




Abstract
Individuals with the neurofibromatosis 2 (NF2) inherited tumor predisposition syndrome are prone to the development of nervous system tumors, including schwannomas and meningiomas. The NF2 tumor suppressor protein, merlin or schwannomin, inhibits cell growth and motility as well as affects actin cytoskeleton-mediated processes. Merlin interacts with several proteins that might mediate merlin growth suppression, including hepatocyte growth factor-regulated tyrosine kinase substrate (HRS or HGS). Previously, we demonstrated that regulated overexpression of HRS in RT4 rat schwannoma cells had the same functional consequences as regulated overexpression of merlin. To determine the functional significance of this interaction, we generated a series of HRS truncation mutants and defined the regions of HRS required for merlin binding and HRS growth suppression. The HRS domain required for merlin binding was narrowed to a region (residues 470–497) containing the predicted coiled-coil domain whereas the major domain responsible for HRS growth suppression was distinct (residues 498–550). To determine whether merlin growth suppression required HRS, we demonstrated that merlin inhibited growth in HRS +/+, but not HRS −/− mouse embryonic fibroblast cells. In contrast, HRS could suppress cell growth in the absence of Nf2 expression. These results suggest that merlin growth suppression requires HRS expression and that the binding of merlin to HRS may facilitate its ability to function as a tumor suppressor.
INTRODUCTION
Neurofibromatosis 2 (NF2) is an autosomal dominant inherited tumor predisposition syndrome in which affected individuals are prone to the development of specific nervous system tumors (1). The hallmark central nervous system tumor in NF2 is the schwannoma, typically involving both eighth cranial nerves (bilateral vestibular schwannomas). Continued growth of these tumors leads to deafness and balance problems. In addition to bilateral vestibular schwannomas, schwannomas can occur on other cranial or peripheral nerves throughout the body. The second most common tumor in NF2 is the meningioma, arising from leptomeningeal cap cells and is seen in 50% of affected individuals. Lastly, ependymomas and, less commonly, astrocytomas are also observed in NF2 patients.
The NF2 gene was identified by positional cloning in 1993 and found to encode a 595 amino acid protein termed merlin or schwannomin (2,3). Merlin contains three predicted structurally important regions, including an amino terminal FERM domain (residues 1–302), a central alpha helical region (residues 303–479) and a unique carboxyl terminus (residues 480–595). Based on this structure, merlin has been classified as a member of the Protein 4.1 subfamily of proteins that includes ezrin, radixin and moesin (ERM proteins) (4). These proteins are proposed to link the actin cytoskeleton to cell surface glycoproteins. While ERM proteins have not been directly implicated in growth regulation, ezrin has been shown to modulate apoptosis in particular cell types (5). Like other ERM proteins, merlin binds to actin and is associated with the actin cytoskeleton (6). However, merlin has a different subcellular distribution than other ERM proteins in peripheral nerve Schwann cells, reinforcing the idea that merlin is a unique member of this Protein 4.1 family of proteins (7,8).
Since individuals with NF2 are at increased risk of developing specific nervous system tumors, the NF2 gene has been hypothesized to function as a tumor suppressor (9). Support for this classification derives from studies demonstrating germline mutations in the NF2 gene in NF2 patients and bi-allelic inactivation of NF2 in NF2-associated tumors, including schwannomas, ependymomas and meningiomas (10–12). Moreover, re-expression of the NF2 gene in merlin-deficient meningioma cells in vitro and schwannoma cells both in vitro and in vivo results in growth suppression (13,14). In contrast, regulated expression of merlin containing missense NF2 patient mutations has no effect on cell growth either in vitro or in vivo (15). Merlin loss is also observed in nearly all sporadic schwannomas and over half of sporadic meningiomas, suggesting that the NF2 tumor suppressor plays a more general role in the molecular pathogenesis of these tumors (16–19).
Clues to the mechanism of action of merlin have derived from several different studies. Mice with a targeted mutation in the Nf2 gene develop malignant tumors that exhibit a highly metastatic phenotype (20). This finding suggested that merlin might normally regulate both cell proliferation and actin cytoskeleton-associated processes important for mediating cell motility. In this regard, human NF2-deficient schwannoma cells have defects in actin cytoskeleton organization that can be restored by the re-expression of wild-type, but not mutant, merlin (21). This phenotypic abnormality can also be partially rescued by modulating Rac/Rho signaling (22). In addition, regulated overexpression of wild-type merlin, but not merlin containing missense NF2 patient mutations, in rat schwannoma cells results in reduced cell motility and alterations in actin cytoskeleton organization during the initial phases of cell spreading in vitro (15,23).
Merlin has been shown to interact with a number of potentially important effectors, including a sodium–hydrogen exchange regulatory factor (24), an actin-binding protein (βII-spectrin) (25), schwannomin interacting protein (26), syntenin (27), the CD44 transmembrane hyaluronic acid binding protein (28,29) and hepatocyte growth factor-regulated tyrosine kinase substrate (HRS/HGS) (30,31). Merlin interacts with HRS through residues in the merlin carboxyl terminal region. Previous studies from our laboratory have shown that regulated overexpression of HRS has similar effects to overexpression of merlin on rat RT4 schwannoma cell proliferation and actin cytoskeleton-associated processes (30). HRS overexpression results in reduced RT4 cell proliferation and motility as well as alterations in actin cytoskeleton organization during the initial phases of cell spreading (31).
HRS was originally identified as a 115 kDa tyrosine-phosphorylated protein in B16-F1 mouse melanoma cells treated with hepatocyte growth factor (HGF) (32). HGF is one of the most potent mitogenic stimuli for Schwann cells and has been shown to promote cell motility in a variety of cell types (33,34). Human HRS contains 777 amino acids with several conserved protein–protein interaction domains, including a FYVE domain, a VHS zinc finger domain, a coiled-coil domain and two proline-rich regions (32). The FYVE and VHS domains have been implicated in the localization of HRS to the early endosome, where HRS functions to modulate endocytosis and exocytosis (35,36). In addition, HRS has been suggested to function in the TGF-β signaling pathway by binding to SARA, a Smad family adaptor protein (37), as well as mediate cell growth regulation by binding to the STAT signal transducing adaptor molecule (STAM) and modulating STAT pathway signaling (38).
To elucidate the relationship between HRS binding and merlin function, we determined the HRS domains required for growth suppression and merlin binding. In this report, we demonstrate that the HRS domains important for merlin binding and HRS growth suppression are distinct. We further show that merlin growth suppression is impaired in cells lacking HRS expression, but that HRS can function as a growth suppressor in the absence of merlin expression. Collectively, these results suggest that merlin growth suppression is dependent on HRS and that HRS may function to transduce the merlin growth suppressor signal.
RESULTS
Truncated HRS fragments are expressed both in vitro and in vivo
In order to study the interaction between merlin and HRS, we generated a series of truncated human HRS fragments as described in the Materials and Methods section. Truncated HRS fragments were designed to serially delete the predicted conserved protein–protein binding domains (Fig. 1A). Two approaches were taken to demonstrate HRS fragment expression. Gene expression in vitro was demonstrated by coupled transcription and translation in vitro (TNT) and proteins were detected with the 9E10 anti-myc monoclonal antibody (Fig. 1B). In vivo expression was demonstrated by transient transfection in RT4 schwannoma cells and detected by western blot using the 9E10 anti-myc monoclonal antibody in cell lysates (Fig. 1C). Each of the HRS fragments produced proteins of the expected sizes both in vitro and in vivo.
The merlin binding site is located within the HRS predicted coiled-coil domain
To determine the region of HRS important for mediating the interaction with merlin, we performed two complementary sets of experiments. First, we employed glutathione-S-transferase (GST) affinity chromatography. GST-fused carboxyl-terminal (C-term) merlin (residues 299–595) protein was used to interact with radioactive TNT products of various HRS fragments. These initial experiments demonstrated that the carboxyl terminus of merlin could interact with HRS (270–777) and HRS (447–777) as well as full-length HRS (not shown), but not with HRS (270–435) and HRS (551–777) (Fig. 2A). Further experiments showed that the carboxyl terminus of merlin also interacts with HRS (447–626) and HRS (270–550) (Fig. 2A). Based on these results, we mapped the minimal region responsible for merlin binding to HRS residues 447–550.
Second, we analysed the interaction between the carboxyl terminus of merlin and different HRS fragments in vivo by co-immunoprecipitation after transient transfection in RT4 cells. These experiments demonstrated that HRS (270–497) and HRS (470–777) interact with the carboxyl terminus of merlin (Fig. 2B). In other experiments, equivalent binding to the carboxyl terminus of merlin was observed with HRS (270–497) and HRS (447–777) (Fig. 4A). In contrast, HRS (270–435) and HRS (498–777) do not bind to the carboxyl terminus of merlin (Fig. 2B). Collectively, these results suggest that merlin binding to HRS required residues 470–497 within the HRS predicted coiled-coil domain (Fig. 2C).
HRS growth suppression requires residues in the carboxyl terminal domain
Next, we wanted to define the domain important for HRS growth suppression to determine whether it was distinct from the merlin-binding region. As we have shown previously, HRS overexpression results in decreased RT4 rat schwannoma cell growth in clonogenic assays as well as by using inducible RT4 cell lines (31). Using the HRS truncation constructs described above, we analysed HRS-mediated RT4 growth suppression defined as a greater than 25% reduction in colony number compared with vector controls. Initially, we observed significant HRS growth suppression with constructs HRS (270–777) and HRS (447–777) (36.53%, P<0.01 and 35.35%, P<0.01, respectively; Fig. 3A). In contrast, no significant growth suppression was observed with HRS (270–435) and HRS (551–777) (6.07%, P>0.10 and 15.27%, P>0.05, respectively; Fig. 3A). To further narrow the minimal growth suppression domain of HRS, we studied additional fragments for their ability to suppress RT4 cell growth. As shown in Figure 3B, HRS (498–777) suppressed cell growth significantly (39.7%, P>0.001), while HRS (270–497) and HRS (551–777) did not (12.09%, P>0.05 and 12.82%, P>0.05, respectively). Growth suppression was also seen with the HRS (470–777) mutant (data not shown). Based on these experiments, the domain required for HRS growth suppression maps between residues 498 and 551, which is distinct from the sequences important for merlin binding (Fig. 3C).
A merlin-binding, but non-growth-suppressing, HRS mutant cannot reverse merlin growth suppression
Since the HRS domains important for merlin binding and HRS growth suppression were distinct and separable, we next evaluated the possibility that exogenous overexpression of an HRS fragment capable of binding to merlin, but not suppressing cell growth, might impair merlin growth suppression by interfering with endogenous merlin–HRS interactions. Using the HRS fragment containing residues 270–497, which does not suppress cell growth but binds merlin (Fig. 4A), we transfected an inducible merlin RT4 cell line with HRS (270–497) as well as other HRS truncation mutants. Whereas merlin induction using doxycycline resulted in an ∼50% reduction in RT4 cell colony number (‘vector’), HRS co-expression resulted in an 80% reduction (Fig. 4B and C). We observed no cooperative effect using the HRS (1–232) mutant that fails to bind merlin and lacks growth suppressor activity. HRS mutants capable of binding merlin and able to suppress growth [HRS (447–777)] exhibited similar growth suppressor properties as full length HRS. HRS (270–497) had no effect on merlin growth suppression, suggesting that interfering with endogenous merlin–HRS binding is not sufficient to reverse merlin growth suppression.
HRS is required for merlin tumor suppressor function
Given our inability to demonstrate a dominant inhibitory effect for growth suppressor defective merlin-binding mutants of HRS, we next sought to determine whether merlin growth suppression required HRS expression. To test this hypothesis, we assayed the ability of merlin to suppress cell growth in HRS-deficient mouse embryonic fibroblasts (Hrs−/− MEFs). The endogenous expression profiles of merlin and HRS were confirmed by Western blot using specific anti-merlin or anti-HRS antibodies (data not shown). In these experiments, merlin could not inhibit the growth of HRS deficient cells. In contrast, the re-introduction of HRS resulted in significant growth suppression (Fig. 5A). In HRS+/+ MEF (Fig. 5B) or NIH3T3 (data not shown) cells, overexpression of either merlin or HRS resulted in growth suppression. Collectively, these data suggest that merlin growth suppression requires HRS expression.
HRS growth suppression does not require merlin expression
Since merlin cannot suppress MEF growth in the absence of HRS expression, we next wished to determine whether HRS growth suppression required merlin expression. Using Nf 2-deficient mouse embryonic cells (Nf 2−/− MEFs), which express endogenous HRS, we demonstrated that re-introduction of either HRS or merlin could inhibit Nf 2−/− MEF colony formation (Fig. 5C). This is in agreement with our previous results demonstrating that HRS is capable of suppressing the growth of RT4 rat schwannoma cells that express nearly undetectable levels of merlin (31). In contrast to merlin, HRS growth suppression does not require merlin expression and suggests that HRS may function downstream of merlin in a potential growth regulatory pathway.
DISCUSSION
Among the known merlin-interacting proteins, HRS is one of the most attractive candidates for a merlin effector protein that transduces the NF2 growth regulatory signal. Our previous studies identified HRS as a unique binding partner for merlin, which suggested that HRS might be involved in mediating merlin growth suppression (30,31). Several lines of evidence support a link between merlin and HRS. First, HRS is a specific merlin interacting protein and does not bind to other Protein 4.1 or ERM molecules (14,30). Second, HRS overexpression in RT4 rat schwannoma cells has the same functional consequences as regulated overexpression of merlin (31). Lastly, HRS functions in the HGF signaling pathway, which has been implicated in the control of Schwann cell growth and motility (33,34), processes that are also modulated by merlin (15). In this report, we define the residues on HRS required for merlin binding and HRS growth suppression and show that these regions are distinct and non-overlapping. We further demonstrate that merlin growth suppression requires HRS, but that HRS growth suppression is not dependent upon merlin expression. Collectively, these results suggest that HRS acts downstream of merlin and might function to transduce the merlin growth suppressor signal.
We were able to define the domain of HRS important for mediating interactions with merlin. This ‘merlin-interaction’ domain maps to residues 470–497 within the predicted coiled-coil domain in human HRS. A number of other HRS interacting proteins also bind to this region, including STAM (human HRS residues 452–570) (38), HRS binding protein (mouse HRS residues 431–499) (39), synaptosome-associated protein of 25 kDa (rat HRS residues 478–562) (40), p21 activated kinase 1 (human HRS residues 451–570) (41) and sorting nexin-1 (rat HRS residues 225–541) (42). Only HBP requires the same HRS binding region as merlin, based on more detailed HRS fragment binding studies. Additional mapping studies will be required to determine whether all the above HRS-binding proteins use the same binding domain as merlin. It is also not known whether HRS binding to these various molecules occurs in a mutually exclusive fashion or whether multi-molecular complexes containing a number of these proteins can exist in cells.
Using a series of HRS truncation mutants, we were able to separate the domains in HRS responsible for merlin binding from those important for growth suppression. Previously, we demonstrated that regulated overexpression of HRS dramatically reduced RT4 rat schwannoma cell proliferation and that combined HRS and merlin overexpression resulted in an additional decrease in cell growth compared with the effects of either alone (31). The domain responsible for HRS growth suppression maps to residues 498–550, which is outside of the predicted coiled-coil domain in human HRS important for merlin binding. Using a merlin inducible cell line, we now show that HRS overexpression further reduced RT4 colony number in concert with merlin overexpression. These results suggest that merlin and HRS overexpression have additive effects on growth suppression, but do not address the requirement of HRS for merlin growth suppression or vice versa. Interestingly, HRS fragments that do not bind merlin are still able to provide this additional growth suppression, arguing that HRS probably functions either downstream of or independent of merlin.
To address the requirement for HRS in merlin growth suppression, we utilized HRS−/− MEFs and demonstrated that merlin was unable to suppress cell growth in the absence of HRS expression. These results suggest that HRS functions downstream of merlin and that it is important for merlin growth suppression. In support of this downstream position, HRS growth suppression was unaffected by merlin expression and HRS was equally effective as a growth regulator in the presence or absence of merlin. Future genetic complementation studies in mice and Drosophila will be required to confidently position HRS and merlin function relative to each other.
Recent studies have elucidated the upstream signals that are important in merlin growth suppression. Proper membrane localization of merlin appears to be critical for merlin function, in that merlin mutants unable to associate with the cell membrane are defective as growth regulators (43,44). One such upstream molecule is the transmembrane hyaluronate receptor, CD44. Merlin binds to CD44 under growth arrest conditions (29). Under these conditions, merlin exists in a relatively hypophosphorylated form. Conversely, in the growth-permissive state, merlin is hyperphosphorylated and exhibits decreased binding to CD44. In this model (45), merlin growth suppression occurs in a specific cellular context and its association with CD44 is partially mediated by phosphorylation events (46,47). Another ‘upstream’ merlin interacting protein is paxillin, which binds to merlin and regulates its density-dependent localization (48). Paxillin binds to merlin residues 50–70 contained within exon 2 and facilitates the localization of merlin to the cell membrane where it can interact with cell surface proteins, like CD44 and β1-integrin (49).
While these studies shed light on the upstream signaling events relevant to merlin growth suppression, comparatively less is known about the pathways and events downstream of merlin that transduce the merlin growth suppressor signal. A number of potential interacting proteins have been identified over the years since the cloning of the NF2 gene. Some of these molecules also bind other ERM proteins (e.g. βII-spectrin) (25), while others have unknown functions (e.g. schwannomin-interacting protein) (26). One interesting merlin interactor that operates within a critical Schwann cell signaling pathway is HRS. Our results positioning HRS downstream of merlin in mammalian cells suggest that HRS may transduce merlin growth suppression. Several possibilities can be envisioned to explain how HRS might propagate merlin's signal. First, merlin may serve to bring HRS to the cell membrane where it can interact with key molecules. We examined the possibility that merlin overexpression might serve to redistribute HRS within cells. In these experiments, we observed no change in HRS subcellular distribution upon merlin induction (C.X.S., unpublished observations). Similarly, we did not see any change in merlin subcellular localization upon HRS overexpression. These results argue that this mechanism is unlikely to explain how HRS functions as a merlin signal transducer. Further studies will be required to determine whether HRS changes the ability of merlin to interact with specific cell membrane proteins.
Alternatively, merlin could bind to HRS and result in activation of HRS by facilitating its interaction with specific HRS effector proteins. While HRS growth suppression is not dependent upon merlin expression, merlin binding to HRS may allow HRS to function more efficiently as a negative growth regulator. In this fashion, merlin-mediated HRS activation would lead to decreased cell growth, perhaps by modulating pathways previously ascribed to HRS, such as endocytosis and exocytosis (50–57), lysosomal trafficking (42), TGF-β:Smad signaling (37), or Jun kinase (JNK) activation (38). The role of HRS in JNK activation is particularly intriguing in light of experiments demonstrating increased JNK signaling in Nf 2−/− mouse embryonic fibroblasts (46). In addition, recent studies have indicated that HRS might participate in receptor tyrosine kinase (RTK) endocytosis (52). In this model, HRS activation might modulate the endocytosis of RTK molecules (36,39,53), like epidermal or hepatocyte growth factor receptor, to affect mitogenic signaling and cell proliferation. Additional studies will be necessary to dissect the mechanism(s) underlying HRS-dependent merlin growth regulation.
MATERIALS AND METHODS
Antibodies, plasmids, and cell lines
The merlin and HRS cDNAs used in these experiments were of human origin as described previously (30,31). HRS fragments were generated with PCR. The primers for PCR are: HRS (1–232), 5′-CTG GAT CCC GGG CGA GGC AGC GGC ACC-3′ and 5′-CTC ACT CAG TGG TGG AAG TGG C-3′; HRS (1–435), 5′-CTG GAT CCC GGG CGA GGC AGC GGC ACC-3′ and 5′-CTC AAC TCT TCA TGC GGT TCA C-3′; HRS (270–435), 5′-CTG GAT CCC CAG TCA GAG GCG GAG GAG-3′ and 5′-CTC AAC TCT TCA TGC GGT TCA C-3′; HRS (270–777), 5′-CTG GAT CCC CAG TCA GAG GCG GAG GAG-3′ and 5′-GTC AGT CGA ATG AAA TGA GCT G-3′; HRS (447–777), 5′-CTG GAT CCC AAC GGC ATG CAC CCG CAG-3′ and 5′-GTC AGT CGA ATG AAA TGA GCT G-3′; HRS (447–626), 5′-CTG GAT CCC AAC GGC ATG CAC CCG CAG-3′ and 5′-CTC ACG CAG TGC TGG GCA TGC T-3′; HRS (270–550), 5′-CTG GAT CCC CAG TCA GAG GCG GAG GAG-3′ and 5′-GTC ACT GCT GCT CCA GCC GCA TC-3′; HRS (270–626), 5′-CTG GAT CCC CAG TCA GAG GCG GAG GAG-3′ and 5′-CTC ACG CAG TGC TGG GCA TGC T-3′; HRS (270–497), 5′-CTG GAT CCC CAG TCA GAG GCG GAG GAG-3′ and 5′-GTC ACC GGC GAA GCT TCT CCC GGT G-3′; HRS (470–777), 5′-CTG GAT CCC CTG CAG GAC AAG CTG GCA C-3′ and 5′-GTC AGT CGA ATG AAA TGA GCT G-3′; HRS (498–777), 5′-CTG GAT CCC GCA GCC GAG GAG GCA GAG C-3′ and 5′-GTC AGT CGA ATG AAA TGA GCT G-3′; HRS (551–777), 5′-CTG GAT CCC AAG CAG ACG GTC CAG ATG C-3′ and 5′-GTC AGT CGA ATG AAA TGA GCT G-3′; HRS (551–777Δ625–693), 5′-CTG GAT CCC AAG CAG ACG GTC CAG ATG C-3′ and 5′-GTC AGT CGA ATG AAA TGA GCT G-3′; HRS (662–777), 5′-CTG GAT CCC TCC TAC CAG CCT ACT CCC ACA-3′ and 5′-GTC AGT CGA ATG AAA TGA GCT G-3′. PCR products containing a BamHI restriction site were initially cloned into pCR2.1 T/A cloning vector (Invitrogen Inc.) and sequenced in their entirety. HRS fragments were next subcloned into a pcDNA3.myc vector generated in our laboratory.
Mouse anti-myc monoclonal antibody (9E10), mouse anti-myc monoclonal antibody agarose conjugates (9E10 AC) and rabbit anti-merlin polyclonal antibody (C18 and A19) were purchased from Santa Cruz Technology. Rabbit anti-HRS polyclonal antibody (Ab10802) was generated as described previously (30). Tubulin (clone DM1A) was purchased from Sigma (St Louis, MO, USA).
The RT4 rat schwannoma cell line was maintained in complete DMEM with 10% FBS. Mouse embryonic fibroblasts (Hrs−/−; Hrs+/+ and Nf2+/+) and NIH3T3 cells were maintained in complete DMEM with 10% FBS. NF2-deficient (Nf2−/−) MEFs were provided by Dr Marco Giovannini (Fondation Jean Dausset, CEPH, France) and maintained in complete DMEM plus 10% FBS and 0.1 mm β-mercaptoethanol. The merlin inducible rtTA.merlin RT4 cells were maintained in complete DMEM with 10% FBS, selected by 500 µg/ml G418, and 1 µg/ml puromycin. Merlin expression was induced by addition of 1 µg/ml doxycycline.
In vitro and in vivo protein expression
To determine the expression of constructed HRS fragments in vitro, a nonradioactive coupled transcription/translation reaction was performed using the TNT® coupled Reticulocyte Lysate System (Promega) according to the recommended protocol. Nonradioactive products were separated by 12% SDS–PAGE and analysed by western blot using the 9E10 myc monoclonal antibody. To analyse the expression of truncated HRS fragments in vivo, RT4 cells were seeded in six-well plates and transfected with various HRS fragments using Lipofectamine according to the manufacturer's recommendations. Cell lysates were harvested 48 h later, separated by SDS–PAGE and analysed by western blot using the 9E10 myc antibody.
Growth suppression assay
Clonogenic assays were performed by transfecting cells with equimolar amounts of vector (pcDNA3.myc), pcDNA3.myc.HRS and various HRS fragments in pcDNA3.myc vector. Cells were selected in 500 µg/ml G418 for 14 days and the number of surviving colonies greater than 1 mm were counted in quadruplicate dishes after staining in 0.5% Crystal violet. In some experiments, transfected cells were selected in 500 µg/ml G418, 1 µg/ml puromycin and 200 µg/ml hygromycin. Merlin induction in rtTA.merlin RT4 cells lines was accomplished using 1 µg/ml doxycycline, as previously reported (15). Each experiment was repeated at least three times with identical results.
To determine merlin or HRS function in genetically-defined MEFs, we transiently transfected equimolar amounts of vector (pcDNA3), pcDNA3.HRS, or pcDNA3.NF2 plus 10-fold less pBABE.PURO plasmid. Cells were selected by 1 µg/ml puromycin for 14 days, and surviving colonies were counted as above. Each experiment was repeated for four times with identical results.
GST protein affinity chromatography
Glutathione-S-transferase (GST)–merlin fusion proteins were generated as previously described (13,58). Briefly, GST.C-term merlin (residues 299–595) was transformed into DE3 (BL21) competent cells for fusion protein production. Bacteria were induced in 0.4 mm IPTG at room temperature for 20–21 hrs and GST fusion protein was collected on glutathione–agarose beads (Sigma) for the interaction experiments.
GST fusion protein was prepared as above for HRS–merlin interaction experiments with in vitro transcribed and translated HRS fragment proteins. In vitro transcribed and translated HRS fragment proteins were synthesized in the presence of [35S]-methionine using the TNT® coupled Reticulocyte Lysate Systems (Promega) and detected by autoradiography. In these experiments, radiolabeled proteins were incubated with equimolar amounts of GST fusion protein immobilized on glutathione–agarose beads for 4 h at 4°C. Supernatants were saved and the beads were washed three times with TEN buffer (10 mm Tris–Cl pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100) and eluted in 2× Laemmli buffer. Supernatants and eluted bound fractions were separated by 12% SDS–PAGE and analysed by autoradiography. Each interaction experiment was repeated at least three times with identical results.
In vivo HRS interaction experiments
RT4 schwannoma cells were transiently transfected with merlin constructs and various HRS fragments using LipofectAMINE (Gibco BRL) and lysates prepared 48 h later using NP-40 lysis buffer (50 mM Tris, pH 7.4; 150 mM NaCl; 3 mM MgCl2; 0.5% NP 40; 1 mM DTT; 1 mM PMSF; 10 µg/ml aprotinin; 10µg/ml leupeptin). Protein lysates were incubated with the myc antibody conjugated to agarose beads (Santa Cruz Biotechnology) for 2 h at 4°C followed by extensive washing with 1× PBS. Eluted proteins were separated by SDS–PAGE and blotted with the C18 anti-merlin polyclonal antibody to detect merlin proteins. Membranes were probed with the 9E10 myc antibody after stripping with ECL stripping buffer (6.25 ml 1 m Tris, pH 6.8; 770 µl β-mercaptoethanol; 10 ml 20% SDS; ddH2O to 100 ml). Each experiment was repeated at least three times with identical results.
ACKNOWLEDGEMENTS
We thank the members of our laboratory for their expert assistance during the execution of this project. These studies were supported by a grant from the National Institutes of Health (NS35848 to D.H.G.).
To whom correspondence should be addressed at: Department of Neurology, Washington University School of Medicine, Box 8111, 660 South Euclid Avenue, St Louis, MO 63110, USA. Tel: +1 3143627379; Fax: +1 3143622388; Email: gutmannd@neuro.wustl.edu
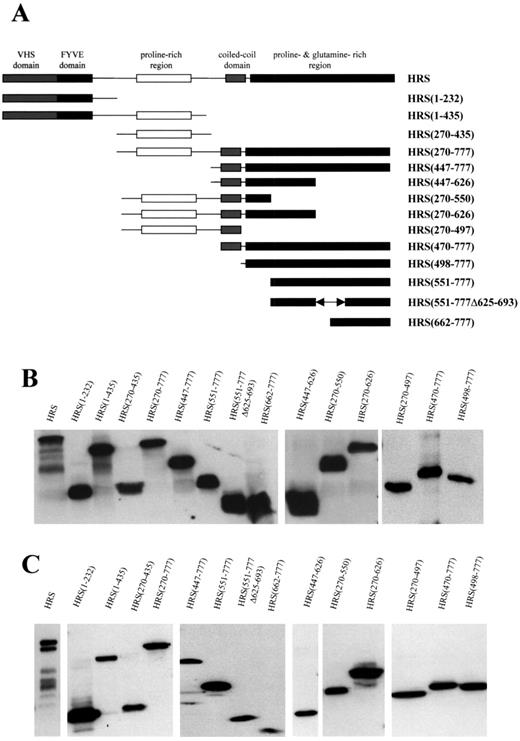
Figure 1. Generation of truncated HRS fragments. (A) Truncated human HRS fragments were generated by PCR to serially delete the predicted protein–protein binding domains. All fragments were cloned into the pcDNA3.myc-tagged vector. (B) The expression of these fragments was detected by in vitro transcription and translation, separation by SDS–PAGE, and western blotting with mouse anti-c-myc monoclonal antibody. (C) The in vivo expression was demonstrated by transiently transfecting fragments into RT4 rat schwannoma cells, separation by SDS–PAGE, and western blotting with c-myc antibodies.
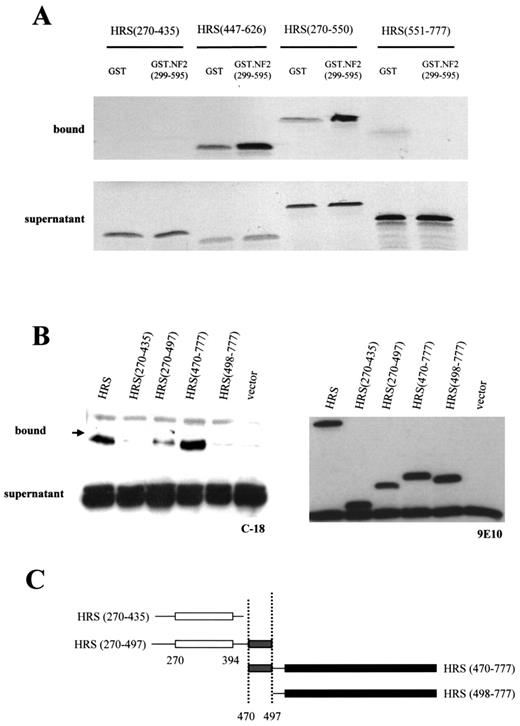
Figure 2. Merlin binding maps to the HRS coiled-coil domain. (A) GST-affinity chromatography experiments demonstrated that HRS (447–626) and HRS (270–550) bind to merlin (299–595), however HRS (270–435) and HRS (551–777) do not bind. (B) To further narrow the HRS domain that mediates binding to merlin, co-immunoprecipitation experiments were performed. The carboxyl terminus of merlin (residues 299–595) and various HRS fragments were co-transfected into RT4 cells. Cell lysates were incubated with c-myc agarose conjugate, separated by SDS–PAGE and western blotted with the C18 rabbit anti-merlin polyclonal antibody. Full length HRS, HRS (270–497) and HRS (470–777) bind to C-term merlin, but HRS (270–435) and HRS (498–777) do not, suggesting that the binding domain maps between L470 and R497, within the HRS predicted coiled-coil domain (C).
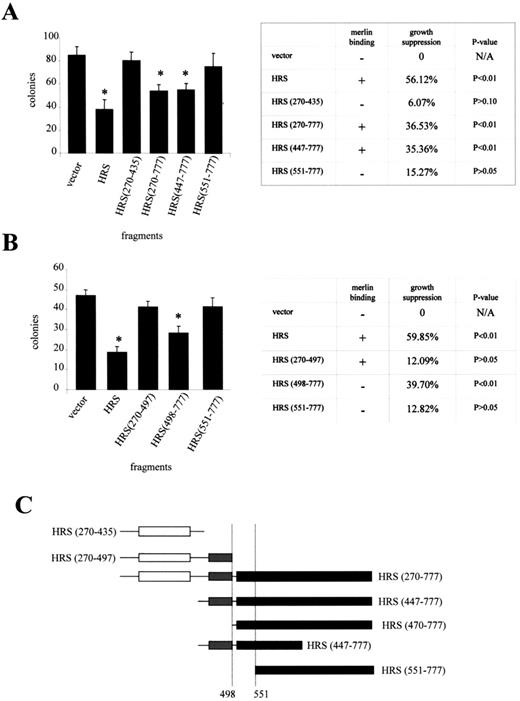
Figure 3. The carboxyl terminus of HRS is required for growth suppression. (A) In order to determine the region of HRS responsible for growth suppression, we performed clonogenic assays with various HRS fragments. HRS, HRS (270–777) and HRS (447–777) were able to suppress RT4 cell growth, whereas HRS (270–435) and HRS (551–777) did not. (B) Further experiments showed that HRS (498–777) also suppressed cell growth, whereas HRS (270–497) and HRS (551–777) had no effect. The growth suppressor domain of HRS maps between A498 and Q551, which does not overlap with the merlin-binding domain (C). Asterisks denote statistically significant growth suppression.
![Figure 4. HRS (270–497) cannot reverse merlin growth suppression. (A) The interaction of merlin and various HRS fragments were performed by co-immunoprecipitation as described in the Materials and Methods section. Merlin interacts with HRS (270–497), HRS (447–777), as well as full-length HRS, but not HRS (1–232). (B) In order to determine whether expressing an HRS mutant capable of binding merlin, but not reducing cell growth, could inhibit merlin growth suppression, we transfected various HRS fragments into the tetracycline-regulatable RT4 rtTA NF2.17 cell line, in which merlin expression is induced upon the addition of doxycycline. In these experiments, HRS (270–497) was not able to reverse merlin growth suppression. HRS fragments with growth suppressor activity [HRS, HRS (498–777), and HRS (447–777)] cooperated with merlin to further reduce colony number. (C) These results are tabulated from at least four independent experiments. Asterisks denote statistically significant growth suppression.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/11/25/10.1093/hmg/11.25.3167/2/m_ddf31304.jpeg?Expires=1716412032&Signature=rHjdyRl3Lopd1O0APKIpGNcC-BvYdqBdcNma4ZhWScZOcRWeL1ZFaTqp1dhiPX~rVQisHDKzyYupo3gtg3lu4p5Awm-0QofmmJ1eVCFZJCRhah~Q-nHQ6C~uiRGlpDjqtELWThRq4QixMAqVxKtmqChwD-lIefdZXmJ48xUXUucIhOP9tfrKf5zmPeySfpYsFQHh6CK63NP7iFU3UKavhvQCEdTQ1UmcYbGWoi2doomEYFLuUBIrLz6AQKczYK8n-QH1TmiYAp93LErXDjyBzeDrnHAAjCOI2foiQFNmQKo9uC8On5pRdyajsFQkNEHu5nXf0gT5eLOw3~SvSMfP~w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 4. HRS (270–497) cannot reverse merlin growth suppression. (A) The interaction of merlin and various HRS fragments were performed by co-immunoprecipitation as described in the Materials and Methods section. Merlin interacts with HRS (270–497), HRS (447–777), as well as full-length HRS, but not HRS (1–232). (B) In order to determine whether expressing an HRS mutant capable of binding merlin, but not reducing cell growth, could inhibit merlin growth suppression, we transfected various HRS fragments into the tetracycline-regulatable RT4 rtTA NF2.17 cell line, in which merlin expression is induced upon the addition of doxycycline. In these experiments, HRS (270–497) was not able to reverse merlin growth suppression. HRS fragments with growth suppressor activity [HRS, HRS (498–777), and HRS (447–777)] cooperated with merlin to further reduce colony number. (C) These results are tabulated from at least four independent experiments. Asterisks denote statistically significant growth suppression.
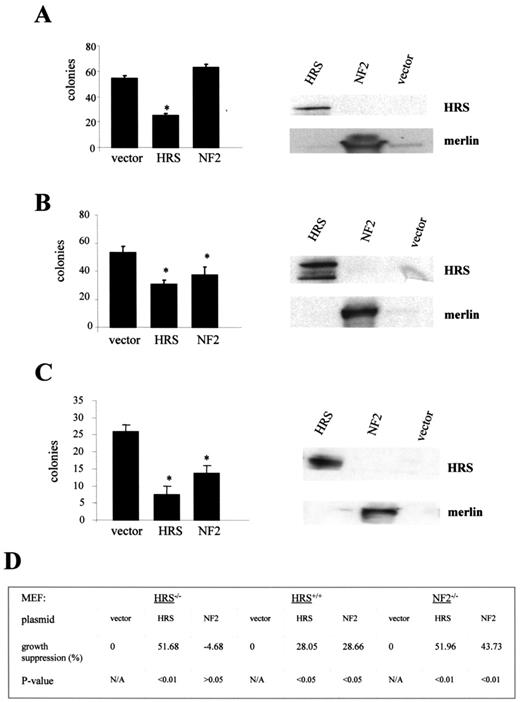
Figure 5. HRS is required for merlin growth suppression. (A) HRS or merlin was transiently transfected into HRS-deficient fibroblasts (Hrs−/− MEFs). Re-introduction of HRS into HRS deficient cells reduces cell growth, whereas merlin has no effect. The right panel shows the overexpression of HRS or merlin in HRS deficient MEFs. (B) Both HRS and merlin suppress Hrs+/+ MEF growth. The right panel shows the overexpression of HRS or merlin in Hrs+/+ MEFs. (C) Both HRS and merlin can suppress Nf 2−/− MEF growth. The right panel shows the overexpression of HRS or merlin in Nf 2−/− MEFs. The results from these experiments are tabulated in (D). Asterisks denote statistically significant growth suppression.
References
Evans, D.G., Huson, S.M., Donnai, D., Neary, W., Blair, V., Newton, V., Strachan, T. and Harris, R. (
Rouleau, G.A., Merel, P., Lutchman, M., Sanson, M., Zucman, J., Marineau, C., Xuan, K.H., Demczuk, S., Desmaze, C., Plougastel, B. et al. (
Trofatter, J.A., MacCollin, M.M., Rutter, J.L., Murrell, J.R., Duyao, M.P., Parry, D.M., Eldridge, R., Kley, N., Menon, A., Pulaski, K. et al. (
Tsukita, S., Yonemura, S. and Tsukita, S. (
Gautreau, A., Poullet, P., Louvard, D., Arpin, M. (
Xu, H.M. and Gutmann, D.H. (
Scherer, S.S. and Gutmann, D.H. (
Scherer, S.S., Xu, T., Crino, P., Arroyo, E.J. and Gutmann, D.H. (
Sainz, J., Huynh, D.P., Figueroa, K., Ragge, N.K., Baser, M.E. and Pulst, S.M. (
Bourn, D., Carter, S.A., Mason, S., Gareth, D., Evans, R. and Strachan, T. (
Huynh, D.P., Mautner, V., Baser, M.E., Stavrou, D. and Pulst, S.M. (
Sherman, L., Xu, H.-M., Geist, R.T., Saporito-Irwin, S., Howells, N., Ponta, H., Herrlich, P. and Gutmann, D.H. (
Gutmann, D.H., Hirbe, A.C., Huang, Z.Y. and Haipek, C.A. (
Gutmann, D.H., Hirbe, A.C. and Haipek, C.A. (
Ruttledge, M.H., Sarrazin, J., Rangaratnam, S., Phelan, C.M., Twist, E., Merel, P., Delattre, O., Thomas, G., Nordenskjöld, M. et al. (
Gutmann, D.H., Giordano, M.J., Fishback, A.S. and Guha, A. (
Stemmer-Rachamimov, A.O., Gonzalez-Agosti, C., Xu, L., Burwick, J.A., Beauchamp, R., Pinney, D., Louis, D.N. and Ramesh, V. (
Deprez, R.H.L., Bianchi, A.B., Groen, N.A., Seizinger, B.R., Hagemeijer, A., van Drunen, E., Bootsma, D., Koper, J.W., Avezaat, C.J.J., Kley, N. and Zwarthoff, E.C. (
McClatchey, A.I., Saotome, I., Mercer, K., Crowley, D., Gusella, J.F., Bronson, R.T. and Jacks, T. (
Bashour, A.M., Meng, J.J., Ip, W., MacCollin, M. and Ratner, N. (
Pelton, P.D., Sherman, L.S., Rizvi, T.A., Marchionni, M.A., Wood, P., Friedman, R.A. and Ratner, N. (
Gutmann, D.H., Sherman, L., Seftor, L., Haipek, C., Hoang, Lu, K. and Hendrix, M. (
Murthy, A., Gonzalez-Agosti, C., Cordero, E., Pinney, D., Candia, C., Solomon, F., Gusella, J. and Ramesh, V. (
Scoles, D.R., Huynh, D.P., Morcos, P.A., Coulsell, E.R., Robinson, N.G., Tamanoi, F. and Pulst, S.M. (
Goutebroze, L., Brault, E., Muchardt, C., Camonis, J. and Thomas, G. (
Jannatipour, M., Dion, P., Khan, S., Jindal, H., Fan, X., Laganiere, J., Chishti, A.H. and Rouleau, G.A. (
Sainio, M., Zhao, F., Heiska, L., Turunen, O., den Bakker, M., Zwarthoff, E., Lutchman, M., Rouleau, G.A., Jaaskelainen, J., Vaheri, A. and Carpen, O. (
Morrison, H., Sherman, L.S., Legg, J., Banine, F., Isacke, C., Haipek, C.A., Gutmann, D.H., Ponta, H. and Herrlich, P. (
Scoles, D.R., Huynh, D.P., Chen, M.S., Burke, S.P., Gutmann, D.H. and Pulst, S.M. (
Gutmann, D.H., Haipek, C.A., Burke, S.P., Sun, C.X., Scoles, D.R. and Pulst, S.M. (
Komada, M. and Kitamura, N. (
Krasnoselsky, A., Massay, M.J., DeFrances, M.C., Michalopoulos, G., Zarnegar, R. and Ratner, N. (
Maulik, G., Shrikhande, A., Kijima, T., Ma, P.C., Morrison, P.T. and Salgia, R. (
Komada, M. and Soriano, P. (
Mao, Y., Nickitenko, A., Duan, X., Lloyd, T.E., Wu, M.N., Bellen, H. and Quiocho, F.A. (
Miura, S., Takeshita, T., Asao, H., Kimura, Y., Murata, K., Sasaki, Y., Hanai, J.I., Beppu, H., Tsukazaki, T., Wrana, J.L. et al. (
Asao, H., Sasaki, Y., Arita, T., Tanaka, N., Endo, K., Kasai, H., Takeshita, T., Endo, Y., Fujita, T. and Sugamura, K. (
Takata, H., Kato, M., Denda, K. and Kitamura, N. (
Kwong, J., Roundabush, F.L., Hutton Moore, P., Montague, M., Oldham, W., Li, Y., Chin, L.S. and Li, L. (
Sasaki, Y. and Sugamura, K. (
Chin, L.S., Raynor, M.C., Wei, X., Chen, H.Q. and Li, L. (
Brault, E., Gautreau, A., Lamarine, M., Callebaut, I., Thomas, G. and Goutebroze, L. (
Schulze, K.M., Hanemann, C.O., Muller, H.W. and Hanenberg, H. (
Sherman, L.S. and Gutmann, D.H. (
Shaw, R.J., Paez, J.G., Curto, M., Yaktine, A., Pruitt, W.M., Saotome, I., O'Bryan, J.P., Gupta, V., Ratner, N., Der, C.J. et al. (
Xiao, G.H., Beeser, A., Chernoff, J. and Testa, J.R. (
Fernandez-Valle, C., Tang, Y., Ricard, J., Rodenas-Ruano, A., Taylor, A., Hackler, E., Biggerstaff, J. and Iacovelli, J. (
Obremski, V.J., Hall, A.M. and Fernandez-Valle, C. (
Komada, M. and Kitamura, N. (
Raiborg, C., Bache, K.G., Gillooly, D.J., Madshus, I.H., Stang, E. and Stenmark, H. (
Raiborg, C., Bache, K.G., Mehlum, A. and Stenmark, H. (
Lloyd, T.E., Atkinson, R., Wu, M.N., Zhou, Y., Pennetta, G. and Bellen, H.J. (
Li, Y., Chin, L.S., Levey, A.I. and Li, L. (
Raiborg, C., Bache, K.G., Mehlum, A., Stang, E. and Stenmark, H. (
Murai, S. and Kitamura, N. (
Urbe, S., Mills, I.G., Stenmark, H., Kitamura, N. and Clague, M.J. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}