





Abstract
C-mannosylation is the attachment of an α-mannopyranose to a tryptophan via a C–C linkage. The sequence WXXW, in which the first Trp becomes mannosylated, has been suggested as a consensus motif for the modification, but only two-thirds of known sites follow this rule. We have gathered a data set of 69 experimentally verified C-mannosylation sites from the literature. We analyzed these for sequence context and found that apart from Trp in position +3, Cys is accepted in the same position. We also find a clear preference in position +1, where a small and/or polar residue (Ser, Ala, Gly, and Thr) is preferred and a Phe or a Leu residue discriminated against. The Protein Data Bank was searched for structural information, and five structures of C-mannosylated proteins were obtained. We showed that modified tryptophan residues are at least partly solvent exposed. A method predicting the location of C-mannosylation sites in proteins was developed using a neural network approach. The best overall network used a 21-residue sequence input window and information on the presence/absence of the WXXW motif. NetCGlyc 1.0 correctly predicts 93% of both positive and negative C-mannosylation sites. This is a significant improvement over the WXXW consensus motif itself, which only identifies 67% of positive sites. NetCGlyc 1.0 is available at http://www.cbs.dtu.dk/services/NetCGlyc/. Using NetCGlyc 1.0, we scanned the human genome and found 2573 exported or transmembrane transcripts with at least one predicted C-mannosylation site.
Introduction
Among posttranslational modifications, protein glycosylation is more abundant and structurally diverse than all the other types combined (Hart 1992; Seitz 2000). Glycosylation is known to affect protein folding, localization, trafficking, solubility, antigenicity, biological activity, and half-life, as well as cell–cell interactions (Varki 1993). An impressive variety of carbohydrate–peptide linkages have been described, which are distributed among glycoproteins found in essentially all living organisms, ranging from eubacteria to eukaryotes (Spiro 2002). In mammals, seven different monosaccharides and six amino acid types participate in these bonds, so that at least 11 sugar–amino acid combinations exist (Ohtsubo and Marth 2006).
C-Mannosylation is the attachment of an α-mannopyranosyl residue to the indole C2 of tryptophan via a C–C link (Hofsteenge et al. 1994; de Beer et al. 1995). The first example of glycosylation of a tryptophan residue (with a hexose of unknown type) was discovered in a neuropeptide from a stick insect (Gade et al. 1992). Since then, numerous C-mannosylation sites have been found in mammalian proteins, of which the first was in RNase 2 (Hofsteenge et al. 1994; Furmanek and Hofsteenge 2000). In all mammalian cases, the glycan has been found to be a single α-mannopyranose. The transfer of mannose to the protein is catalyzed by the enzyme C-mannosyltransferase, and this probably occurs in the endoplasmic reticulum (ER) (Doucey et al. 1998; Perez-Vilar et al. 2004). C-Mannosyltransferase activity toward peptides derived from human RNase has been found in Caenorhabditis elegans, amphibians, birds, and mammals, but not in Escherichia coli, insects, or yeast (Krieg et al. 1997; Doucey et al. 1998; Furmanek and Hofsteenge 2000). At present, little is known about the function of C-mannosylation, but two recent studies indicate that it is probably required for proper folding of Cys subdomains in two mucins (Perez-Vilar et al. 2004) and that it may have a pathological role in diabetic complications under hypoglycemic conditions (Ihara et al. 2005).
A study involving site-directed mutagenesis of RNase 2 showed that the sequence WXXW, in which the first Trp becomes mannosylated, is the specificity determinant for C-mannosylation (Krieg et al. 1998). In thrombospondin repeats, containing the motif WXXWXXWXXC (in some cases with one or two of the tryptophan residues substituted by other amino acids), C-mannosylation was found on one, two or all three tryptophans (Hofsteenge et al. 1999). The shortest peptide still valid as a substrate for C-mannosyltransferase found so far is WAKW (Hartmann and Hofsteenge 2000). However, in two particular thrombospondin repeats (from complement component C6 and C7), the first tryptophan is mutated to phenylalanine or tyrosine respectively, (Hofsteenge et al. 1999), and two recently discovered C-mannosylation sites in bovine lens fiber membrane intrinsic protein show no relationship at all to the WXXW motif (Ervin et al. 2005). This indicates that although the WXXW motif seems to be a sufficient requirement for C-mannosylation, it does not seem to be a necessary one.
According to estimates based on the Swiss–Prot database, more than half of all proteins are glycosylated (Apweiler et al. 1999). However, despite the fact that human proteins are the most studied of all and that only proteins with some experimental verification are present in Swiss–Prot, only approximately 1.7% of human Swiss–Prot entries have experimentally verified glycosylation site information. To bridge the enormous gap between an exponential increase in gene sequences in databases and a linear increase in proteins investigated for posttranslational modifications, prediction methods are needed. Prediction of glycosylation sites is a valuable tool when trying to characterize a new protein, e.g. for the interpretation of mass spectrometry results. Further, prediction of glycosylation sites is one of the important features when predicting orphan protein function (Jensen et al. 2003). Since glycosylation may affect the structure of the protein and occurs primarily in surface-exposed regions, predicted glycosylation sites may be used to improve protein structural prediction as well. Prediction can also be useful in protein engineering to incorporate or abolish glycosylation sites and to design competitive inhibitors of glycosyltransferases (Hansen et al. 1998).
We have analyzed experimentally verified C-mannosylation sites with respect to sequence and structure. We have trained a predictor method, NetCGlyc 1.0, which correctly predicts 93% of both positive and negative C-mannosylation sites. This is a significant improvement over the WXXW consensus motif, which identifies only 67% of the positive sites. NetCGlyc 1.0 is publicly available at http://www.cbs.dtu.dk/services/NetCGlyc/. Using NetCGlyc 1.0, we scanned the human genome for predicted C-mannosylation sites.
Results
Sequence analysis
From the literature, we gathered a dataset of 12 native proteins and 27 naturally occurring or engineered mutants/peptides that contain a total of 69 experimentally verified C-mannosylation sites and 88 nonmodified sites. The sequence neighborhood around the sites can be illustrated using sequence logos based on Shannon information content (Schneider and Stephens 1990) (Figure 1A) or Kullback–Leibler information content (Kesmir et al. 2003) (Figure 1B). The Shannon information logo (Figure 1A) is based only on the occurrence of residues in different positions in positive sites and shows that the strongest discrimination is clearly at position +3, where mostly tryptophan and cysteine are accepted. Our analysis also indicates a strong preference for small and/or polar residues such as serine, alanine, glycine, and threonine at position +1, not previously reported. A repetition pattern where a tryptophan adjacent to a serine/glycine is repeated every three residues on either side of the glycosylation site is also evident. This probably arises from the C-mannosylation sites located in thrombospondin repeats that contain WXXWXXWXXC motifs where one, two, or all three of the tryptophans are glycosylated.
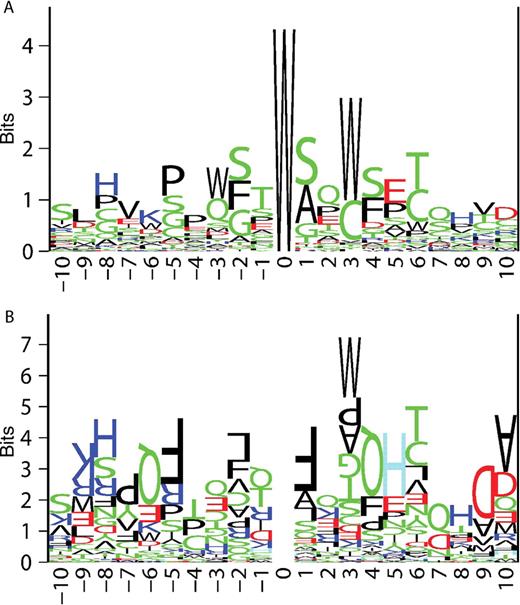
Sequence logos for C-mannosylation sites. Position 0 denotes the location of the glycosylated tryptophan residue. Amino acids are represented by their one-letter code and the letters are colored according to the following scheme: hydrophobic residues in black, polar residues in green, acidic residues in red, and basic residues in blue. (A) The Shannon logo shows the frequencies of amino acid residues at each position in positive sites, as the relative heights of letters, along with the degree of sequence conservation as the total height of a stack of letters. (B) The Kullback–Leibler logo shows the differences in frequencies of amino acid residues at each position in positive sites compared with negative sites. Amino acids over-represented in positive sites are shown as regular letters; those over-represented in negative sites are shown as upside-down letters. Histidines over-represented in negative sites (upside-down H's) are shown in light blue for clarity. The larger the skew is, the larger the letter will be.
The Kullback–Leibler information logo (Figure 1B) is based on both positive and negative sites. Residues over-represented in positive sites are shown as normal letters and those that are over-represented in negative sites are shown as upside-down letters. Note that the modified tryptophan residue in the middle is entirely cancelled out since both positive and negative sites have a tryptophan at that position. Not surprisingly, the strongest preference is again found at position +3, where tryptophan and, to some extent, cysteine is preferred and most other residues are discriminated against. We found that phenylalanine and leucine, both large and hydrophobic, are not tolerated at position +1 of the positive sites. We also found a number of residues at different positions, even surprisingly far away from the attachment site, that seem to be inconsistent with C-mannosylation: arginine/lysine at position −9, glutamine at positions −6 and 4, phenylalanine at position −5, histidine at position 5, aspartic acid at position 9, and alanine at position 10. Whether these are true reflections on the requirements for C-mannosylation or a result of insufficient sequence sampling in the dataset is hard to say at this point.
Structural analysis
Using FeatureMap3D (Wernersson et al. 2006), we were able to identify five nuclear magnetic resonance (NMR) or X-ray structures in the worldwide Protein Data Bank (Berman et al. 2006) showing the structure of C-mannosylated proteins (Table I). Two of the structures (1SZL and 1LSL) show the structure of thrombospondin repeats. The fold of a thrombospondin repeat contains two β strands along with a third, fairly extended, but not hydrogen-bonded stretch running parallel to the β sheet (Figure 2B) (Tan et al. 2002; Paakkonen et al. 2006). The three, potentially glycosylated, tryptophans are situated in the non-β stretch. The aromatic rings of the three tryptophans are parallel to each other at a Cα–Cα distance of 8.3–8.5 Å, which is too long to allow aromatic stacking (π–π interactions). In two particular thrombospondin repeats (from complement components C6 and C7), C-mannosylation is found in this structural context without the presence of a true WXXW motif. Instead, the first tryptophan is mutated to phenylalanine or tyrosine, respectively.
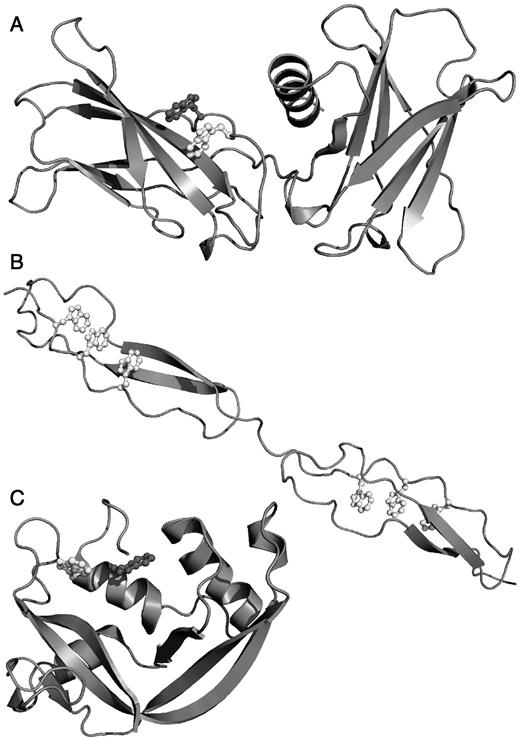
Protein structures of C-mannosylated proteins. Figures were prepared using PyMol (http://www.pymol.sourceforge.net/). Glycosylated tryptophans are shown in white, WXXW nonglycosylated tryptophans in dark gray. (A) Human erythropoietin receptor (1EER) (Syed et al. 1998). (B) Thrombospondin repeats (1LSL) (Tan et al. 2002). (C) Eosinophil-derived neurotoxin (2BZZ) (Baker et al. 2006).
Structural context of C-mannosylation sites
| Swiss–Prot | Protein name | PDB entry | Identity (%)a | Resol. | TSRb | Sitec | Secondary structured |
|---|---|---|---|---|---|---|---|
| P35446 | Spondin-1 (F-spondin) | 1SZL | 100 | NMR | Yes | 420(Trp)448 | C |
| 423(Trp)451 | C | ||||||
| P29460 | Interleukin-12 subunit β | 1F42 | 97 | 2.50 | No | 297(Trp)297 | C |
| P13671 | Complement component C6 | 1LSL | 44 | 1.90 | Yes | 547(Trp)420 | C |
| 550(Trp)423 | C | ||||||
| 553(Trp)426 | C | ||||||
| P10643 | Complement component C7 | 1LSL | 43 | 1.90 | Yes | 481(Trp)477 | C |
| 484(Trp)480 | C | ||||||
| 487(Trp)483 | C | ||||||
| P07357 | Complement component C8 α chain | 1LSL | 44 | 1.90 | Yes | 522(Trp)420 | C |
| 525(Trp)423 | C | ||||||
| 528(Trp)426 | C | ||||||
| P07358 | Complement component C8 β chain | 1LSL | 52 | 1.90 | Yes | 519(Trp)423 | C |
| 522(Trp)426 | C | ||||||
| P14753 | Erythropoietin receptor | 1EER | 82 | 1.90 | No | 208(Trp)209 | C |
| P10153 | Ribonuclease 2 | 2BZZ | 99 | 0.98 | No | 11(Trp)1007 | H |
| Swiss–Prot | Protein name | PDB entry | Identity (%)a | Resol. | TSRb | Sitec | Secondary structured |
|---|---|---|---|---|---|---|---|
| P35446 | Spondin-1 (F-spondin) | 1SZL | 100 | NMR | Yes | 420(Trp)448 | C |
| 423(Trp)451 | C | ||||||
| P29460 | Interleukin-12 subunit β | 1F42 | 97 | 2.50 | No | 297(Trp)297 | C |
| P13671 | Complement component C6 | 1LSL | 44 | 1.90 | Yes | 547(Trp)420 | C |
| 550(Trp)423 | C | ||||||
| 553(Trp)426 | C | ||||||
| P10643 | Complement component C7 | 1LSL | 43 | 1.90 | Yes | 481(Trp)477 | C |
| 484(Trp)480 | C | ||||||
| 487(Trp)483 | C | ||||||
| P07357 | Complement component C8 α chain | 1LSL | 44 | 1.90 | Yes | 522(Trp)420 | C |
| 525(Trp)423 | C | ||||||
| 528(Trp)426 | C | ||||||
| P07358 | Complement component C8 β chain | 1LSL | 52 | 1.90 | Yes | 519(Trp)423 | C |
| 522(Trp)426 | C | ||||||
| P14753 | Erythropoietin receptor | 1EER | 82 | 1.90 | No | 208(Trp)209 | C |
| P10153 | Ribonuclease 2 | 2BZZ | 99 | 0.98 | No | 11(Trp)1007 | H |
aSequence identity between query protein and PDB sequence.
bC-Mannosylation is located in a thrombospondin repeat or not.
cThe location of the C-mannosylation site. The number before the parentheses refers to the numbering in the mature query protein and the number after the parentheses refers to the numbering in the PDB entry.
dDSSP secondary structure. “H” is α-helix, “C” is random coil.
Structural context of C-mannosylation sites
| Swiss–Prot | Protein name | PDB entry | Identity (%)a | Resol. | TSRb | Sitec | Secondary structured |
|---|---|---|---|---|---|---|---|
| P35446 | Spondin-1 (F-spondin) | 1SZL | 100 | NMR | Yes | 420(Trp)448 | C |
| 423(Trp)451 | C | ||||||
| P29460 | Interleukin-12 subunit β | 1F42 | 97 | 2.50 | No | 297(Trp)297 | C |
| P13671 | Complement component C6 | 1LSL | 44 | 1.90 | Yes | 547(Trp)420 | C |
| 550(Trp)423 | C | ||||||
| 553(Trp)426 | C | ||||||
| P10643 | Complement component C7 | 1LSL | 43 | 1.90 | Yes | 481(Trp)477 | C |
| 484(Trp)480 | C | ||||||
| 487(Trp)483 | C | ||||||
| P07357 | Complement component C8 α chain | 1LSL | 44 | 1.90 | Yes | 522(Trp)420 | C |
| 525(Trp)423 | C | ||||||
| 528(Trp)426 | C | ||||||
| P07358 | Complement component C8 β chain | 1LSL | 52 | 1.90 | Yes | 519(Trp)423 | C |
| 522(Trp)426 | C | ||||||
| P14753 | Erythropoietin receptor | 1EER | 82 | 1.90 | No | 208(Trp)209 | C |
| P10153 | Ribonuclease 2 | 2BZZ | 99 | 0.98 | No | 11(Trp)1007 | H |
| Swiss–Prot | Protein name | PDB entry | Identity (%)a | Resol. | TSRb | Sitec | Secondary structured |
|---|---|---|---|---|---|---|---|
| P35446 | Spondin-1 (F-spondin) | 1SZL | 100 | NMR | Yes | 420(Trp)448 | C |
| 423(Trp)451 | C | ||||||
| P29460 | Interleukin-12 subunit β | 1F42 | 97 | 2.50 | No | 297(Trp)297 | C |
| P13671 | Complement component C6 | 1LSL | 44 | 1.90 | Yes | 547(Trp)420 | C |
| 550(Trp)423 | C | ||||||
| 553(Trp)426 | C | ||||||
| P10643 | Complement component C7 | 1LSL | 43 | 1.90 | Yes | 481(Trp)477 | C |
| 484(Trp)480 | C | ||||||
| 487(Trp)483 | C | ||||||
| P07357 | Complement component C8 α chain | 1LSL | 44 | 1.90 | Yes | 522(Trp)420 | C |
| 525(Trp)423 | C | ||||||
| 528(Trp)426 | C | ||||||
| P07358 | Complement component C8 β chain | 1LSL | 52 | 1.90 | Yes | 519(Trp)423 | C |
| 522(Trp)426 | C | ||||||
| P14753 | Erythropoietin receptor | 1EER | 82 | 1.90 | No | 208(Trp)209 | C |
| P10153 | Ribonuclease 2 | 2BZZ | 99 | 0.98 | No | 11(Trp)1007 | H |
aSequence identity between query protein and PDB sequence.
bC-Mannosylation is located in a thrombospondin repeat or not.
cThe location of the C-mannosylation site. The number before the parentheses refers to the numbering in the mature query protein and the number after the parentheses refers to the numbering in the PDB entry.
dDSSP secondary structure. “H” is α-helix, “C” is random coil.
Two structures show similar local structures around the C-mannosylation site compared with the thrombospondin repeats, 1EER (Figure 2A) and 1F42 (not shown). Again, the glycosylated tryptophan is situated in a fairly extended, non-hydrogen-bonded stretch running parallel to a β strand (Syed et al. 1998; Yoon et al. 2000). The aromatic rings are parallel to each other at a Cα–Cα distance of 8.6 and 8.7 Å, respectively.
One structure shows an entirely different local structure, 2BZZ (Figure 2C). The two tryptophans are located in an α-helix and rotated so that the aromatic rings are face to edge at a Cα–Cα distance of 5.1 Å, indicating aromatic stacking between the rings (Baker et al. 2006). The protein has been co-crystallized with a ligand (not shown), but a ligand-free structure not available in the Protein Data Bank shows very similar orientations of the tryptophan rings (Mosimann et al. 1996). Unfortunately, no structure was found for the only protein where the C-mannosylation sites are completely unrelated to the WXXW motif, lens fiber membrane intrinsic protein.
On the basis of the available structures, we found that the accessible surface according to DSSP (digital shape sampling and processing) is 30–147 Å2 (mean, 71 Å2) for glycosylated tryptophans and 0–85 Å2 (mean, 39 Å2) for nonglycosylated tryptophans, showing that modified tryptophans are, on average, more solvent exposed, and all of them are solvent exposed to a certain extent.
Prediction of C-mannosylation sites
Before developing a predictor using machine learning, we investigated what prediction performance is obtained when searching for the simple consensus pattern suggested: WXXW, where the first tryptophan would be glycosylated (Krieg et al. 1998). This is the approach used so far and must ultimately be out-performed for a more complex machine learning approach to be worthwhile. In our dataset consisting of 69 positive and 88 negative sites, the consensus pattern predictor correctly identifies 67% of the positive sites and 93% of the negative sites (see Table II). This means that the consensus rule does not apply for as much as one-third of the positive sites in our data set. Since most experimental studies have so far been directed toward sites that follow the WXXW rule, our data set is, if anything, biased toward sites that do follow it. The number of true sites missed when using the consensus pattern predictor could therefore be much higher. As a test we trained neural networks based only on the information of whether the WXXW pattern was present or not. Not surprisingly, these networks all had predictive performances identical to the consensus predictor itself.
Performance of the NetCGlyc predictor
| Method | Ca | Sn,posb (%) | Spc (%) | Sn,negd (%) |
|---|---|---|---|---|
| WXXW pattern search | 0.63 | 66.7 | 88.5 | 93.2 |
| NetCGlyc | 0.86 | 92.8 | 91.4 | 93.2 |
| Method | Ca | Sn,posb (%) | Spc (%) | Sn,negd (%) |
|---|---|---|---|---|
| WXXW pattern search | 0.63 | 66.7 | 88.5 | 93.2 |
| NetCGlyc | 0.86 | 92.8 | 91.4 | 93.2 |
aMatthews correlation coefficient.
bPositive site sensitivity (the fraction of positive sites correctly predicted).
cSpecificity (the fraction of all positive predictions that are correct).
dNegative site sensitivity (the fraction of negative sites correctly predicted).
Performance of the NetCGlyc predictor
| Method | Ca | Sn,posb (%) | Spc (%) | Sn,negd (%) |
|---|---|---|---|---|
| WXXW pattern search | 0.63 | 66.7 | 88.5 | 93.2 |
| NetCGlyc | 0.86 | 92.8 | 91.4 | 93.2 |
| Method | Ca | Sn,posb (%) | Spc (%) | Sn,negd (%) |
|---|---|---|---|---|
| WXXW pattern search | 0.63 | 66.7 | 88.5 | 93.2 |
| NetCGlyc | 0.86 | 92.8 | 91.4 | 93.2 |
aMatthews correlation coefficient.
bPositive site sensitivity (the fraction of positive sites correctly predicted).
cSpecificity (the fraction of all positive predictions that are correct).
dNegative site sensitivity (the fraction of negative sites correctly predicted).
To develop a more complex predictor, we used a neural network strategy developed for the prediction of mucin-type glycosylation sites (Julenius et al. 2005). We transformed the sequence information (letters) in various ways into numbers that the neural network predictor can understand, to learn what type of encoding would work best for this particular predictor problem. We used sparse encoding (the standard way), profile encoding (the corresponding row in the BLOSUM62 matrix), PSI–BLAST profile encoding (the corresponding row in the profile computed from PSI–BLAST) and amino acid composition. We also trained networks based only on sequence-derived features: predicted secondary structure, predicted surface accessibility, and predicted disorder (three different definitions). The window size presented to the network varied up to 21 residues, with the possibly glycosyated tryptophan in the middle. Initially, we trained neural network predictors with five hidden neurons for all possible networks involving single features. The complexity of the neural network architecture, and therefore the number of parameters that needs to be learned, increases with the window size and the number of hidden neurons used. For these predictors, the Matthews correlation coefficient was calculated using a cross-validation scheme (see the Materials and methods section) and the results are shown in Figure 3. The consensus pattern search performance (0.63) is shown as a thin black line. Of the four different ways to present the sequence, profile encoding was the most successful, with correlation coefficients >0.80 for window sizes 7 and 11. Of the sequence-derived features, disorder prediction according to the DSSP loop–coil definition, and surface accessibility were the most successful, with correlation coefficients >0.63 for many window sizes.
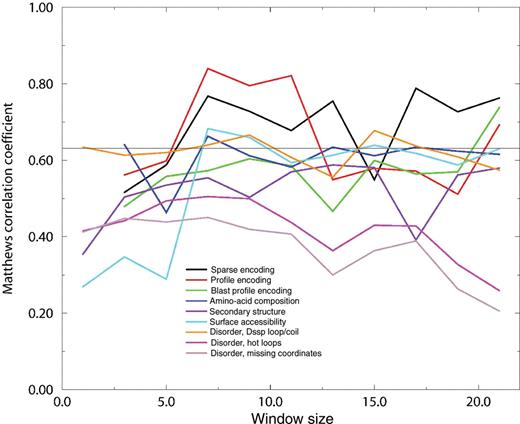
Cross-validation performance of neural networks trained on different features using five hidden neurons. The window size is the number of amino acids for which the information in question is presented to the network, with the tryptophan in question located in the center of the window. See the Materials and methods section and the Supplementary data for a detailed description of the different features. Sparse, profile, and blast profile encoding are three different ways of representing the amino acid sequence. Amino-acid composition is the frequency of amino acid residues within the window. Secondary structure, surface accessibility, and disorder (three different definitions) are predicted from the amino acid sequence. The thin black line denotes the performance of a WXXW consensus pattern search.
To find the best possible combination of features, we used a greedy strategy, trying to combine what appeared to be good input information when training the single feature networks. We also combined the information on the presence/absence of the WXXW motif. For feature combinations that seemed promising, networks with a varying number of hidden neurons (different network complexity) were trained. The very best combination was sparse encoding in a 21-residue window, and information on the presence/absence of the WXXW motif, using eight hidden neurons. This network correctly identifies 93% of both the positive and the negative sites (see Table II). Figure 4 shows the trade-off between making many positive predictions, of which some are false, and making fewer predictions and thereby missing some. A curve reaching far up into the upper left corner is to be preferred and completely random designation would perform along the diagonal. ROC (receiver operating curves) curves are widely used in describing the quality of a classification method such as a predictor or a medical diagnostic tool. For comparison, the performance of the consensus pattern search is marked with X.
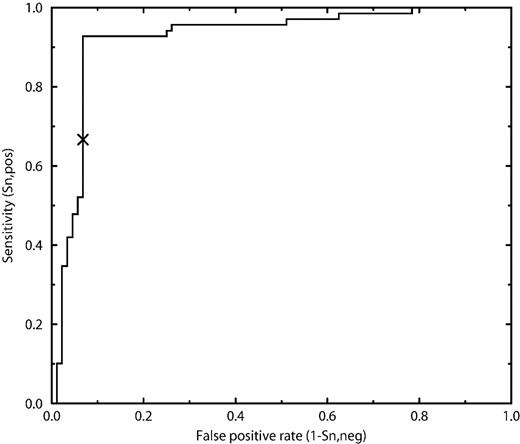
ROC curve showing predictor performance of NetCGlyc. The sensitivity is the fraction of positive sites correctly predicted. The false positive rate is the fraction of negative sites wrongly predicted to be positive. A predictor making random guesses would perform along the diagonal and a perfect predictor along the y-axis. The performance of the consensus pattern search (WXXW) is marked with an X.
Scanning the human genome
All human transcripts with signal peptides and/or transmembrane helices were scanned with NetCGlyc 1.0 for predicted C-mannosylation sites. Since C-mannosylation occurs in the ER, only tryptophans either in extracellular proteins or on the extracellular side of membrane proteins can be mannosylated. Of the 14 554 downloaded transcripts, 2573 (18%) were predicted to contain at least one C-mannosylation site. These proteins were investigated for gene ontology (GO) annotation, and the results are shown in Table III. An enrichment factor >1 indicates that the term is over-represented for the C-mannosylated proteins. Of the 3713 predicted sites, 1366 were located at the first tryptophan in a WXXW motif, 214 were located at the second tryptophan in a WXXW motif, and 2133 were found in different sequence contexts.
GO annotations for human proteins predicted to be C-mannosylated
| Occurrence | Enrichment factor | GO term | GO annotation |
|---|---|---|---|
| 442 | 1.29 | GO:0004872 | Receptor activity |
| 257 | 1.00 | GO:0005515 | Protein binding |
| 227 | 0.92 | GO:0007165 | Signal transduction |
| 195 | 1.34 | GO:0005509 | Calcium ion binding |
| 152 | 1.32 | GO:0006810 | Transport |
| 148 | 0.84 | GO:0007186 | G-protein-coupled receptor protein signaling pathway |
| 129 | 1.25 | GO:0016740 | Transferase activity |
| 127 | 1.15 | GO:0006811 | Ion transport |
| 119 | 1.51 | GO:0005524 | ATP binding |
| 110 | 1.01 | GO:0007155 | Cell adhesion |
| 109 | 1.28 | GO:0005215 | Transporter activity |
| 108 | 1.09 | GO:0006508 | Proteolysis |
| 102 | 1.34 | GO:0008152 | Metabolism |
| 101 | 1.48 | GO:0000166 | Nucleotide binding |
| 88 | 1.28 | GO:0016787 | Hydrolase activity |
| 83 | 1.10 | GO:0001584 | Rhodopsin-like receptor activity |
| 77 | 1.18 | GO:0008270 | Zinc ion binding |
| 75 | 0.94 | GO:0046872 | Metal ion binding |
| 74 | 0.98 | GO:0006955 | Immune response |
| 71 | 1.15 | GO:0005554 | Molecular function unknown |
| 65 | 1.35 | GO:0007275 | Development |
| 65 | 1.19 | GO:0005216 | Ion channel activity |
| 62 | 1.23 | GO:0000004 | Biological process unknown |
| 57 | 1.41 | GO:0005529 | Sugar binding |
| 52 | 1.08 | GO:0006118 | Electron transport |
| 49 | 2.29 | GO:0016887 | ATPase activity |
| 49 | 0.63 | GO:0050896 | Response to stimulus |
| 48 | 1.90 | GO:0004930 | G-protein-coupled receptor activity |
| 47 | 4.09 | GO:0004896 | Hematopoietin/interferon-class (D200-domain) cytokine receptor activity |
| 47 | 1.48 | GO:0006468 | Protein amino acid phosphorylation |
| 47 | 1.47 | GO:0006814 | Sodium ion transport |
| 46 | 1.21 | GO:0007399 | Nervous system development |
| 45 | 1.72 | GO:0006812 | Cation transport |
| 43 | 1.78 | GO:0006816 | Calcium ion transport |
| 43 | 1.39 | GO:0016757 | Transferase activity, transferring glycosyl groups |
| 42 | 1.26 | GO:0005975 | Carbohydrate metabolism |
| 42 | 1.30 | GO:0006629 | Lipid metabolism |
| 42 | 1.26 | GO:0030154 | Cell differentiation |
| 41 | 1.84 | GO:0004222 | Metalloendopeptidase activity |
| Occurrence | Enrichment factor | GO term | GO annotation |
|---|---|---|---|
| 442 | 1.29 | GO:0004872 | Receptor activity |
| 257 | 1.00 | GO:0005515 | Protein binding |
| 227 | 0.92 | GO:0007165 | Signal transduction |
| 195 | 1.34 | GO:0005509 | Calcium ion binding |
| 152 | 1.32 | GO:0006810 | Transport |
| 148 | 0.84 | GO:0007186 | G-protein-coupled receptor protein signaling pathway |
| 129 | 1.25 | GO:0016740 | Transferase activity |
| 127 | 1.15 | GO:0006811 | Ion transport |
| 119 | 1.51 | GO:0005524 | ATP binding |
| 110 | 1.01 | GO:0007155 | Cell adhesion |
| 109 | 1.28 | GO:0005215 | Transporter activity |
| 108 | 1.09 | GO:0006508 | Proteolysis |
| 102 | 1.34 | GO:0008152 | Metabolism |
| 101 | 1.48 | GO:0000166 | Nucleotide binding |
| 88 | 1.28 | GO:0016787 | Hydrolase activity |
| 83 | 1.10 | GO:0001584 | Rhodopsin-like receptor activity |
| 77 | 1.18 | GO:0008270 | Zinc ion binding |
| 75 | 0.94 | GO:0046872 | Metal ion binding |
| 74 | 0.98 | GO:0006955 | Immune response |
| 71 | 1.15 | GO:0005554 | Molecular function unknown |
| 65 | 1.35 | GO:0007275 | Development |
| 65 | 1.19 | GO:0005216 | Ion channel activity |
| 62 | 1.23 | GO:0000004 | Biological process unknown |
| 57 | 1.41 | GO:0005529 | Sugar binding |
| 52 | 1.08 | GO:0006118 | Electron transport |
| 49 | 2.29 | GO:0016887 | ATPase activity |
| 49 | 0.63 | GO:0050896 | Response to stimulus |
| 48 | 1.90 | GO:0004930 | G-protein-coupled receptor activity |
| 47 | 4.09 | GO:0004896 | Hematopoietin/interferon-class (D200-domain) cytokine receptor activity |
| 47 | 1.48 | GO:0006468 | Protein amino acid phosphorylation |
| 47 | 1.47 | GO:0006814 | Sodium ion transport |
| 46 | 1.21 | GO:0007399 | Nervous system development |
| 45 | 1.72 | GO:0006812 | Cation transport |
| 43 | 1.78 | GO:0006816 | Calcium ion transport |
| 43 | 1.39 | GO:0016757 | Transferase activity, transferring glycosyl groups |
| 42 | 1.26 | GO:0005975 | Carbohydrate metabolism |
| 42 | 1.30 | GO:0006629 | Lipid metabolism |
| 42 | 1.26 | GO:0030154 | Cell differentiation |
| 41 | 1.84 | GO:0004222 | Metalloendopeptidase activity |
GO annotations for human proteins predicted to be C-mannosylated
| Occurrence | Enrichment factor | GO term | GO annotation |
|---|---|---|---|
| 442 | 1.29 | GO:0004872 | Receptor activity |
| 257 | 1.00 | GO:0005515 | Protein binding |
| 227 | 0.92 | GO:0007165 | Signal transduction |
| 195 | 1.34 | GO:0005509 | Calcium ion binding |
| 152 | 1.32 | GO:0006810 | Transport |
| 148 | 0.84 | GO:0007186 | G-protein-coupled receptor protein signaling pathway |
| 129 | 1.25 | GO:0016740 | Transferase activity |
| 127 | 1.15 | GO:0006811 | Ion transport |
| 119 | 1.51 | GO:0005524 | ATP binding |
| 110 | 1.01 | GO:0007155 | Cell adhesion |
| 109 | 1.28 | GO:0005215 | Transporter activity |
| 108 | 1.09 | GO:0006508 | Proteolysis |
| 102 | 1.34 | GO:0008152 | Metabolism |
| 101 | 1.48 | GO:0000166 | Nucleotide binding |
| 88 | 1.28 | GO:0016787 | Hydrolase activity |
| 83 | 1.10 | GO:0001584 | Rhodopsin-like receptor activity |
| 77 | 1.18 | GO:0008270 | Zinc ion binding |
| 75 | 0.94 | GO:0046872 | Metal ion binding |
| 74 | 0.98 | GO:0006955 | Immune response |
| 71 | 1.15 | GO:0005554 | Molecular function unknown |
| 65 | 1.35 | GO:0007275 | Development |
| 65 | 1.19 | GO:0005216 | Ion channel activity |
| 62 | 1.23 | GO:0000004 | Biological process unknown |
| 57 | 1.41 | GO:0005529 | Sugar binding |
| 52 | 1.08 | GO:0006118 | Electron transport |
| 49 | 2.29 | GO:0016887 | ATPase activity |
| 49 | 0.63 | GO:0050896 | Response to stimulus |
| 48 | 1.90 | GO:0004930 | G-protein-coupled receptor activity |
| 47 | 4.09 | GO:0004896 | Hematopoietin/interferon-class (D200-domain) cytokine receptor activity |
| 47 | 1.48 | GO:0006468 | Protein amino acid phosphorylation |
| 47 | 1.47 | GO:0006814 | Sodium ion transport |
| 46 | 1.21 | GO:0007399 | Nervous system development |
| 45 | 1.72 | GO:0006812 | Cation transport |
| 43 | 1.78 | GO:0006816 | Calcium ion transport |
| 43 | 1.39 | GO:0016757 | Transferase activity, transferring glycosyl groups |
| 42 | 1.26 | GO:0005975 | Carbohydrate metabolism |
| 42 | 1.30 | GO:0006629 | Lipid metabolism |
| 42 | 1.26 | GO:0030154 | Cell differentiation |
| 41 | 1.84 | GO:0004222 | Metalloendopeptidase activity |
| Occurrence | Enrichment factor | GO term | GO annotation |
|---|---|---|---|
| 442 | 1.29 | GO:0004872 | Receptor activity |
| 257 | 1.00 | GO:0005515 | Protein binding |
| 227 | 0.92 | GO:0007165 | Signal transduction |
| 195 | 1.34 | GO:0005509 | Calcium ion binding |
| 152 | 1.32 | GO:0006810 | Transport |
| 148 | 0.84 | GO:0007186 | G-protein-coupled receptor protein signaling pathway |
| 129 | 1.25 | GO:0016740 | Transferase activity |
| 127 | 1.15 | GO:0006811 | Ion transport |
| 119 | 1.51 | GO:0005524 | ATP binding |
| 110 | 1.01 | GO:0007155 | Cell adhesion |
| 109 | 1.28 | GO:0005215 | Transporter activity |
| 108 | 1.09 | GO:0006508 | Proteolysis |
| 102 | 1.34 | GO:0008152 | Metabolism |
| 101 | 1.48 | GO:0000166 | Nucleotide binding |
| 88 | 1.28 | GO:0016787 | Hydrolase activity |
| 83 | 1.10 | GO:0001584 | Rhodopsin-like receptor activity |
| 77 | 1.18 | GO:0008270 | Zinc ion binding |
| 75 | 0.94 | GO:0046872 | Metal ion binding |
| 74 | 0.98 | GO:0006955 | Immune response |
| 71 | 1.15 | GO:0005554 | Molecular function unknown |
| 65 | 1.35 | GO:0007275 | Development |
| 65 | 1.19 | GO:0005216 | Ion channel activity |
| 62 | 1.23 | GO:0000004 | Biological process unknown |
| 57 | 1.41 | GO:0005529 | Sugar binding |
| 52 | 1.08 | GO:0006118 | Electron transport |
| 49 | 2.29 | GO:0016887 | ATPase activity |
| 49 | 0.63 | GO:0050896 | Response to stimulus |
| 48 | 1.90 | GO:0004930 | G-protein-coupled receptor activity |
| 47 | 4.09 | GO:0004896 | Hematopoietin/interferon-class (D200-domain) cytokine receptor activity |
| 47 | 1.48 | GO:0006468 | Protein amino acid phosphorylation |
| 47 | 1.47 | GO:0006814 | Sodium ion transport |
| 46 | 1.21 | GO:0007399 | Nervous system development |
| 45 | 1.72 | GO:0006812 | Cation transport |
| 43 | 1.78 | GO:0006816 | Calcium ion transport |
| 43 | 1.39 | GO:0016757 | Transferase activity, transferring glycosyl groups |
| 42 | 1.26 | GO:0005975 | Carbohydrate metabolism |
| 42 | 1.30 | GO:0006629 | Lipid metabolism |
| 42 | 1.26 | GO:0030154 | Cell differentiation |
| 41 | 1.84 | GO:0004222 | Metalloendopeptidase activity |
Investigating proteins with more than five predicted sites, we found that proteins with thrombospondin repeats are highly over-represented (e.g. semaphorins, brain-specific angiogenesis inhibitors, ADAMTS's) as would be expected. More surprisingly, we also found many proteins related to low-density lipoprotein (LDL)-receptor. Looking more closely at this class of proteins, we find that a substantial number of LDL-receptor class B repeats, also called YWTD repeats, have an additional tryptophan, making the repeated sequence YWTDW. According to PROSITE (http://www.expasy.org/prosite/), there are 47 such YWTDW repeats in the human proteome, and our predictor predicts most of these to be positive for C-mannosylation. There are three available crystal structures (PDB ID 1IJQ, 1NPE, and 1N7D) of LDL-receptor class B repeats from two different proteins (human LDL-receptor and mouse nidogen 1). In both proteins, six repeats are packed very closely together in a six-bladed β-propeller (Jeon et al. 2001; Rudenko et al. 2002; Takagi et al. 2003). Because of close contact with a hydrophobic residue on the preceding repeat (phenylalanine in both structures), the first tryptophan of the YWTDW sequence is inaccessible to the solvent. If all LDL-receptor class B repeats fold into six-bladed β-propellers, C-mannosylation at these sites would be highly unlikely. However, in the case of two of the YWTDW repeats, an additional tryptophan precedes the YWTDW repeat, making the sequence WMYWTDW. Judging from the available structures in which the corresponding position is occupied by a phenylalanine and an asparagine, respectively, the first tryptophan is more solvent accessible and this residue is therefore a more likely C-mannosylation site.
One of the characteristic structural features of type I cytokine receptors is a WSXWS motif in the C-terminal domain (Bazan 1990). This has, at least in the case of the erythropoietin receptor, been shown to be C-mannosylated (Furmanek et al. 2003). We extracted 29 human protein sequences with annotated WSXWS motifs from Swiss–Prot and performed prediction of C-mannosylation sites using NetCGlyc 1.0. Twenty-seven of 29 proteins have at least one predicted site. The two exceptions were growth hormone receptor (P10912) and interleukin-3 receptor alpha chain (P26951), both with degenerated motifs (YGEFS and LSAWS respectively). Interestingly, several receptors contain more than one predicted C-mannosylation site. Interleukin-6 receptor subunit beta (P40189), leptin receptor (P48357), and leukemia inhibitory factor receptor (P42702) each contain as many as four predicted sites and what seem to be two WSXWS motifs. Type I cytokine receptors are classified as GO:0004896 (hematopoietin/interferon class cytokine receptor activity), which explains the high enrichment factor (4.09) of this GO term among the human transcripts predicted to be C-mannosylated (Table III).
Discussion
The structural analysis indicates that aromatic stacking may play a role in the substrate recognition of C-mannosyltransferase, at least in the case of substrates that contain the WXXW motif. Modified tryptophan residues are typically at least partly solvent exposed, whereas nonmodified tryptophans may be completely buried in the interior of the protein. Previous studies have shown C-mannosylation to take place very early, probably even before the folding of the protein (Doucey et al. 1998; Perez-Vilar et al. 2004). To explain the differences in solvent accessibility of different tryptophans, we suggest that the modification probably, at least in some proteins, affects the folding of the protein. It would be interesting to investigate what prevents C-mannosylation of the YWTDW motifs of LDL receptor class B repeats before folding, since this would then prevent the correct folding into six-bladed β-propellers.
The results of the training on predicted features (Figure 3) are in agreement with the results of the structural analysis. The fact that the predicted surface accessibility proved to be good input information for the network method can be explained by the fact that glycosylated tryptophans are more solvent accessible than the tryptophans that are not modified. Predicted disorder according to DSSP loop–coil definition was much better input information than either of the other two predicted disorder measures. In four of the five available structures, the glycosylated tryptophan is located in a fairly extended, non-hydrogen-bonded stretch. These stretches are classified as loop or coil according to the DSSP definition, but are not particularly disordered according to the two other definitions, which require the loop–coil to have elevated temperature factor, “hot loops”, or atom coordinates to be missing in the structure. It is hardly surprising that the prediction of a disorder definition that seems to apply to a large part of glycosylated tryptophans is good input information to the predictor network.
We were able to develop a predictor that predicts more sites than the WXXW consensus rule (higher sensitivity) without making any additional false predictions. Obtaining higher sensitivity without loss of specificity is usually very difficult, but can probably be explained by the fact that there is a lot of sequence information in various positions of the aligned sites (Figure 1) in addition to the tryptophan in position +3. Our method is able to use these additional sites in an optimal way. We would like to point out that although this is the case, NetCGlyc 1.0 will work best on WXXW-related sites since most of the sites in the training examples were of this type. If future experiments show that C-mannosylation is common in other sequence contexts as well, NetCGlyc will be retrained to accommodate this.
By training a predictor, NetCGlyc 1.0, and making it publicly available among our other predictors for different types of glycosylation sites at our web page, www.cbs.dtu.dk/services, we hope to bring attention to this newly discovered type of glycosylation. The glycan is very small, only one hexose, which is probably why the modification was left undiscovered for so long. One hexose would not change the migration rate on sodium dodecylsulphate–polyacrylamide gel electrophoresis enough to attract attention to its presence, compared with the large glycans of N-glycosylation and proteoglycans, or the numerous glycans of a mucin. The two newly discovered sites in lens fiber membrane intrinsic protein (Ervin et al. 2005) indicate that although tryptophan is the rarest of the amino acid residues, its modification with α-mannopyranose does not require the presence of a WXXW motif and may actually be more common than we think.
Materials and methods
Dataset
Experimentally verified mammalian C-mannosylation sites were extracted from O-GlycBase v6.00 (www.cbs.dtu.dk/databases/OGLYCBASE/) (Gupta et al. 1999), Swiss–Prot (Boeckmann et al. 2003), and through literature searches. We also found one protein reported to have no C-mannosylation sites. Twelve native proteins and 27 naturally occurring or engineered mutants/peptides were gathered in this way. The original articles were checked for the protein region investigated for glycosylation sites in each case. Tryptophans located in the investigated regions and not reported as positive or partial sites were used as negative sites. Partly glycosylated tryptophans were used as positive sites. No tryptophans located in signal peptides were used. The dataset consisted of 69 positive and 88 negative sites.
Neural network training
For readability, this section was shortened to suit the average reader of Glycobiology. The Supplementary data provide details of sequence encoding, feature encoding, and neural networks.
A neural network does not understand letters, so the amino acid sequence and different features must be translated into numbers. This is called encoding, and can be done in a number of ways. Each number that is presented to the neural network constitutes what is called an input neuron. The goal is to provide the network with as much information as possible while still keeping the number of input neurons as low as possible.
Sparse encoding (Qian and Sejnowski 1988; Hertz et al. 1991) is the conventional way to convert an amino acid sequence into numerical form.
With profile encoding, the input for each amino acid consisted of the corresponding row in the BLOSUM62 matrix (Henikoff and Henikoff 1992).
With PSI–BLAST encoding, the input for each amino acid consisted of the corresponding row in the position-specific scoring matrix computed from three cycles of PSI–BLAST (Altschul et al. 1997).
The amino acid composition was calculated for a sequence window around each particular site.
Surface accessibility was predicted using a neural network method called surfg (Hansen et al. 1998).
Secondary structure was predicted using PSIPRED (Jones 1999; McGuffin et al. 2000) using position-specific scoring matrices computed from three cycles of PSI–BLAST (Altschul et al. 1997).
Protein disorder was predicted using DisEMBL (Linding et al. 2003). DisEMBL predicts disorder according to three different definitions: (1) loops–coils as defined by DSSP (Kabsch and Sander 1983); (2) hot loops, being loops according to DSSP with a high degree of mobility as determined from Cα temperature factors; and (3) missing coordinates in X-ray structures.
The neural networks were of the two-layer feed-forward type, trained by standard back propagation. Network complexity was varied by changing the number of neurons in the input layer as well as in the hidden layer to find the optimal complexity for this particular prediction problem. This is important, since a network with too little complexity (too few neurons) will lack the ability to learn the training examples, and a network with too much complexity (too many neurons) will learn the examples too well and lose the ability to make predictions for examples that were not in the training set (the ability to generalize). This second problem is sometimes called over-training and is one of the reasons why it is so important to make sure that the examples in the test set are different from and unrelated to the examples in the training set. If the sets are unrelated to each other, the performance on the test set will decrease when over-training occurs and if the problem can be detected, it can also be avoided. The risk of over-training increases with decreasing data set size.
The predictive performance was monitored using the Matthews correlation coefficient (Matthews 1975) during training and test of the networks
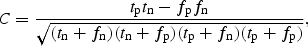
where tp is the number of correctly predicted positive sites (true positives), tn the number of correctly predicted negative sites (true negatives), fn the number of sites falsely predicted to be negative (false negatives), and fp the number of sites falsely predicted to be positive (false positives). The Matthews correlation coefficient will always be a value between −1 and 1, where a predictor that is always wrong will have a correlation coefficient of −1, one that is always right will have a correlation coefficient of 1, and one that makes random guesses will have a correlation coefficient of 0. It takes into account the performance on both the positive and the negative sites and is widely used for classification problems such as this one.
The fraction of positive sites correctly predicted, the positive site sensitivity, Sn,pos, was computed as
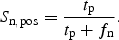
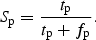
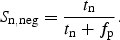
A region of 21 residues around each (positive or negative) site was extracted (10 amino acids on each side of the tryptophan). The sites were aligned according to the central tryptophan and an unrooted neighbor-joining tree was constructed using CLUSTAL X (Thompson et al. 1997). From this tree, groups of closely related sites were identified. One or more of these groups were collected into larger sets, in total six, each containing both positive and negative sites and of roughly equal size. Between sites belonging to different sets, sequence identity did not exceed 50%. The six sets were used so that every network was trained six times, using five sets as training sets and one set as the test set. The reported cross-validation performance is the joint performance of the six resulting networks on their respective test sets.
Scanning the humane genome
Sequences and their GO annotations for all human protein transcripts (build NCBI36) with either signal peptide and/or transmembrane helices were downloaded from http://www.ensembl.org using the EnsMart system. Looking at GO annotations, “cellular component” terms were ignored. We compared the occurrences of different GO terms of the proteins predicted to be C-mannosylated with the occurrence of the different GO terms of all the protein transcripts, since some GO terms are more frequently occurring than others. The enrichment factor was calculated as the ratio between the occurrence of the term for the C-mannosylated sequences and the occurrence of the term for a random sample of the same size. An enrichment factor >1 indicates that the term is over-represented for the C-mannosylated proteins.
Proteins with annotated WSXWS motifs were extracted from Swiss–Prot by searching for the term “WSXWS motif.” in the “features” section of all entries. Human proteins were identified using the last part of the entry name, which is “_HUMAN” for human proteins. In total, 29 human type I cytokine receptors were identified in this way.
Supplementary data
Supplementary data are available at Glycobiology online (http://www.glycob.oxfordjournals.org/).
Acknowledgments
The author thanks Kristoffer Rapacki for technical assistance in making the web predictor operational, Anne Mølgaard for help with the analysis of protein structures, and Timo Pikkarainen for critical reading of the manuscript. This work was supported by the Knut and Alice Wallenberg foundation.
Conflict of interest statement
None declared.
Abbreviations
- DSSP
digital shape sampling and processing
- ER
endoplasmic reticulum
- GO
gene ontology
- LDL
low-density lipoprotein
- NMR
nuclear magnetic resonance
- PSI–BLAST
position specific iterative–basic local alignment search tool
- RNase 2
human ribonuclease 2
- ROC curve
receiver operating characteristics curve.

{kind=link}
{kind=link}
{kind=link}
{kind=link}