence that was classified as canonical H2A by HMMER-based algorithm was classified as H2A.X if it had this motif at its C-terminus. The described above classification algorithm can be also applied to classify any sequence of interest (see section ‘3.3. Custom sequence annotation’). The numbers of curated and automatically annotated histone sequences are shown in Table 1.
Histone features and annotations
Every histone type and variant in the database has three types of manually collected annotations: (i) a brief description of the type/variant, (ii) a set of structural and functional features and their locations along the sequence, (c) a list of related references. The features and annotations were extracted from the literature and were inferred from the analysis of variant nucleosome structures. The positions of structural and functional features along the sequence are provided with respect to the representative histone sequence of the corresponding variant. The locations of features on other histone sequences or multiple sequence alignments (MSA) are inferred automatically by performing a global sequence alignment of the representative sequence with the sequence of interest or with the consensus sequence of MSA.
Website overview
The database website provides extensive functionality to: (i) browse histone variants, their annotations, features and sequences, (ii) analyze phylogenetic trees of histone variants, (iii) perform multiple sequence alignments of various sequences and browse them together with annotations, (iv) study the taxonomic distribution of histone variants in the automatically extracted sequence set, (e) classify sequences provided by the user and find the closest matches in the HistoneDB 2.0 database.
Browsing the variants
The front page allows choosing one of the five histone types, by selecting a color coded 3D model for each variant. After a histone type is chosen, the user is redirected to the histone type summary page that contains a list of variants with their alternate names, taxonomic spans, and sequence counts in curated and automatically extracted sequence sets (Figure 3a). One can also see a phylogenetic tree, which shows if variants have mono- or polyphyletic origins and depicts the relationship between different variants of the same histone type. A click on the species name within the tree will redirect the user to the ‘Curated sequences’ (see below) tab of the respective variant with the selected sequence highlighted in the table. By clicking on the histone variant name, the user is redirected to the histone variant summary page (Figure 3b), which includes its description, a preview of the histone sequence (for human if available) with the highlighted features, feature legends with descriptions, and a list of related references. Both histone type and histone variant pages have four other tabs described below.

A summary of HistoneDB 2.0 web site. (a) information page for a given histone type, (b) a summary page for a typical histone variant, (c) ‘Curated sequences’ tab of the histone variant page, (d) a table view of HMMER scores used to classify the selected sequences from the automatically extracted set (access via ‘Advanced’ menu, ‘Score against all HMMs’ button).
‘Curated sequences’ tab displays a table with a set of manually curated sequences with their corresponding identifiers, sequence descriptions and taxonomic classification (Figure 3c). By clicking on the row of this table, the user can select a variant’s sequence to be aligned to the canonical sequence of the same histone type. This alignment will be displayed above the table and allows highlighting the variant-specific features. If one or more sequences are selected, there are options to view a multiple sequence alignment, export it in FASTA format or add it to a basket to combine sequences from different variants.
‘Curated alignments’ tab depicts a multiple sequence alignment of all curated sequences for the selected histone type or variant. Alignments are annotated with the key sequence, structural and functional features, such as secondary structures, key arginines, acidic patches and features specific for a given variant.
The ‘Automatically extracted sequences’ tab contains a list of all sequences automatically extracted (see previous section) and assigned to a given variant type. For each sequence, Genbank identifiers, variant’s name, taxonomy, a short description, HMMER bitscore and E-value are provided. If one or more sequences are selected, there are options to view a multiple sequence alignment or add them to a basket. For more advanced users, there is an option to view the scores for selected sequences against all HMM models (‘Advanced’-‘Score against all HMMs’) (3d). This option might be useful for examining if a sequence is similar to several models or if there is a classification error. We urge the users to examine these scores before drawing ultimate conclusions about the sequence in question, especially if the automatically assigned variant type is known to be among the group of closely related variants (e.g. canonical H3, H3.3, H3.5 which all differ only by several positions). It is also possible to filter the sequences by taxonomy, sequence headers, sequence motifs, GI identifiers and sequences found in RefSeq database (34).
The last tab ‘Inferred Taxonomic Distribution’ allows browsing the taxonomy of automatically extracted sequences. The color of each sector in the ‘sunburst’ representation of taxonomy shows the average HMMER score of all sequences in the automatically extracted set for a given taxon scored with the corresponding variant model.
Database search options
There are two types of search options for sequence entries in the database, a basic and an advanced one. Both are accessible at the upper right corner of the website. The basic search allows the user to find sequences based on the keywords while an advanced option provides opportunities to look for specific histone types with particular sequence motifs and to show only unique sequences from a given organism. The quick search field is also implemented on the top of the table while viewing any list of sequences. In this case the search is limited to the content of the respective table. Another helpful feature of the website is ‘Basket’. While viewing a set of sequences, the user can mark entries of interest in the table and press the button ‘Add to basket’. The cumulative list of sequences can be then viewed at the basket page accessible from the website header.
Custom sequence annotation
The user can enter a sequence in FASTA format to annotate it through the ‘Analyze Your Sequence’ utility. A sequence is first classified using HMMER against all variant models and the most likely variant or variants are reported. The scores against all HMM models are available via ‘View scores against all HMMs’ button. In addition to that the query sequence is compared using BLAST against all curated sequences in the HistoneDB 2.0 and the list of similar sequences sorted by BLAST E-values are given in an interactive table. Clicking on the corresponding row of the table automatically updates the alignment view between the query sequence and the selected sequence.
Methods, internal structure and software
The HistoneDB 2.0 is written in Django, a high-level Python Web Framework, with a MySQL backend. The project has two applications, ‘browse’ and ‘djangophyolcore.’ Browse contains the HistoneDB models (equivalent to database tables), views (python functions to render each page), and templates (HTML files) and was developed specifically for the current project. The database schema implied by this application is outlined in Supplementary Figure S3. Djangophylocore is a previously developed django application to store the NCBI taxonomy database in a Django relational database using an algorithm similar to Modified Preorder Tree Traversal (35). The website layout is based on Twitter Bootstrap, with four important pages: Main browse of all histone types, Individual histone type browse, Variant browse, Analyze and Search.
Hidden Markov Model construction and search relies on HMMER 3.1b2 (33). Phylogenetic trees are displayed using jsPhyloSVG software (36). A tree is created by aligning all curated sequences for a given histone type using MUSCLE v3.8.31 (23) and by further applying the Neighbor-Joining procedure implemented in CLUSTALW 2.1 (37). The trees are converted to PhyloXML using BioPython (38) and are edited to add colors and variant and taxonomy labels in jsPhyloSVG. Alignments are displayed using BioJS MSA Viewer (39).
Conclusions
Despite the considerable progress in sequencing and understanding the functions of histone variants, many of them remain poorly annotated in public databases. Moreover, the specific molecular mechanisms of variants’ action, deposition and eviction are still unknown. One of the reasons is that the histone variants are highly specific and at the same time multi-functional and context dependent. The importance of studying the histone variants is difficult to overestimate; they are involved in regulation of many cellular processes and represent emerging key players in cancer (24, 40). To analyze known histone variants, compare them and interpret their functions would be challenging and time consuming. To fulfill this goal, we designed the HistoneDB 2.0 in order: to organize histones by variant; to provide reference alignments for each variant; to offer curated annotations of variant specific features; to study how variants evolved; to find likely orthologs of variants in other species; to classify histone-like sequences, and finally, to promote the new histone variant nomenclature (16). This database aids finding variant sequences from different organisms and comparing histone variants and corresponding canonical histones. It will help to understand the origin of functional specificity of variants and to guide the 3D modeling of variant nucleosomes. The database can be easily extended to include new variants and to annotate them based on the similarity to the existing annotated variants.
Acknowledgements
We would like to thank Franco Simonetti and Alexander Goncearenco for discussions about Django and the IT support team at NCBI for the troubleshooting the server related issues.
Funding
This work was supported by the Intramural Research Program of the National Library of Medicine, NIH. A.S. was supported in part by the US-Russia Collaboration in the Biomedical Sciences NIH visiting fellows program.
Conflict of interest. None declared.
References
Author notes
These authors contributed equally to this work.
Citation details: Draizen,E.J., Shaytan,A.K., Mariño-Ramírez,L. et al. HistoneDB 2.0: a histone database with variants—an integrated resource to explore histones and their variants. Database (2016) Vol. 2016: article ID baw014; doi:10.1093/database/baw014

{kind=link}
{kind=link}
{kind=link}