





Abstract
Atherosclerosis is a chronic inflammatory disease. Pathophysiological similarities between chronic infections and atherosclerosis triggered interest in a clinical association between these conditions. Various infectious microbes have been linked to atherosclerotic vascular disease in epidemiological studies. However, this association failed to satisfy the Koch’s postulates of causation with multiple clinical trials demonstrating inefficacy of anti-infective therapies in mitigating atherosclerotic cardiovascular events. Identification of underlying pathophysiological mechanisms and experience with vaccination against various infectious agents has ushered a new avenue of efforts in the development of an anti-atherosclerotic vaccine. Studies in animal models have identified various innate and adaptive immune pathways in atherosclerosis. In this review, we discuss the patho-biological link between chronic infections and atherosclerosis, evaluate existing evidence of animal and human trials on the association between infections and cardiovascular disease and introduce the concept of an anti-atherosclerotic vaccine.
Introduction
Atherosclerosis is now considered a chronic inflammatory disease. Various stimuli that trigger and sustain this state of heightened inflammation have been identified. Chronic infections represent one such stimulus that share the common pathophysiological milieu of chronic inflammation. Various bacteria and viruses have been shown to have a direct effect on the vascular endothelium as well as an indirect effect by systemic cytokine release, both of which contribute to accelerated atherosclerosis. Infectious agents that have been linked to atherosclerotic disease include, but not limited to Chlamydia pneumoniae, Porphyromonas gingivalis, Helicobacter pylori, influenza A virus, hepatitis C virus, cytomegalovirus (CMV), and human immunodeficiency virus (HIV). However, there are significant differences in the strength of the data supporting their association. Emerging evidence supporting the role of immunity in atherosclerosis and the discovery of molecular mimicry between oxidized low density lipoprotein cholesterol (ox-LDL) and bacterial cell wall polysaccharide has opened new avenues in the search for an anti-atherosclerotic vaccine. This has led to interest in targeting infective agents in an effort to mitigate the atherosclerotic process. Initial data from animal studies has fuelled multiple human clinical trials of anti-infective therapies to alleviate atherosclerosis.
In this review, we discuss the patho-biological link between chronic infections and atherosclerosis, evaluate existing evidence of animal and human trials on the association between infections and cardiovascular disease and highlight key features concerning the development of an anti-atherosclerotic vaccine.
Pathobiologic mechanisms of atherosclerosis development
Infections have long been implicated in the initiation, progression, and rupture (plaque instability and thrombosis) of atherosclerotic plaque. The ‘response to injury’ hypothesis conceptualizes atherosclerosis as a pathway of chronic inflammation and lipid accumulation in response to endothelial injury.1 Although sero-epidemiologic studies demonstrating an association between infectious agents and clinical atherosclerosis are conflicting, one of the strongest arguments in favour of the ‘infection hypothesis’ is based on the common substrate of chronic inflammation.2 Many studies propose the ‘direct’ (invasion of cells causing accelerated plaque growth through local effects) and ‘indirect’ mechanisms (systemic expression of inflammatory cytokines which catalyze development of plaque) by which infectious agents contribute to atherogenesis. Here, we review some of the key mechanisms implicated in the ‘infection hypotheses’ of atherosclerosis.
Local effects
Numerous studies have shown the presence of bacterial and viral microorganisms in the atherosclerotic plaque. Pathogens may be harboured in a latent state or replicate in cells such as macrophages creating a chronic inflammatory environment. Most implicated organisms are intracellular microbes which exert their effects from within the cell, evading the body’s immune mechanisms.3 , 4 Major local pathogenic effects of various microbes are listed in Table 1.
Summary of local pro atherosclerotic mechanisms in chronic infections
| Phase of atherogenesis | Pathogen | Pathogenic effect |
|---|---|---|
Lesion formation
|
|
|
Lesion progression
|
|
|
Lesion instability
|
|
|
| Phase of atherogenesis | Pathogen | Pathogenic effect |
|---|---|---|
Lesion formation
|
|
|
Lesion progression
|
|
|
Lesion instability
|
|
|
Summary of postulated local effects of infectious agents in atherogenesis.
CMV, cytomegalovirus; Cp, Chlamydia pneumoniae; Pg, Porphyromonas gingivalis; HIV, human immunodeficiency virus; HCV, hepatitis C virus; Hp, Helicobacter pylori; HSV, herpes simplex virus; MCP-1, macrophage chemoattractant protein-1; LDL, low density lipoprotein; ox-LDL, oxidized LDL; MMPs, matrix metalloproteinases.
Summary of local pro atherosclerotic mechanisms in chronic infections
| Phase of atherogenesis | Pathogen | Pathogenic effect |
|---|---|---|
Lesion formation
|
|
|
Lesion progression
|
|
|
Lesion instability
|
|
|
| Phase of atherogenesis | Pathogen | Pathogenic effect |
|---|---|---|
Lesion formation
|
|
|
Lesion progression
|
|
|
Lesion instability
|
|
|
Summary of postulated local effects of infectious agents in atherogenesis.
CMV, cytomegalovirus; Cp, Chlamydia pneumoniae; Pg, Porphyromonas gingivalis; HIV, human immunodeficiency virus; HCV, hepatitis C virus; Hp, Helicobacter pylori; HSV, herpes simplex virus; MCP-1, macrophage chemoattractant protein-1; LDL, low density lipoprotein; ox-LDL, oxidized LDL; MMPs, matrix metalloproteinases.
Lesion formation: activation of endothelium, migration of leucocytes, and formation of lipid core
Insult to the endothelial cell monolayer by shear stress, excess of ox-LDL or hyperglycaemia results in production of reactive oxygen species (ROS), chemoattractant cytokines and induction of adhesion molecules which enable entry and migration of inflammatory cells into the sub-intimal space. Multiple infectious agents, including C. pneumonia, CMV, periodontal pathogens, HIV, HCV, Influenza, have been shown to induce expression of adhesion molecules on endothelial cells.5 , 6 Heat shock protein 60 (HSP 60) isolated from P. gingivalis has been shown to induce a toll-like receptor (TLR) 4 mediated increase in Intercelluar adhesion molecule-1 (ICAM-1), Vascular cell adhesion molecule-1 (VCAM-1), and ox-LDL receptor (LOX-1) expression in mouse vessels.7 An increased production of monocyte chemoattractant protein-1 (MCP-1), has been found to be induced by many pathogens, including C. pneumoniae, periodontal organisms, CMV, HIV, and Influenza A virus.8 , 9
Lipid accumulation in the sub-endothelial layers occurs by internalization of ox-LDL, largely mediated by specialized receptors such as lectin-like ox-LDL receptor-1 (LOX-1), scavenger receptors (SR)-AI/II, SRBI, CD36 and TLR.10 Chlamydia pneumoniae has been shown to bind to and activate TLRs in vitro and upregulate the expression of LOX-1 in vivo.11 Increase in ox-LDL in turn increases the production of E- selectin, I-CAM-1, VCAM-1 in endothelial cells. Cytomegalovirus has been shown to mediate this through increased mRNA expression of class A SRs and expression of CD-36.12 HIV-1 Nef protein has been demonstrated to impair ATP-binding cassette transporter A1-dependent cholesterol efflux from human macrophages, and promote formation of foam cells.13 HIV-1 virus is also known to promote transendothelial migration as well as inhibit reverse transendothelial migration. This is further enhanced by increased levels of TNF-α in HIV+ serum.14 Lipopolysaccharide from P. gingivalis also increases expression of granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-1b, IL-10, and IL-12 in both macrophages and foam cells.
Lesion progression: migration and proliferation of smooth muscle cells and other key cells
Various infections have been associated with increased smooth muscle cell (SMC) proliferation in the vessel wall. Cytomegalovirus has been shown to increase SMC proliferation and inhibit apoptosis through suppression of tumour suppressor gene p53.15 In addition, CMV infection leads to enhanced levels of several growth factors including transforming growth factor β1, fibroblast growth factors, and epidermal growth factors which contribute to endothelial proliferation. Chlamydia pneumoniae infected cells have also been shown to be resistant to apoptosis.16 Gingipain, a cysteine protease of P. gingivalis has been shown to increase proliferation of aortic SMCs while another recent study showed it to increase angiopoetin 2 expression within the vessel layers and stimulate migration of SMCs.17 , 18 Together, these changes might be expected to lead to accumulation of infected SMCs in the vessel wall and contribute to plaque growth.
Lesion rupture: thrombosis and plaque instability
Impaired vasomotor tone .i.e. decreased vasodilator and increased constrictor activity is believed to contribute to acute coronary events. Liuba et al . 19 demonstrated in mice and swine infected with C. pneumoniae alone or coinfected with H. pylori that these infections alter vasomotor tone mediated through nitric oxide.20 Another mechanism that has been long recognized is the switch from anticoagulant to procoagulant milieu seen in human vascular endothelial cells infected with C. pneumoniae, CMV, and HSV. Vercellotti et al.21 reported that HSV infection of endothelium resulted in a marked increase in thrombin-induced platelet adhesion with a concomitant decrease in prostacyclin secretion in response to thrombin, while CMV infected endothelial cells may exhibit a procoagulant response via activation of pro-inflammatory cascade.22 Monocytes infected with CMV and C. pneumoniae have enhanced clotting mediated via increased generation of tissue factor and plasminogen activator inhibitor (PAI-1) secretion.23 Influenza virus infection also increases PAI-1 levels and decreases endothelial nitric oxide synthase expression. Matrix metalloproteinases (MMPs) have been implicated in thinning of fibrous cap and plaque destabilization. C. pneumoniae, through upregulation of LOX-1, increases levels of MMP-2 and MMP-9. While the glycoprotein 120 component of HIV-1 has been shown to increase levels of MMP-2 and MMP-9 in brain microvessel cells, the Tat protein has been shown to upregulate MMP-9 in monocytes.24 We have previously reported that patients with HCV have increased acute coronary events compared with HCV negative patients, despite similar angiographic coronary disease burden.25 Similar results have been reported in HIV patients as well.26 Enhanced plaque instability resulting from infection induced increase in MMP activity could partly explain these clinical findings. These studies implicate pathways by which incidence of vascular events is increased in patients with chronic infections.
Systemic effects
Potentiation of systemic inflammation
Regardless of pathogens found in situ in the plaque, infections induce systemic inflammation via release of cytokines and activate the immune system, both innate (TLR, HSP, and inflammasome signalling) as well as the adaptive arm (Th1, Th17 activation). Interferon alpha, which is produced in response to viral infections, is a potent inflammatory amplifier in the context of atheromatous plaque. Studies have shown that Hepatitis C viral proteins initiate a pro-inflammatory state by promoting an imbalanced T helper (Th)1/Th2 cytokine ratio.27 Other viruses, such as CMV and influenza, have also been shown to increase the plasma levels of interferon-γ, ILs and TNF-α and enhance monocyte differentiation.28 Recent studies show that periodontal pathogens such as P. gingivalis and Actinomycetes comitans promote TH17 subset responses both within the spleen and within the atherosclerotic lesions, which in turn increases the release of a host of potent cytokines such as IL-1β, IL-6, and IL-17.29 , 30
Inflammasomes are multi-protein cytosolic complexes that integrate several pathogen-triggered signalling cascades ultimately leading to caspase-1 activation and generation of pro-inflammatory cytokines including interleukin IL-18 and IL-1β. Recently, P. gingivalis was found to increase the aortic induction of NLRP3 inflammasome receptor gene.31 HIV-1 as well as HCV have been found to activate inflammasomes in human monocytes resulting in the above mentioned effects.32 C. pnemoniae and Influenza have been shown to decrease the anti -inflammatory properties of HDL, contributing to indirect progression.33
Molecular mimicry and role of heat shock proteins
There is increasing evidence for the role of molecular mimicry in modulating immune responses to infectious agents in atherosclerosis. Protein components of microbial agents ‘mimic’ host proteins, and mount an immune response directed against host proteins which are homologous to the pathogenic antigens. Heat shock proteins have also been implicated in this process. Heat shock proteins are proteins that are phylogenetically highly conserved in mammalian and bacterial species, and the shared homology lead to extensive immunological cross-reactivity. Augmented expression of HSPs occurs on the cell surface in times of stress including infection and inflammation. Human atherosclerotic plaques express a variety of inflammatory cytokines, most of which can enhance HSP expression.34 Serum antibodies to HSP60 have been seen to cross-react with those of human HSP, C. pneumoniae HSP, Escherichia. coli HSP (GroEL), and that of H. pylori.35 Cross reactivity between anti-CMV antibodies and HSP60 has also been recently described.
Antibodies against bacterial HSP can hence induce and exacerbate the established inflammatory milieu thereby promoting plaque progression and instability.36 Plasma levels of anti-HSP60 antibodies have been correlated with atherosclerosis burden and occurrence of acute coronary syndromes.37 Kol et al.38 demonstrated that Chlamydial HSP60 mediates the activation of endothelial cells and SMCs, and induce TNF-α and MMP production by macrophages in the atherosclerotic plaques. Chlamydial HSP60 also was demonstrated to promote a Th1 immune response via maturation of monocyte derived dendritic cells.39
Thus, various chronic infections have been implicated in their association with atherosclerosis, both by epidemiological and pathophysiological association. Local and systemic effects of infections mimic and perhaps contribute to various steps in the atherosclerotic process (Figure 1).
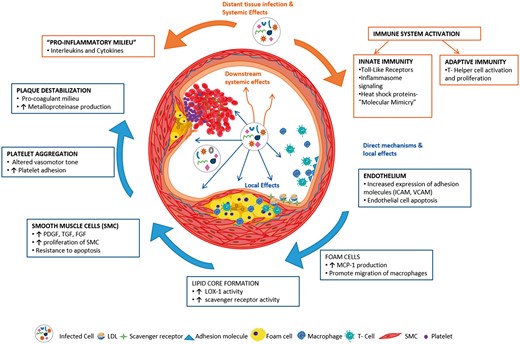
Schematic of various pro-atherosclerotic pathways activated in chronic infection. The blue arrows denote direct effects of the microorganism on vascular endothelium and the orange arrows depict systemic effects that are pro-atherosclerotic.
Anti-infective therapy and atherosclerosis
To test the hypothesis that eradicating infectious stimuli would diminish the chronic inflammatory response and hence reduce cardiovascular events, several trials of anti-infective therapy have been conducted in animal models of atherosclerotic disease as well as in humans.
Animal studies
The vast majority of anti-infective studies have focused on infection with C. pneumonia, which is ubiquitous and often subclinical, so a course of targeted antibiotics could theoretically treat an infection in a large population. In addition, macrolide antibiotics which target C. pneumonia are thought to have anti-inflammatory properties, and could theoretically contribute to plaque stability. Muhlestein et al. 40 inoculated rabbits with C. pneumoniae (or placebo) and treated these animals with azithromycin, and evaluated aortic atherosclerosis and intimal thickening. They found that C. pneumoniae infection accelerated intimal wall thickening and the extent of atherosclerosis; importantly, treatment with azithromycin decreased the extent of atherosclerosis. These data were contradicted by Rothstein et al. 41 who reported no effect of azithromycin on aortic lesion size in mice infected with C. pneumoniae even though aortic lesion size was larger compared with placebo prior to antibiotic therapy.42 Madan et al. 43 studied the effect of treatment with doxycycline in P. gingivalis-associated atherosclerosis in an ApoE heterozygote murine model. They found that doxycycline decreased circulating pro-inflammatory cytokine levels as well as atherosclerotic lesion development. Others evaluated the effect of metronidazole treatment prior to P. gingivalis inoculation and demonstrated a significant decrease in circulating inflammatory markers and atherosclerotic lesion size in mice that were pretreated.44 Ayada et al . 45 studied the effect of H. pylori infection in mice. These investigators were careful to only inoculate via the GI tract, in order to replicate the natural route of infection, as there are questions as to the amount of systemic inflammation that H. pylori can induce when administered by parenteral route. Helicobacter pylori infection was associated with increased size and thickness of aortic lesions, compared with non-infected controls. These authors then extrapolated their results to show that treatment with triple drug therapy (lasonoprazole, amoxicillin, clarithromycin) could decrease the atherogenic effects of gastrointestinal H. pylori infection. These findings from animal studies have fuelled interest in studying the effect of anti-infective therapies in humans.
Human studies
The possible link between anti-infective therapy and development or progression of atherosclerosis in animal studies led to several human studies to investigate the possible connection between anti-infective therapy and development of atherosclerosis in humans. The results of some of the major trials are summarized in Table 2. As evident, most human trials of anti-infective therapies have not shown any significant cardio-protection. However, all trials were heterogeneous in treatment choices, duration and cardiovascular end points. Initial human trials used short treatment courses of macrolide antibiotics. The largest trials of antibiotic therapy for atherosclerosis were PROVE IT TIMI (Pravastatin or Atorvastatin Evaluation and Infection Therapy - Thrombolysis in Myocardial Infarction) and ACES (Azithromycin and Coronary Events Study), and enrolled more than 4,000 patients each. The ACES trial used azithromycin, while PROVE IT-TIMI trial used gatifloxacin. There was no difference in end-points of coronary heart disease events, or cardiac death in either of these trials.
Summary of antibiotic interventions in human clinical trials of atherosclerosis46–55
| Study | Sample size | Antibiotic | Treatment duration | Follow-up duration | Clinical setting | Outcome |
|---|---|---|---|---|---|---|
| In support of anti-infective therapy preventing atherosclerosis | ||||||
| CLARIFY | 148 | Clarithromycin | 3 months | 18 months | ACS (Non-Q) | Reduced risk of ischaemic CV events |
| Against anti-infective therapy preventing atherosclerosis | ||||||
| ACADEMIC | 302 | Azithromycin | 3 months | 2 years | CHD | No reduction in CV events |
| ACES | 4012 | Azithromycin | 12 months | 4 years | CHD | No reduction in cardiac events |
| ANTIBIO | 872 | Roxithromycin | 6 weeks | 1 year | AMI | No reduction in cardiac events |
| AZACS | 1439 | Azithromycin | 5 days | 6 months | ACS | No reduction in ischaemic CV events |
| ISAR-3 | 1020 | Roxithromycin | 1 month | 6–12 months | Post-PCI | No prevention of re-stenosis in patients with low C. pneumoniae titers;Reduced risk of restenosis in patients with high Chlamydia pneumoniae titers |
| PROVE IT TIMI | 4162 | Gatifloxacin | 18 months | 2 years | ACS | No reduction in cardiac events |
| ROXIS | 202 | Roxithromycin | 30 days | 6 months | ACS (Non-Q) | Reduced risk of death and reinfarction |
| WIZARD | 7724 | Azithromycin | 3 months | 3 years | CHD | No reduction in cardiac events |
| Study | Sample size | Antibiotic | Treatment duration | Follow-up duration | Clinical setting | Outcome |
|---|---|---|---|---|---|---|
| In support of anti-infective therapy preventing atherosclerosis | ||||||
| CLARIFY | 148 | Clarithromycin | 3 months | 18 months | ACS (Non-Q) | Reduced risk of ischaemic CV events |
| Against anti-infective therapy preventing atherosclerosis | ||||||
| ACADEMIC | 302 | Azithromycin | 3 months | 2 years | CHD | No reduction in CV events |
| ACES | 4012 | Azithromycin | 12 months | 4 years | CHD | No reduction in cardiac events |
| ANTIBIO | 872 | Roxithromycin | 6 weeks | 1 year | AMI | No reduction in cardiac events |
| AZACS | 1439 | Azithromycin | 5 days | 6 months | ACS | No reduction in ischaemic CV events |
| ISAR-3 | 1020 | Roxithromycin | 1 month | 6–12 months | Post-PCI | No prevention of re-stenosis in patients with low C. pneumoniae titers;Reduced risk of restenosis in patients with high Chlamydia pneumoniae titers |
| PROVE IT TIMI | 4162 | Gatifloxacin | 18 months | 2 years | ACS | No reduction in cardiac events |
| ROXIS | 202 | Roxithromycin | 30 days | 6 months | ACS (Non-Q) | Reduced risk of death and reinfarction |
| WIZARD | 7724 | Azithromycin | 3 months | 3 years | CHD | No reduction in cardiac events |
ACS, acute coronary syndrome; CHD, coronary heart disease; AMI, acute myocardial infarction; PCI, percutaneous coronary intervention.
Summary of antibiotic interventions in human clinical trials of atherosclerosis46–55
| Study | Sample size | Antibiotic | Treatment duration | Follow-up duration | Clinical setting | Outcome |
|---|---|---|---|---|---|---|
| In support of anti-infective therapy preventing atherosclerosis | ||||||
| CLARIFY | 148 | Clarithromycin | 3 months | 18 months | ACS (Non-Q) | Reduced risk of ischaemic CV events |
| Against anti-infective therapy preventing atherosclerosis | ||||||
| ACADEMIC | 302 | Azithromycin | 3 months | 2 years | CHD | No reduction in CV events |
| ACES | 4012 | Azithromycin | 12 months | 4 years | CHD | No reduction in cardiac events |
| ANTIBIO | 872 | Roxithromycin | 6 weeks | 1 year | AMI | No reduction in cardiac events |
| AZACS | 1439 | Azithromycin | 5 days | 6 months | ACS | No reduction in ischaemic CV events |
| ISAR-3 | 1020 | Roxithromycin | 1 month | 6–12 months | Post-PCI | No prevention of re-stenosis in patients with low C. pneumoniae titers;Reduced risk of restenosis in patients with high Chlamydia pneumoniae titers |
| PROVE IT TIMI | 4162 | Gatifloxacin | 18 months | 2 years | ACS | No reduction in cardiac events |
| ROXIS | 202 | Roxithromycin | 30 days | 6 months | ACS (Non-Q) | Reduced risk of death and reinfarction |
| WIZARD | 7724 | Azithromycin | 3 months | 3 years | CHD | No reduction in cardiac events |
| Study | Sample size | Antibiotic | Treatment duration | Follow-up duration | Clinical setting | Outcome |
|---|---|---|---|---|---|---|
| In support of anti-infective therapy preventing atherosclerosis | ||||||
| CLARIFY | 148 | Clarithromycin | 3 months | 18 months | ACS (Non-Q) | Reduced risk of ischaemic CV events |
| Against anti-infective therapy preventing atherosclerosis | ||||||
| ACADEMIC | 302 | Azithromycin | 3 months | 2 years | CHD | No reduction in CV events |
| ACES | 4012 | Azithromycin | 12 months | 4 years | CHD | No reduction in cardiac events |
| ANTIBIO | 872 | Roxithromycin | 6 weeks | 1 year | AMI | No reduction in cardiac events |
| AZACS | 1439 | Azithromycin | 5 days | 6 months | ACS | No reduction in ischaemic CV events |
| ISAR-3 | 1020 | Roxithromycin | 1 month | 6–12 months | Post-PCI | No prevention of re-stenosis in patients with low C. pneumoniae titers;Reduced risk of restenosis in patients with high Chlamydia pneumoniae titers |
| PROVE IT TIMI | 4162 | Gatifloxacin | 18 months | 2 years | ACS | No reduction in cardiac events |
| ROXIS | 202 | Roxithromycin | 30 days | 6 months | ACS (Non-Q) | Reduced risk of death and reinfarction |
| WIZARD | 7724 | Azithromycin | 3 months | 3 years | CHD | No reduction in cardiac events |
ACS, acute coronary syndrome; CHD, coronary heart disease; AMI, acute myocardial infarction; PCI, percutaneous coronary intervention.
Among viral infections, HCV and HIV have been established to have a significant association with atherosclerotic heart disease. Recent advances in anti-viral therapies have led to the possibility of a complete cure in HCV. Whether achieving a sustained virological response to anti-HCV therapy leads to a reduction in atherosclerotic cardiovascular end points is an area of active research. In the era of interferon-based therapy, studies have shown a reduction in incidence of ischaemic stroke [hazard ratio (HR) 0.62] and acute coronary events (HR 0.77) in patients who achieved a sustained virological response over an 8-year follow up.56 , 57 Impact of newer anti-viral regimens which have a much higher virological clearance rate, on cardiovascular end points is yet to be explored. In HIV patients, effect of anti-retroviral therapy on cardiovascular outcomes has remained controversial. In the randomized Strategies for Management of Antiretroviral Therapy trial, interruption of anti-retroviral therapy was associated with increased levels of pro inflammatory cytokines and incident myocardial infarction, suggesting that viral suppression might lead to decreased inflammatory signalling and reduced adverse cardiac events.58 However, a later large prospective study of more than 20 000 HIV patients on anti-retroviral therapy showed that the risk of incident myocardial infarction was significantly higher in patients who received protease inhibitors, even after adjusting for traditional cardiac risk factors.59 This elevated cardiac risk was attributed to protease inhibitor induced metabolic dysregulation. Whether, protease inhibitors currently approved for HCV therapy have similar cardiovascular risk profile is yet to be determined.
Overall, in most clinical trials of anti-infective agents, antibiotic choices, treatment regimens and therapeutic duration were adequate but remained ineffective. Negative results of these trials highlight the paucity in our understanding of the complex biological interactions in the pathobiology of atherosclerosis. Pathogens are perhaps not integral culprits in the progression of atherosclerosis, but rather co-exist and are incidentally noted in the atherosclerotic plaques. If bacterial pathogens were integral components, one would have seen a clear and strong correlation with anti-infective therapy slowing the progression of disease. However, several arguments explaining the relative inefficacy of antibiotic trials in reducing atherosclerotic endpoints can be justified. First, presence of persistent low grade infection that is not responsive to antibiotic therapy could explain persistent low grade inflammation. Secondly, end points in most the clinical trials were cardiovascular endpoints in patients with established vascular disease, which may or may not be modified by antibiotic therapy. Thirdly, the issue of antibiotic resistance with chronic treatment cannot be properly addressed. In summary, albeit animal experiments and a few small human clinical trials showed promise in the role of antibiotic therapy, larger studies have generally been negative.
Vaccination and atherosclerosis
As discussed above, various infections have been linked to atherosclerosis. This ‘infection hypothesis’ lends itself to possible alteration of the atherosclerotic process through treatment or prevention of infections. Given this interaction between infections and atherosclerosis, several investigators have looked at the effects of vaccinations against these organisms on the atherosclerotic process.60 In animal models, exposure to P. gingivalis accelerated atherosclerosis while immunization against it induced attenuation of atherosclerosis.61 Koizumi et al . 62 employed nasal immunization using the outer membrane protein of P. gingivalis and demonstrated a significant reduction of atherosclerotic lesion size in the aortic sinuses of mice, along with a decrease in concentration of circulating inflammatory cytokines. Streptococcus pneumoniae is another bacterium that has been linked to atherosclerosis, perhaps driven by its molecular mimicry with oxLDL.63 Immunization against S. pneumoniae results in the production of protective anti-phosphorylcholine (PC) antibodies. These possibly result in cross-reactivity with ox-LDL, as suggested by the presence of a naturally found IgM that recognizes epitopes in both ox-LDL and the PC head groups on the surface of apoptotic cells, thus inhibiting their uptake by macrophages.64 In one study, when homozygous LDL-receptor knockout (LDL − / −) mice were actively immunized against S. pneumoniae, it resulted in increased ox-LDL-specific antibodies and reduced atherosclerosis.65 Another group used a PC conjugate to immunize Apolipoprotein E homozygous knockout (ApoE−/−) mice. Similar to the previous study, this resulted in an increase in both anti-PC and ox-LDL antibodies, with reduced atherosclerosis.
Further elucidation of immunological mechanisms of atherosclerosis (Figure 2) has fuelled interest in vaccination as a potential preventive strategy. Encouraging results in animal studies have led to clinical evaluation of vaccines as a means of reducing atherosclerotic cardiac events. In initial single centre studies, pneumococcal vaccination was associated with a decrease of more than 50% in the rate myocardial infarction at two year follow up.66 However, data from the large prospective California Mens Health study of 84,170 participants failed to reveal a beneficial impact of pneumococcal vaccination on outcomes of acute stroke or myocardial infarction.67 The most exciting effect that an established routine vaccine has had on cardiovascular outcomes is the effect of the influenza vaccine. An increased rate of death and myocardial infarction (MI) have been noted during the influenza season.68 While the mechanism of action is unknown, several studies suggest that influenza vaccination leads to reduction in cardiovascular events such as ischaemia, rehospitalizations after MI, and cardiovascular death.69–71 A meta-analysis provided further support for the use of influenza vaccination in the secondary prevention of cardiovascular events.72 The cardiovascular effect of this vaccine could be related to the effect of antibody mediated inactivation of the influenza virus, which has been shown to directly invade vascular tissue and accelerate atherosclerosis by locally induced inflammation. There is continued mounting evidence in favour of annual influenza vaccination leading to reduced cardiovascular morbidity, stroke, and all-cause mortality in patients with known cardiovascular disease; in addition, there is no evidence of negative effects of the vaccine in the recipients.73 , 74 The American Heart Association and the American College of Cardiology now advocate its use for secondary prevention in individuals with known cardiovascular disease (Class I, Level B).75
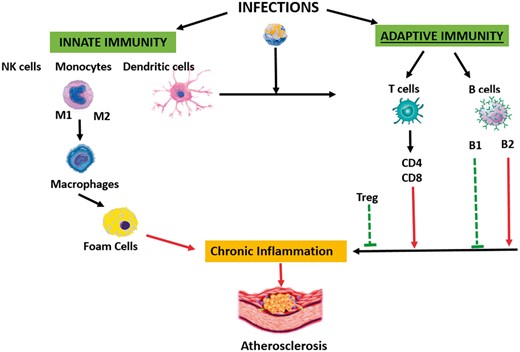
Schematic depicting various immune pathways triggered by infections. Modulations of these pathways with vaccination hold promise. Red arrows denote atherogenic pathways, green arrows denote natural anti-atherogenic pathways, and the broken blue arrows denote pathways that can to be modulated by immunization. (Treg—regulator T cells).
Anti-atherosclerosis vaccine
Given the complex interactions between immune response (innate and adaptive) and atherogenesis, modulation of this immune response towards an athero-protective path makes intuitive sense in the battle against atherosclerosis. One of the biggest challenges in this regard has been the identification of key antigens which when targeted will favourably alter the atherosclerotic process.76 To that effect, many studies have been conducted and others are underway. Unlike vaccinations against the exogenous antigens found on microbes described earlier, numerous studies exploring vaccinations against endogenous antigens have been performed. The most commonly studied antigens have included native or modified homologous LDL since LDL has the strongest causative link with atherosclerosis. Immunization of experimental animals with homologous native or modified LDL (oxidized or MDA modified) has generally resulted in a reduction in atherosclerosis. As a major component of LDL, ApoB100 was an attractive initial target for the development of an anti-atherosclerotic vaccine and has fuelled significant research in this area which is beyond the scope of the current review. We would like to refer the interested reader to recently published literature on this topic elsewhere.60 , 77
Summary
There is a strong epidemiological association between infections and cardiovascular disease. Chronic infections and atherosclerosis share multiple biological mechanisms with ample experimental evidence supporting the pro-atherosclerotic nature of various bacterial and viral infections (Central Illustration). Despite pathophysiological correlation, anti-infective therapy has failed to satisfy Koch’s postulate of a causative link with multiple human trials demonstrating negative results. Identifying the appropriate target pathway has been the Achilles heel is in our efforts to mitigate the atherosclerotic response. Is it the microorganism per se or the myriad of signalling pathways that are activated in chronic infection that are pro-atherogenic? With negative clinical trial results and increasing data on adverse effects of prolonged antibiotic therapy on both cardiac and non-cardiac outcomes, enthusiasm towards designing more anti-infective clinical trials is dwindling. In this conjecture, development of an atherosclerosis vaccine that targets various signalling pathways related to infections may hold promise.
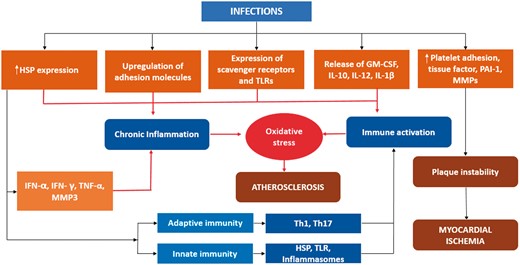
Schematic of the pathogenic mechanisms induced by chronic infections and role of anti-atherosclerosis vaccination (IL, Interleukins, IFN, Interferon, TNF, tumour necrosis factor, TLRs, toll like receptors, MMP, matrix metalloproteinase, HSP, heat shock protein, Th, T helper cells, PAI, Plasminogen activator inhibitor, GM-CSF, granulocyte monocyte colony stimulating factor).
Conflict of interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
Comments