




Abstract
Congenital nephrotic syndrome (CNS) is clinically and genetically heterogeneous, with mutations in WT1, NPHS1 and NPHS2 accounting for part of cases. We recently delineated a new autosomal recessive entity comprising CNS with diffuse mesangial sclerosis and distinct ocular anomalies with microcoria as the leading clinical feature (Pierson syndrome). On the basis of homozygosity mapping to markers on chromosome 3p14–p22, we identified homozygous or compound heterozygous mutations of LAMB2 in patients from five unrelated families. Most disease-associated alleles were truncating mutations. Using immunohistochemistry and western blotting we could demonstrate that the respective LAMB2 mutations lead to loss of laminin β2 expression in kidney and other tissues studied. Laminin β2 is known to be abundantly expressed in the glomerular basement membrane (GBM) where it is thought to play a key role in anchoring as well as differentiation of podocyte foot processes. Lamb2 knockout mice were reported to exhibit congenital nephrosis in association with anomalies of retina and neuromuscular junctions. By studying ocular laminin β2 expression in unaffected controls, we detected the strongest expression in the intraocular muscles corresponding well to the characteristic hypoplasia of ciliary and pupillary muscles observed in patients. Moreover, we present first clinical evidence of severe impairment of vision and neurodevelopment due to LAMB2 defects. Our current data suggest that human laminin β2 deficiency is consistently and specifically associated with this particular oculorenal syndrome. In addition, components of the molecular interface between GBM and podocyte foot processes come in the focus as potential candidates for isolated and syndromic CNS.
INTRODUCTION
Congenital nephrotic syndrome (CNS) constitutes a heterogeneous group of conditions having in common the disruption of normal glomerular permselectivity (1). The glomerular filtration barrier comprises three major components: the fenestrated capillary endothelium, the glomerular basement membrane (GBM) and the podocytes which by their interdigitating foot processes form filtration slits bridged by a membrane-anchored, extracellular meshwork of proteins, the slit diaphragm (1,2). Hitherto known molecular causes of CNS are mutations of the genes encoding nephrin in Finnish type CNS (MIM 256300) and podocin in autosomal recessive steroid-resistant NS (MIM 600995). Both proteins play essential roles in the formation of the slit diaphragm. Their close functional interrelationship is further evidenced by the recent finding of digenic inheritance of CNS (3). Additionally, autosomal dominant WT1 mutations can cause CNS and diffuse mesangial sclerosis in patients with Denys–Drash syndrome (MIM 256370). The pathogenesis of NS caused by specific WT1 mutations has been debated but is not completely understood so far (4). A considerable proportion of patients with CNS, however, do not belong to either of these entities (1), suggesting that additional, hitherto unidentified genes play a role in its pathogenesis. Aside from isolated renal disease, CNS can occur in association with various non-renal symptoms. A definite classification of CNS has not yet been established.
In two unrelated consanguineous families with a total of 11 affected offspring, we recently delineated a new autosomal recessive entity comprising severe CNS with diffuse mesangial sclerosis and distinct eye abnormalities clinically characterized by microcoria (fixed narrowing of the pupils), but obviously representing a complex ocular maldevelopment including hypoplasia of iris and ciliary body, lenticonus posterior and corneal and retinal anomalies (Fig. 1) (5). The condition was first described by Pierson et al. (6) in 1963, and a small number of cases with similar manifestations has been reported, so far (7–10). However, the disorder that we proposed to term Pierson syndrome had not been recognized as a separate entity before. In all previous reports, the disorder was uniformly associated with early lethality. Herein we describe the molecular basis of this disease.
RESULTS
Linkage analysis and identification of the candidate gene
Homozygosity mapping in the two families reported earlier (5) identified a candidate region on chromosome 3p flanked by markers D3S1768 and D3S1766 (Fig. 2). Multipoint lod score analysis generated a lod of 3.5 for locus D3S2409 (Table 1). The candidate region derived from gross mapping still encompassed a segment of ∼17 cM. Linkage refinement, however, was not attempted because LAMB2 (MIM 150325), which is located within the candidate region close to D3S2409, represented a perfect candidate on the basis of the findings that lamb2−/− mice display CNS (11).
Mutational analysis
Patients and obligate heterozygotes from five unrelated families with at least one child affected by Pierson syndrome were subjected to mutational screening by bidirectional direct sequencing of the entire coding region, including the exon–intron boundaries of LAMB2 (GenBank accession no. AAB34682.2). The families included two in whom linkage analysis had been performed as well as one unreported and two previously reported families (8,12) with similar renal and ocular manifestations in affected children (Table 2). In patients from four families we found homozygous or compound heterozygous LAMB2 mutations predicting a truncation of the protein. Two disease-associated alleles were nonsense mutations at the codons 1507 and 1562 of the protein, which comprises 1798 amino acid residues. Two alleles were 1 bp deletions leading to translational stop codons at positions 1150 and 1520, and another was a 1 bp insertion creating a premature termination codon at position 1760 (Fig. 3A–C and E; Table 2). In the remaining family (family 4), both affected children had a homozygous missense mutation (Fig. 3D) predicting the substitution of a highly conserved arginine by tryptophan (Fig. 4). By sequencing of the respective exons in all available members of each family, we found full segregation of the mutations. None of the mutations was detected among 200 control chromosomes. In 12 unrelated individuals with CNS and variable associated manifestations, who had earlier been negative for mutational screening in the nephrin, podocin and WT1 genes, no LAMB2 mutations were detected, indicating that LAMB2 defects are specifically associated with Pierson syndrome.
Immunohistology and western blotting
To study the consequences of the observed LAMB2 mutations on protein expression, we examined kidney, skeletal muscle and ocular tissues from different patients by immunofluorescence and western blotting using laminin β2 antibodies directed against the N-terminal region. In individuals with the homozygous nonsense mutation (family 2), laminin β2 immunoreactivity of the GBM was virtually absent (Fig. 5A–D), and lack of laminin β2 could also be demonstrated by western blotting of muscle proteins (Fig. 6). Similar immunofluorescence results were obtained in kidney tissues of affected individuals from family 3 and family 5 (data not shown). In frozen kidney tissue of one patient with the homozygous missense mutation R246W (family 4), significant reduction but not a complete lack of laminin β2 could be demonstrated by western blotting (Fig. 6). By densitometry, a 7-fold reduction of laminin β2 relative to laminin β1 expression was calculated. Accordingly, immunostaining for laminin β2 was absent in eye sections of one aborted fetus homozygous for the mutation R1562X (family 2; data not shown). Eye sections of normal controls revealed that specific laminin β2 immunoreactivity is present not only in the retina and lens capsule as already known (data not shown), but also most prominently in the ciliary and iris muscles (Fig. 5E–H).
DISCUSSION
We demonstrate here that LAMB2 mutations can be consistently found in patients with Pierson syndrome, a newly delineated entity characterized by CNS and distinct ocular anomalies. All but one mutant LAMB2 allele predict a C-terminal truncation of the protein owing to premature translational stop codons. Even with the least extensive truncation of the protein by only 39 amino acids (family 5), no significant laminin β2 immunoreactivity of the GBM could be demonstrated. We assume that these truncating mutations disturb the assembly of the mutated protein into laminin heterotrimers, as it was shown that a peptide sequence of approximately 50 amino acid residues forming the C-terminal alpha-helical domain of the laminin chains represents the critical site for the initiation of laminin chain assembly (13). In the kidney tissue of a patient with the homozygous missense mutation R246W, significant reduction of laminin β2 could be demonstrated by western blotting (Fig. 6). The mutation is located within a highly conserved sequence motif of domain VI that is crucial for laminin polymerization to form BMs (14). One might suggest that laminin β2 deficiency as evidenced by western blotting results from increased degradation of the protein either due to decreased stability within the BM network or related to primary misfolding of the protein chain. Preservation of some residual function, on the other hand, might be an explanation for a somewhat milder phenotype with prolonged survival in this family (Table 2). Taken together, these findings clearly indicate that Pierson syndrome is caused by the absence or severe quantitative reduction in laminin β2 expression rather than a dysfunctional protein.
Laminin β2 is one component of the laminins, a family of heterotrimeric extracellular glycoproteins consisting of variable assemblies of α-, β- and γ-chains and representing major BM constituents that play important roles in cell adhesion, proliferation, differentiation and migration (15,16). This report describes only the third human phenotype resulting from mutations in members of the laminin family, after Herlitz-type epidermolysis bullosa including two variants (MIM 245660, 226650 and 226700) that can be caused by mutations in the genes encoding one of the laminin-5 components (LAMA3, LAMB3 and LAMC2) and congenital muscular dystrophy due to merosin deficiency (MIM 607855, LAMA2). Laminin β2 (formerly s-laminin) was initially defined by an antiserum to an extract of lens capsule, which stained synaptic BM of the neuromuscular junction (17), but it was subsequently found to be also abundant in some non-muscle BMs, like perineurial, glomerular and arterial BMs (18). In laminin β2-deficient mice, a lethal phenotype of massive early-onset proteinuria was observed, suggesting a critical role in proper maturation or function of the renal filtration apparatus (11). Laminin-11, the major laminin of the mature GBM and composed of α5-, β2- and γ1-chains (19), was shown to promote podocyte process development in cell culture (20). In addition to the renal alterations, the knockout mouse model exhibited abnormalities of the neuromuscular junctions, proposing laminin β2 to be a muscle-derived regulator of nerve-terminal differentiation (21). Specific expression patterns of laminin β2 were described in the human ciliary body (22) and corneal BL (23). Regarding the role of laminin β2 in ocular development, however, most research focused on the significance for retinal differentiation. In laminin β2-deficient mice, shortened inner and outer segments of photoreceptors and disrupted synaptic connections between photoreceptors and second order cells were demonstrated (24). Moreover, laminin β2 has also been found at distinct sites of the developing central nervous system in rats (25). In contrast to the renal phenotype of laminin β2 deficiency, the functional consequences of developmental deficiencies in neural, retinal and other tissues remained unclear. Knockout mice were reported to display no apparent neurologic phenotype at birth, but it was suggested that the observed presynaptic defects might contribute to the weakness and eventual death of the mutants occurring between days 15 and 30 (21). In addition, abnormal electroretinograms consistent with synaptic disruption in the outer retina were documented (24).
The human phenotype corresponds well to the known expression patterns of laminin β2 and the manifestations in laminin β2 deficient knockout mice. The findings presented here thus provide the missing link between the data derived from experimental studies and the significance of the protein in human development. However, aside from the functions inferred from the animal model, our investigations add some new aspects on the potential physiological role of laminin β2 in humans. Specifically, they point out the importance of laminin β2 for the proper development of structures of the anterior eye segment, particularly the intraocular muscles and lens, demonstrating that a distinct pattern of ocular maldevelopment is associated with human laminin β2 deficiency. The renal histomorphological phenotype (diffuse mesangial sclerosis) in human laminin β2 deficiency differs from the histological findings reported in knockout mice where only minimal changes were observed. This discrepancy, however, may reflect interspecies differences or the natural evolution of renal damage, as we reported earlier that the changes are milder in fetuses than in newborns (5). Additionally, although most patients known to the authors died in the newborn period without being recognized to have an obvious neurological phenotype, the clinical findings in the two previously reported sibs (8) and in particular one patient who is first reported here and who survived until the age of 1.8 years under chronic dialysis (family 3, Table 2; see Materials and Methods: Study population) now provide preliminary evidence that laminin β2 deficiency is associated with significant neurological and developmental deficits. These may reflect the functional importance of the protein at the neuromuscular junction and in the central nervous system. These issues will have to be addressed by future research and should be considered before the decision is made to rescue affected children from renal insufficiency by kidney replacement therapy.
Previously identified molecular causes of CNS emphasized the pathogenetic significance of impaired podocyte–podocyte interactions at the slit diaphragm which represents a highly specific renal structure. In contrast, we report on CNS asso-ciated with defects of a protein which has more pleiotropic effects. Our current understanding suggests that human laminin β2 deficiency is consistently and specifically associated with the particular oculorenal syndrome described here. Our findings, however, place components of the molecular interface between GBM and podocyte foot processes in the focus of future research for candidates for isolated and, in particular, syndromic NS. Remarkably, the significance of laminins in the extracellular matrix as triggers for process formation is not the only mechanism shared by podocytes and neuronal cells (26). Therefore, defects in other genes involved in podocyte morphogenesis should particularly be searched for in disorders presenting with CNS and neurological features.
MATERIALS AND METHODS
Study population
Patients were identified as having Pierson syndrome in the presence of CNS and microcoria, the clinical hallmarks of this disorder. Most affected individuals died in the newborn period from renal insufficiency. Survival until the age of 8 months in both affected siblings from a family reported before (8) is considered to reflect a somewhat milder expression. Only one patient survived infancy owing to chronic dialysis. By the time of her death at 1.8 years she exhibited severe neurological deficits previously not described in Pierson syndrome: marked muscular hypotonia, psychomotor retardation and apparent blindness (Table 2; detailed clinical report in preparation). In addition to the patients with definite Pierson syndrome, 12 unrelated individuals with CNS and variable associated manifestations, in whom no mutations had been detected in the nephrin, podocin and WT1 genes, were included for LAMB2 mutational screening.
The study was approved by the Institutional Review Boards. After obtaining informed consent, blood samples were drawn from all available members of the two families included in the linkage studies. DNAs from deceased patients and aborted fetuses were obtained from stored biopsy specimens, Guthrie cards and abortion material. Stored frozen tissue samples from the affected siblings of the family described in a previous publication (8) served for molecular genetic and western blot analyses. Tissues used in this study were derived from stored autopsy or biopsy specimens and were obtained after informed parental consent had been given.
Linkage analysis
A genome scan using microsatellite markers was initiated in one of the families (family 1), a large inbred sibship of Lebanese origin (Fig. 1), and terminated upon finding evidence of linkage after typing of 124 markers covering approximately one-third of the genome (R. Fenski and A. Reis, unpublished data). The locus on chromosome 3p was confirmed and refined in the second consanguineous family (family 2) using a panel of microsatellite markers from chromosome 3p from the Weber panel (Research Genetics). Two-point lod score calculations were performed by the LINKAGE program package (27) with the help of the computer programs LINKRUN and MKS, using an autosomal recessive fully penetrant model.
Mutational analysis
We developed primers flanking all 32 exons of LAMB2 from the genomic sequence. Oligonucleotide sequences and PCR conditions are available on request. We carried out bidirectional direct sequencing using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems) on an ABI 3730 capillary sequencer (Applied Biosystems) and evaluated sequences using the DNAStar software package.
Antibodies
We obtained antibodies against collagen IV and smooth muscle actin commercially (DakoCytomation, clones 1A4 and CIV 22, respectively). The polyclonal antibody against human laminin β1 (H-300) was purchased from Santa Cruz Biotechnology. The polyclonal anti-laminin β2 antibody 409 was a gift from Lydia Sorokin (Experimental Pathology, Lund University, Lund, Sweden). It is directed against recombinant laminin β2 domain VI corresponding to amino acids 30–250 (S. Agrawal, M. Durbeej, M. Sixt, H. Korner, I. Nelissen, G. Opdenakker and L.M. Sorokin, submitted for publication).
Western blot analysis
We carried out western blot analysis on deep frozen muscle and kidney tissues according to standard procedures. The tissues had been stored at −70°C until examination. Equal amounts of protein were separated by SDS–PAGE, transferred to nitrocellulose membranes and probed with antibodies, as previously reported (28). Placental laminins containing both laminin β1 and laminin β2 were used as a positive control. They were derived from placenta extract by immunoaffinity chromatography, as described previously (29). The antibodies 409 and anti-laminin β1 were diluted 1 : 500 and 1 : 1000, respectively. Immunoreactivities were detected by ECL reaction in a Lumi-Imager F1 (Roche, Mannheim, Germany) according to the manufacturer's instructions. Densitometry was performed using the software LumiAnalyst, version 3.1 (Roche, Mannheim, Germany).
Immunofluorescence and immunohistochemistry analysis
Tissue samples were fixed with 10% formalin and embedded in paraffin. Sections of 4 µm were used for immunolocalization studies for detecting endogenous laminin β2 and collagen IV. All primary antibodies were applied at 4°C in a moist chamber overnight. Secondary antibodies were labeled either with Cy-2 or Cy-5 (Dianova, Hamburg, Germany). Nuclear counterstain was performed with DAPI; 70% glycerine was used as mounting medium. For double-labeling immunofluorescence using anti-collagen IV and anti-laminin β2 antibodies, tissue sections were pretreated with hyaluronidase (2 mg/ml in PBS, pH 5 for 60 min at 37°C) and pronase (2 mg/ml in TB, PBS 7.3; Sigma, Taufkirchen, Germany, for 60 min at 37°C) and incubated with a mixture of both antibodies with anti-laminin β2 antibody used at a 10× higher concentration. The anti-laminin β2 antibodies were detected using Cy-2-labeled goat anti-rabbit antibody (Dianova). For the detection of the anti-collagen IV antibodies, sections were first incubated with biotin-labeled goat-anti-mouse antibodies (Dianova) and then with peroxidase-labeled streptavidin. Subsequently, the tyramide amplification system (PerkinElmer, Boston, MA, USA) was used. Finally, the signals were detected using Cy-5-labeled streptavidin (Dianova). Nuclear staining was again performed using DAPI.
To show the distribution of smooth muscle in sagittal eye sections from unaffected individuals, we subjected sections to immunohistochemistry with a monoclonal anti-human smooth muscle actin antibody (DakoCytomation) as described previously (30) using alkaline phosphatase as detection enzyme and 3-hydroxy-2-naphtylacid 2,4-dimethylanilide as substrate. Nuclei were counterstained with hematoxylin.
The images were visualized with an Olympus AX70 fluorescence microscope and digitally captured using AnalySIS Docu software (SoftImaging, Stuttgart, Germany).
ACKNOWLEDGEMENTS
We thank Lydia Sorokin for generously providing antibodies. We thank all the families of affected children for participation in our study, Gudrun Nürnberg for lod score calculations and Angelika Diem for excellent technical assistance.
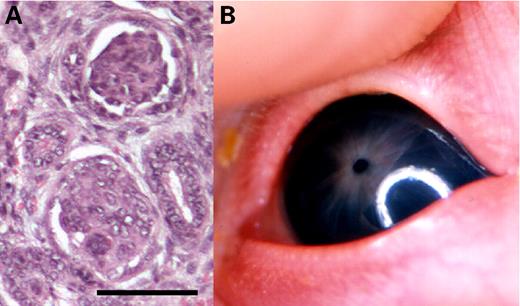
Figure 1. Main clinical features of Pierson syndrome: (A) diffuse mesangial sclerosis in a kidney biopsy (bar: 100 µm) and (B) clinical appearance of the ocular anomalies in an affected newborn.
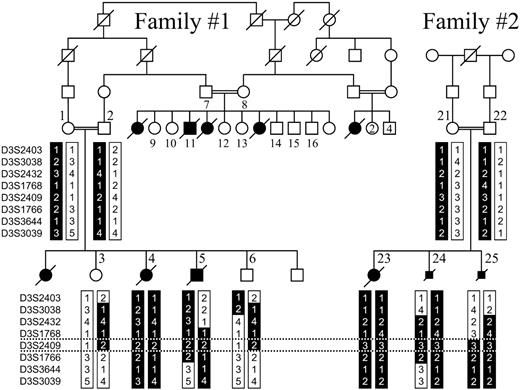
Figure 2. Pedigree of the large family originating from Lebanon with multiple offspring affected by Pierson syndrome (family 1) and the results of haplotype analysis of eight polymorphic microsatellite markers on 3p in one branch of this family and a consanguineous Turkish couple with three affected children (family 2). Numbered family members (1–16,21–25) were available for linkage studies. The shared disease-associated haplotypes are represented by a black background. The homozygous interval in family 1 and recombination in patient 25 define the centromeric and telomeric boundaries, respectively (indicated by dotted lines).
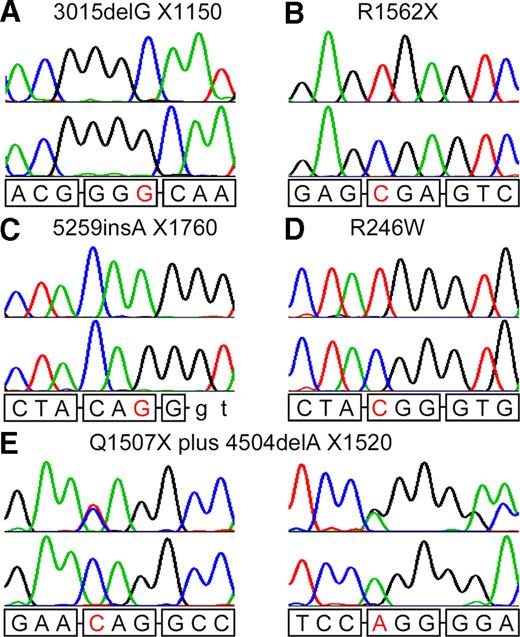
Figure 3. Results of sequence analysis showing mutant sequences from affected individuals of each family (upper lane) compared with the respective sequence of a normal control (lower lane). Normal sequences are indicated at the bottom of the respective chromatograms with boxes indicating triplet codons and letters in red highlighting the sequence deviations.
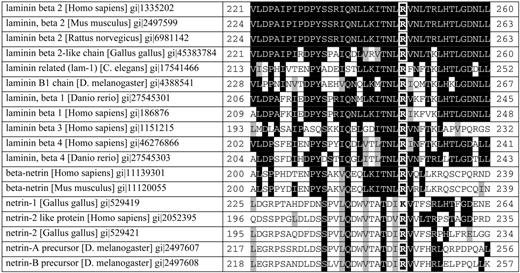
Figure 4. Alignment of human laminin β2 and laminin β2 orthologs from various species, as well as the human β-laminin paralogs and members of the laminin-like netrins shows high conservation of the arginine residue (white frames) at position 246 of laminin β2 and the surrounding sequence motif. Conserved amino acids are shown with black background and similar amino acids with a gray background. Alignments created according to outputs of BLAST (http://www.ncbi.nlm.nih.gov/BLAST) and the Conserved Domain Database (http://www.ncbi.nlm.nih.gov/Structure/cdd; CD: smart00136.10, LamNT).
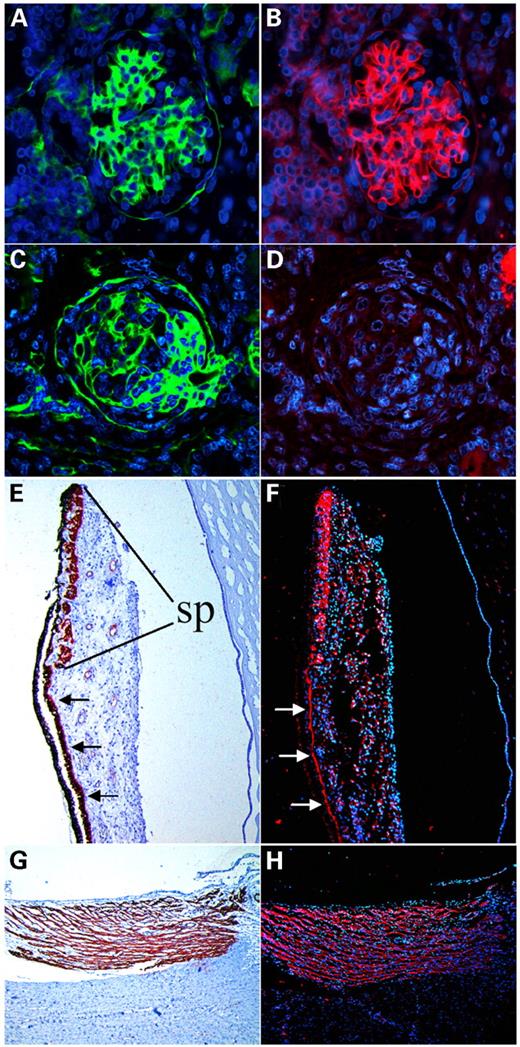
Figure 5. Immunohistology: Fixed, paraffin-embedded tissue sections analyzed by simultaneous indirect immunofluorescence using antibodies against collagen IV as a control protein for glomerular BL (green; A and C) and against laminin β2 (red; B, D, F, H). Nuclear stain with DAPI (blue) is displayed as background. (A and B) Normal neonatal kidney; (C and D) kidney tissue from a newborn patient with the mutation R1562X showing a structurally alterated glomerulus with positive staining for collagen IV (C) but lack of laminin β2 immunoreactivity (D). (E–H) Corresponding eye sections of a newborn control analyzed by immunoperoxidase using antibodies against smooth muscle actin (E and G) and by immunofluorescence using laminin β2 antibodies (F and H). Specific expression of laminin β2 surrounding muscle cells of sphincter pupillae (sp) (F) and ciliary muscle (H); bandlike laminin β2 expression at the basal site of the anterior layer of the posterior iris epithelium that forms the dilatator pupillae (arrows).
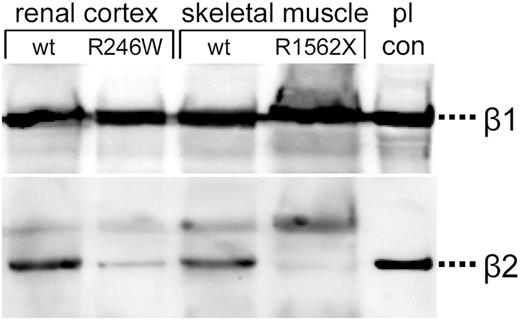
Figure 6. Western blotting of proteins from renal cortex of an affected infant with the mutation R246W and from skeletal muscle of an affected fetus carrying the mutation R1562X compared with tissues of age-matched controls (wt). Placental laminins (pl con) served as a positive control. Consecutive staining of the blot for laminin β2 (bottom) and laminin β1 (top) showing complete lack of laminin β2 immunoreactivity in the mutant R1562X and severe reduction in the mutant R246W.
Two point lod scores calculated for eight 3p microsatellite markers
| Marker/𝛉 | 0.00 | 0.05 | 0.10 | 0.20 | 0.30 | Zmax | 𝛉max | Position on the chromosome (cM) |
|---|---|---|---|---|---|---|---|---|
| D3S2403 | −∞ | −1.098 | −0.359 | 0.098 | 0.152 | 0.330 | 0.065 | 37.20 |
| D3S3038 | −∞ | −0.221 | 0.447 | 0.604 | 0.361 | 0.163 | 0.630 | 44.81 |
| D3S2432 | −∞ | 1.541 | 1.601 | 1.169 | 0.629 | 0.077 | 1.622 | 57.92 |
| D3S1768 | −∞ | 0.423 | 0.704 | 0.665 | 0.421 | 0.134 | 0.739 | 61.52 |
| D3S2409 | 3.495 | 3.063 | 2.632 | 1.790 | 1.019 | 0.000 | 3.495 | 70.60 |
| D3S1766 | 0.662 | 2.654 | 2.449 | 1.721 | 0.929 | 0.043 | 2.659 | 78.64 |
| D3S3644 | −∞ | 0.603 | 0.957 | 0.814 | 0.446 | 0.130 | 0.704 | 91.18 |
| D3S3039 | −∞ | −1.301 | −0.442 | 0.078 | 0.126 | 0.259 | 0.134 | 103.72 |
| Marker/𝛉 | 0.00 | 0.05 | 0.10 | 0.20 | 0.30 | Zmax | 𝛉max | Position on the chromosome (cM) |
|---|---|---|---|---|---|---|---|---|
| D3S2403 | −∞ | −1.098 | −0.359 | 0.098 | 0.152 | 0.330 | 0.065 | 37.20 |
| D3S3038 | −∞ | −0.221 | 0.447 | 0.604 | 0.361 | 0.163 | 0.630 | 44.81 |
| D3S2432 | −∞ | 1.541 | 1.601 | 1.169 | 0.629 | 0.077 | 1.622 | 57.92 |
| D3S1768 | −∞ | 0.423 | 0.704 | 0.665 | 0.421 | 0.134 | 0.739 | 61.52 |
| D3S2409 | 3.495 | 3.063 | 2.632 | 1.790 | 1.019 | 0.000 | 3.495 | 70.60 |
| D3S1766 | 0.662 | 2.654 | 2.449 | 1.721 | 0.929 | 0.043 | 2.659 | 78.64 |
| D3S3644 | −∞ | 0.603 | 0.957 | 0.814 | 0.446 | 0.130 | 0.704 | 91.18 |
| D3S3039 | −∞ | −1.301 | −0.442 | 0.078 | 0.126 | 0.259 | 0.134 | 103.72 |
The position on the chromosome was determined according to the Marshfield data (http://research.marshfieldclinic.org/genetics).
Two point lod scores calculated for eight 3p microsatellite markers
| Marker/𝛉 | 0.00 | 0.05 | 0.10 | 0.20 | 0.30 | Zmax | 𝛉max | Position on the chromosome (cM) |
|---|---|---|---|---|---|---|---|---|
| D3S2403 | −∞ | −1.098 | −0.359 | 0.098 | 0.152 | 0.330 | 0.065 | 37.20 |
| D3S3038 | −∞ | −0.221 | 0.447 | 0.604 | 0.361 | 0.163 | 0.630 | 44.81 |
| D3S2432 | −∞ | 1.541 | 1.601 | 1.169 | 0.629 | 0.077 | 1.622 | 57.92 |
| D3S1768 | −∞ | 0.423 | 0.704 | 0.665 | 0.421 | 0.134 | 0.739 | 61.52 |
| D3S2409 | 3.495 | 3.063 | 2.632 | 1.790 | 1.019 | 0.000 | 3.495 | 70.60 |
| D3S1766 | 0.662 | 2.654 | 2.449 | 1.721 | 0.929 | 0.043 | 2.659 | 78.64 |
| D3S3644 | −∞ | 0.603 | 0.957 | 0.814 | 0.446 | 0.130 | 0.704 | 91.18 |
| D3S3039 | −∞ | −1.301 | −0.442 | 0.078 | 0.126 | 0.259 | 0.134 | 103.72 |
| Marker/𝛉 | 0.00 | 0.05 | 0.10 | 0.20 | 0.30 | Zmax | 𝛉max | Position on the chromosome (cM) |
|---|---|---|---|---|---|---|---|---|
| D3S2403 | −∞ | −1.098 | −0.359 | 0.098 | 0.152 | 0.330 | 0.065 | 37.20 |
| D3S3038 | −∞ | −0.221 | 0.447 | 0.604 | 0.361 | 0.163 | 0.630 | 44.81 |
| D3S2432 | −∞ | 1.541 | 1.601 | 1.169 | 0.629 | 0.077 | 1.622 | 57.92 |
| D3S1768 | −∞ | 0.423 | 0.704 | 0.665 | 0.421 | 0.134 | 0.739 | 61.52 |
| D3S2409 | 3.495 | 3.063 | 2.632 | 1.790 | 1.019 | 0.000 | 3.495 | 70.60 |
| D3S1766 | 0.662 | 2.654 | 2.449 | 1.721 | 0.929 | 0.043 | 2.659 | 78.64 |
| D3S3644 | −∞ | 0.603 | 0.957 | 0.814 | 0.446 | 0.130 | 0.704 | 91.18 |
| D3S3039 | −∞ | −1.301 | −0.442 | 0.078 | 0.126 | 0.259 | 0.134 | 103.72 |
The position on the chromosome was determined according to the Marshfield data (http://research.marshfieldclinic.org/genetics).
Compilation of clinical data on the affected families
| Family no. | Ethnic origin | Parental consan-guinity | No. of affected children | Renal manifestations | Ocular manifestations | Others | Age of death | Mutation | Exon | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Lebanese | Yes | 8 | CNS with DMS, early-onset RF | Non-reactive miosis, ‘buphthalmos’a | ASD and Ebstein anomaly in one affected child | 1–44 days | 3015delG homozygous | 21 | (5) |
| 2 | Turkish | Yes | 3 | CNS with DMS, early-onset RF | Microcoria, lenticonus, corneal and retinal anomalies | — | Prenatal–32 days | R1562X homozygous | 28 | (5) |
| 3 | German | Nob | 1 | CNS with DMS, early-onset RF | Non-reactive miosis, ‘Rieger anomaly’, spherophakia | Severe muscular hypotonia and psychomotor delay, blindness | 1.8 yearsc | 5259insA homozygous | 30 | — |
| 4 | Portuguese | Yes | 2 | NS starting at 1–5 months, DMS | Miosis, nystagmus, cataracts | Muscular hypotonia, hemiparesis, abnormal movements | 8 months | R246W homozygous | 7 | (8) |
| 5 | Polish | No | 1 | CNS with DMS, early-onset RF | Unilateral microphthalmia, contralateral ‘glaucoma’ with pinhole fixed pupil, cataract | — | 75 days | 4504delA Q1507X compound heterozygous | 27 | (12) |
| Family no. | Ethnic origin | Parental consan-guinity | No. of affected children | Renal manifestations | Ocular manifestations | Others | Age of death | Mutation | Exon | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Lebanese | Yes | 8 | CNS with DMS, early-onset RF | Non-reactive miosis, ‘buphthalmos’a | ASD and Ebstein anomaly in one affected child | 1–44 days | 3015delG homozygous | 21 | (5) |
| 2 | Turkish | Yes | 3 | CNS with DMS, early-onset RF | Microcoria, lenticonus, corneal and retinal anomalies | — | Prenatal–32 days | R1562X homozygous | 28 | (5) |
| 3 | German | Nob | 1 | CNS with DMS, early-onset RF | Non-reactive miosis, ‘Rieger anomaly’, spherophakia | Severe muscular hypotonia and psychomotor delay, blindness | 1.8 yearsc | 5259insA homozygous | 30 | — |
| 4 | Portuguese | Yes | 2 | NS starting at 1–5 months, DMS | Miosis, nystagmus, cataracts | Muscular hypotonia, hemiparesis, abnormal movements | 8 months | R246W homozygous | 7 | (8) |
| 5 | Polish | No | 1 | CNS with DMS, early-onset RF | Unilateral microphthalmia, contralateral ‘glaucoma’ with pinhole fixed pupil, cataract | — | 75 days | 4504delA Q1507X compound heterozygous | 27 | (12) |
CNS, congenital nephrotic syndrome; DMS, diffuse mesangial sclerosis; RF, renal failure; ASD, atrial septal defect.
aOcular anomalies not documented in all affected individuals.
bBoth parents originate from a German isolate in Romania.
cSurvived under chronic peritoneal dialysis from the age of 6 weeks and died after kidney transplantation.
Compilation of clinical data on the affected families
| Family no. | Ethnic origin | Parental consan-guinity | No. of affected children | Renal manifestations | Ocular manifestations | Others | Age of death | Mutation | Exon | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Lebanese | Yes | 8 | CNS with DMS, early-onset RF | Non-reactive miosis, ‘buphthalmos’a | ASD and Ebstein anomaly in one affected child | 1–44 days | 3015delG homozygous | 21 | (5) |
| 2 | Turkish | Yes | 3 | CNS with DMS, early-onset RF | Microcoria, lenticonus, corneal and retinal anomalies | — | Prenatal–32 days | R1562X homozygous | 28 | (5) |
| 3 | German | Nob | 1 | CNS with DMS, early-onset RF | Non-reactive miosis, ‘Rieger anomaly’, spherophakia | Severe muscular hypotonia and psychomotor delay, blindness | 1.8 yearsc | 5259insA homozygous | 30 | — |
| 4 | Portuguese | Yes | 2 | NS starting at 1–5 months, DMS | Miosis, nystagmus, cataracts | Muscular hypotonia, hemiparesis, abnormal movements | 8 months | R246W homozygous | 7 | (8) |
| 5 | Polish | No | 1 | CNS with DMS, early-onset RF | Unilateral microphthalmia, contralateral ‘glaucoma’ with pinhole fixed pupil, cataract | — | 75 days | 4504delA Q1507X compound heterozygous | 27 | (12) |
| Family no. | Ethnic origin | Parental consan-guinity | No. of affected children | Renal manifestations | Ocular manifestations | Others | Age of death | Mutation | Exon | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Lebanese | Yes | 8 | CNS with DMS, early-onset RF | Non-reactive miosis, ‘buphthalmos’a | ASD and Ebstein anomaly in one affected child | 1–44 days | 3015delG homozygous | 21 | (5) |
| 2 | Turkish | Yes | 3 | CNS with DMS, early-onset RF | Microcoria, lenticonus, corneal and retinal anomalies | — | Prenatal–32 days | R1562X homozygous | 28 | (5) |
| 3 | German | Nob | 1 | CNS with DMS, early-onset RF | Non-reactive miosis, ‘Rieger anomaly’, spherophakia | Severe muscular hypotonia and psychomotor delay, blindness | 1.8 yearsc | 5259insA homozygous | 30 | — |
| 4 | Portuguese | Yes | 2 | NS starting at 1–5 months, DMS | Miosis, nystagmus, cataracts | Muscular hypotonia, hemiparesis, abnormal movements | 8 months | R246W homozygous | 7 | (8) |
| 5 | Polish | No | 1 | CNS with DMS, early-onset RF | Unilateral microphthalmia, contralateral ‘glaucoma’ with pinhole fixed pupil, cataract | — | 75 days | 4504delA Q1507X compound heterozygous | 27 | (12) |
CNS, congenital nephrotic syndrome; DMS, diffuse mesangial sclerosis; RF, renal failure; ASD, atrial septal defect.
aOcular anomalies not documented in all affected individuals.
bBoth parents originate from a German isolate in Romania.
cSurvived under chronic peritoneal dialysis from the age of 6 weeks and died after kidney transplantation.
References
Jalanko, H., Kääriäinen, H. and Norio, R. (
Somlo, S. and Mundel, P. (
Koziell, A., Grech, V., Hussain, S., Lee, G., Lenkkeri, U., Tryggvason, K. and Scambler, P. (
Patek, C.E., Fleming, S., Miles, C.G., Bellamy, C.O., Ladomery, M., Spraggon, L., Mullins, J., Hastie, N.D. and Hooper, M.L. (
Zenker, M., Tralau, T., Lennert, T., Pitz, S., Mark, K., Madlon, H., Doetsch, J., Reis, A., Müntefering, H. and Neumann, L.M. (
Pierson, M., Cordier, J., Hervouuet, F. and Rauber, G. (
Braga, S., Monn, E., Zimmermann, A. and Oetliker, O. (
Glastre, C., Cochat, P., Bouvier, R., Colon, S., Cottin, X., Giffon, D., Wright, C., Dijoud, F. and David, L. (
Nielsen, K.F. and Steffensen, G.K. (
Schneller, M., Braga, S.E., Moser, H., Zimmermann, A. and Oetliker, O. (
Noakes, P.G., Miner, J.H., Gautam, M., Cunningham, J.M., Sanes, J.R. and Merlie, J.P. (
Swietlinski, J., Maruniak-Chudek, I., Niemir, Z.I., Wozniak, A., Wilinska, M. and Zacharzewska, J. (
Nomizu, M., Utani, A., Beck, K., Otaka, A., Roller, P.P. and Yamada, Y. (
Colognato, H., Winkelmann, D.A. and Yurchenco, P.D. (
Tunggal, P., Smyth, N., Paulsson, M. and Ott, M.C. (
Yurchenco, P.D., Amenta, P.S. and Patton, B.L. (
Sanes, J.R. and Hall, Z.W. (
Hunter, D.D., Shah, V., Merlie, J.P. and Sanes, J.R. (
Kobayashi, N., Mominoki, K., Wakisaka, H., Shimazaki, Y. and Matsuda, S. (
Noakes, P.G., Gautam, M., Mudd, J., Sanes, J.R. and Merlie, J.P. (
Wang, T.H., Lindsey, J.D. and Weinreb, R.N. (
Ljubimov, A.V., Burgeson, R.E., Butkowski, R.J., Michael, A.F., Sun, T.T. and Kenney, M.C. (
Libby, R.T., Lavallee, C.R., Balkema, G.W., Brunken, W.J. and Hunter, D.D. (
Hunter, D.D., Llinas, R., Ard, M., Merlie, J.P. and Sanes, J.R. (
Kobayashi, N. (
Lathrop, G.M. and Lalouel, J.M. (
Sorokin, L.M., Conzelmann, S., Ekblom, P., Battaglia, C., Aumailley, M. and Timpl, R. (
Sixt, M., Hallmann, R., Wendler, O., Scharffetter-Kochanek, K. and Sorokin, L.M. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}