




Abstract
Cystathionine β-synthase (CBS) deficiency is a recessive genetic disorder in humans characterized by elevated levels of total plasma homocysteine (tHcy) and frequent thrombosis in humans. The I278T mutation is the most common mutation found in human CBS-deficient patients. The T424N mutation was identified as a mutation in human CBS that could restore function to I278T in Saccharomyces cerevisiae. In this report, we have engineered mice that express human I278T and I278T/T424N proteins from a metallotheinein-driven transgene. These transgene-containing mice were then bred to CBS knockout animals (Cbs-) to generate mice that express only human I278T or I278T/T424N protein. Both the I278T and the I278T/T424N transgenes are able to entirely rescue the previously described neonatal mortality phenotype despite the animals having a mean tHcy of 250 µm. The transgenic Cbs−/− animals exhibit facial alopecia, have moderate liver steatosis and are slightly smaller than heterozygous littermates. In contrast to human CBS deficiency, these mice do not exhibit extreme methioninemia. The mutant proteins are stable in the liver, kidney and colon, and liver extracts have only 2–3% of the CBS enzyme activity found in wild-type mice. Surprisingly, the I278T/T424N enzyme had exactly the same activity as the I278T enzyme indicating that T424N is unable to suppress I278T in mice. Our results show that elevated tHcy per se is not responsible for the neonatal lethality observed in Cbs−/− animals and suggests that CBS protein may have a function in addition to its role in homocysteine catabolism. These transgenic animals should be useful in the study of homocysteine related human disease.
INTRODUCTION
Cystathionine β-synthase (CBS) deficiency or classical homocystinuria is a recessive genetic disorder characterized by extremely elevated levels of total plasma homocysteine (tHcy, Fig. 1A). It is the most common inherited metabolic disorder involving sulfur metabolism (1). Patients with CBS deficiency have non-methionine loaded tHcy levels of 200–300 µm, compared with ∼10 µm in normal individuals. They also have extreme elevations in plasma methionine, as expected due to the conversion of excess homocysteine to methionine. Individuals with untreated homocystinuria develop arterial occlusions and venous thromboembolisms, often in childhood. Additional phenotypes include osteoporosis, mental retardation, psychiatric disorders and dislocated lenses.
Over the past decade more than 130 CBS mutations have been characterized from patients (2). The vast majority are missense mutations which are predicted to produce full-length mutant CBS protein. Less than 20% of the mutations in CBS-deficient patients are nonsense or deletion alleles and almost all CBS-deficient patients have at least one CBS allele that would be predicted to code for a mutant CBS protein. Most alleles are rare alleles only observed in one or two specific individuals. More frequent mutations tend to be geographically localized. For example, the 919 G>A (G307S) allele is extremely frequent in Ireland, while the R266K mutation is common in Norway (3,4).
By far the most frequently observed mutation is the pan-ethnic 833 T>C (I278T) allele (2). This allele accounts for ∼25% of all the reported mutations found in CBS-deficient patients. The I278T mutation probably arose from an incorrect excision of a 68 bp repeat polymorphism within the CBS gene that is present in ∼5% of all Caucasian chromosomes (5,6). Studies in the yeast Saccharomyces cerevisiae indicate that I278T has only ∼2.4% of the enzyme activity of wild-type human CBS and is unable to complement the growth phenotype of a CBS-deficient (cys4Δ) mutant (7). A genetic screen for cis-acting suppressors of I278T identified a second mutation, T424N, that when combined with I278T results in a protein that has significant enzyme activity and can complement a cys4Δ mutant (8). Molecular modeling of the T424N mutation indicates that it occurs in the CBS domain, which is found in a wide variety of proteins of diverse biological function. Recent studies suggest that this domain may be important for regulation by adenosine-containing molecules (9). In the case of CBS, this domain functions as an inhibitor of enzyme function and this inhibition is relieved by the allosteric regulator S-adenosylmethionine (10). These findings have led to the hypothesis that it might be possible to restore CBS function in CBS-deficient patients by targeting the C-terminal domain (8).
Watanabe et al. (11) created a knockout mouse that was deficient in CBS. Animals homozygous for the knockout allele (Cbstm1Unc; we will refer as Cbs−) rarely survived past 4 weeks of age. The surviving animals had tHcy levels >200 µm and had steatosis in the liver. The few surviving homozygous animals also have elevated tissue levels of S-adenosylhomocysteine (SAH) resulting in decreased DNA methylation in some tissues (12). Recent characterization of the Cbs homozygotes have found that they have hyperkeratosis of the skin, skeletal defects including kyphoscoliosis and evidence of oxidative stress in the liver (13–15).
The motivation for the current study was to determine if the T424N mutation could suppress the effects of the I278T mutation in mice in the same manner it did in yeast. Here, we describe the creation of two transgenic mouse lines, one that expresses I278T and one that expresses I278T+T424N. Surprisingly, we found that although both enzymes are largely inactive, they appear to possess the ability to rescue the neonatal lethality associated with homozygosity for the Cbs− allele. Our findings suggest that CBS may have additional in vivo functions beyond its role as a homocysteine metabolizing enzyme.
RESULTS
Transgene characterization
We used a plasmid containing the human CBS cDNA driven by the mouse metallotheinein promoter as the basis for our experiments (Fig. 1B). We had previously used this construct to express wild-type human CBS (16). Two different constructs were created. One contained the 833T>C mutation in the CBS cDNA such that it would express I278T, while a second had 833T>C and an additional mutation, 1271C>A, that would express a double mutant enzyme I278T/T424N. The constructs were designated Tg-278 and Tg-278/424, respectively. The metallotheinein promoter (mMT-I) was chosen because its expression can be regulated by the addition of zinc to drinking water, and it is expressed in high levels in liver and kidney, mirroring endogenous CBS expression (17). The hemagglutinin A (HA) epitope tag was inserted in the N-terminus of CBS to distinguish between endogenous mouse CBS and the human CBS proteins. Our construct also contained the 5′ and 3′ locus controlling regions of mMT-I to help minimize expression differences due to different sites of integration (18). For each construct approximately 200 injected C57B6/C3H hybrid embryos were implanted into pseudo-pregnant females. We obtained seven founders for the Tg-278 construct and a single founder line for the Tg-278/424 construct.
The founder animals were backcrossed to C57BL6 animals and the offspring were assessed for transgene status. Five of the seven Tg-278 founders transmitted the transgene, as well as the single Tg-278-424 founder. CBS protein level was assessed by immunoblot in the liver of a zinc-treated offspring of each line to confirm expression (Fig. 2A and data not shown). We found significant levels of human CBS in offspring from all six of the lines examined and chose a single I278T founder line and a single I278T+T424N line for future studies.
Tg-I278T and Tg-I278T+T424N rescues neonatal lethality
Watanabe et al. (11) created a knockout allele for CBS (Cbs−), and found that animals homozygous for this allele showed severe homocystinuria, severe growth retardation and rarely survived beyond 6 weeks of age. Previously, we confirmed these findings in our own colony of Cbs− mice (16). We found that at the time of genotyping (4–6 weeks of age), only 3/258 (1.2%) offspring of Cbs+/−×Cbs+/− mating were homozygous for Cbs−, versus an expected number of 64.5 (25%). We also demonstrated that expression of wild-type human CBS from the mMT-I promoter could entirely reverse this lethality.
We first determined if the expression of I278T transgene could also rescue the neonatal lethality associated with homozygosity for Cbs−. Our initial hypothesis was that the Tg-278 transgene would not be able to rescue because in S. cerevisiae it failed to exhibit significant enzyme activity and could not complement a yeast CBS deletion. We mated our Tg-278 animals to Cbs+/− heterozygotes and obtained Tg-278 Cbs+/− offspring. These animals were then put on water containing 25 mm zinc sulfate to induce transgene expression and backcrossed a second time to Cbs+/− mice. If the transgene could rescue lethality, we would expect 1/8 (12.5%) of the offspring to be both homozygous for Cbs− and hemizygous for the transgene. Furthermore, we would also expect to see significantly more viable homozygous animals with the transgene than without the transgene. If the transgene failed to rescue we would expect to see very few (1–2%) Cbs−/− survivors and these would be of equal numbers in regards to transgene status. Surprisingly, we found that out of 179 offspring born, 21 were homozygous for the knockout allele and contained the transgene (11.7%), consistent with the idea that the I278T expressing transgene could rescue lethality. We also observed a highly skewed ratio of transgene positive homozygotes (n=21) compared with transgene negative homozygotes (n=8, P=0.02). These results show that the I278T expressing transgene is able to rescue the lethality associated with homozygosity for Cbs−.
We also examined if the Tg-278/424 construct could rescue viability. In this case, to expedite the generation of Tg-278/424 Cbs−/− animals we set up crosses in which Tg-I278/424 N Cbs+/− animals were intercrossed to sibling animals of the same genotype. From this cross, we would expect that 18.75% of the offspring would be positive for the transgene (either one or two copies) and homozygous for the Cbs− allele, if there was total rescue of the neonatal lethality. We found that 17 of 77 (22%) offspring were Tg-278/424 Cbs−/−, which was not significantly different from the expected number (14.4 expected versus 17 observed). In contrast, we obtained only one animal that was Cbs−/− and lacked the transgene (4.8 expected versus one observed). In addition, we performed a cross in which male Tg-278/424 Cbs−/− animals were crossed with female Tg-278/424 Cbs+/− animals. From this cross 12 of 24 of the offspring were Tg-278/424 positive and homozygous for the Cbs− allele. These results show that the Tg-278/424 transgene is able to rescue the neonatal lethality associated with homozygosity for Cbs−.
Analysis of Tg-278 and Tg-278/424 Cbs−/− animals
Since both of the transgene constructs could rescue the neonatal lethality phenotype, we wanted to determine if this rescue was due to decreased levels of tHcy. Therefore, we measured the tHcy levels in a number of the transgene rescued Cbs−/− homozygous animals (Fig. 3A). The mean tHcy for the Tg-278 Cbs−/− animals was 250 µm, while the Tg-278/424 Cbs−/− animals had an average of 274 µm. These levels of tHcy were at least 60-fold higher than that observed in wild-type control animals. The level of tHcy in the transgenic animals were essentially identical to that found in the few surviving non-transgene-containing Cbs−/− animals. These results show that neither of the mutant proteins is capable of reversing the homocystinuria in Cbs−/− animals.
We also examined plasma methionine levels in these animals because in human CBS deficiency elevations in plasma methionine are often more severe than the elevations in plasma homocysteine. We found that both the Tg-278 and Tg-278/424 Cbs−/− animals had only slightly elevated plasma methionine levels relative to the wild-type control animals. The wild-type animals had a mean plasma methionine of 52 µm compared with a mean of 72 µm for the Tg-278 Cbs−/− (P<0.0015) and 85 µm for Tg-278/424 Cbs−/− (P<0.0001). As with tHcy, transgenic Cbs−/− animals had plasma methionine levels that were similar to those observed in non-transgene containing Cbs−/− animals. The very modest increase in plasma methionine levels in these mice contrasts sharply with the extreme hypermethionemia observed in human patients with CBS deficiency (19).
We also examined levels of tHcy and methionine in whole liver extracts made from some of the mice. As shown in Figure 3B, we saw that both Tg-278 Cbs−/− and surviving non-transgenic Cbs−/− animals had elevated tHcy levels in liver extracts compared with wild-type control extracts. The Tg-278 Cbs−/− animals had about double the average tHcy level of the control animals (2.4 versus 1.3 nmol/mg, P=0.12) although this difference was not statistically significant. The surviving non-transgenic Cbs−/− animals had even higher tHcy levels (4.3 versus 1.3 nmol/mg, P=0.04). Interestingly, although liver tHcy was elevated in the Cbs−/− animals compared with the wild-type control animals the fold increase was much less than that observed in the serum. We did not observe any significant differences with regard to methionine levels in the liver extracts.
The fact that the mutant transgene rescued neonatal lethality, but still had extremely elevated tHcy, was surprising. We therefore examined these animals closely to determine if there might be other phenotypes associated with elevated tHcy. We found two noticeable physical differences. One consistent phenotype (Fig. 4) is that animals lacking CBS had a characteristic alopecia around the nose and eyes. This was true in both transgene rescued and in the non-transgenic survivors. The alopecia phenotype is quite striking and can be used to identify Cbs−/− animals with near perfect accuracy. We do not believe this was the result of ‘barbering’ by other animals, as this occurred even in animals that were kept apart. It is possible that the loss of hair is due to some sort of self-grooming behavior, but we did not observe any indications of obsessive grooming in the routine handling and observation of these animals. In general the Cbs−/− homozygotes had coats that were less shiny and full compared with heterozygous and wild-type siblings.
A second phenotype is that Cbs−/− survivors were also somewhat smaller than heterozygous and wild-type littermates (Fig. 5). On average Cbs−/− animals were ∼10% smaller than their wild-type or heterozygous littermates, although the range of weights tended to be similar. The same difference was observed in both the Cbs−/− animals containing the mutant transgene or in the non-transgene containing animals.
We also examined fixed sections of the liver, kidney and brain of eight Tg-278/424 Cbs−/− animals and eight littermate controls for pathological abnormalities. We observed slight to moderate focal steatosis in liver sections of four of the eight Tg-278/424 Cbs−/− animals and in one of the eight control animals (P=0.14, Fisher's exact, one-sided; see Fig. 4C). In the regions of steatosis, hepatocytes and hepatocytes nuclei appeared to be enlarged as previously noted by Watanabe (11) (Fig. 4). No differences were observed in either the kidneys or brains of the Tg-278/424 Cbs−/− animals and sibling controls. Although we did not examine Tg-278 Cbs−/− animals, we did find similar levels of steatosis with Tg-hCBS Cbs−/− that had been maintained on regular water to induce elevated tHcy (data not shown).
Transgene expression
We wanted to determine the expression of the transgenes especially since the I278T/T424N transgene did not appear to lower tHcy in mice. We first examined expression of I278T and I278T/T424N protein in mouse livers by western blot analysis. As shown in Figure 2B, both the I278T and the I278T/T424N mutant enzymes are present in the liver at levels similar to that observed for endogenous mouse CBS. This finding shows that the I278T and I278T/T424N enzymes are stable and not rapidly degraded in vivo. We also examined expression of the enzymes in various tissues including liver, brain, colon and kidney (Fig. 2C). We observed high levels of transgene expression in the liver, kidney and colon, but saw either little or no expression in the brain. These findings indicate that the mutant proteins are made and are stable in multiple tissues.
We then measured CBS enzyme activity in mouse liver extracts from various Cbs mice (Fig. 6). Liver extracts from wild-type mice had about 1115 units of activity while extract from a Cbs heterozygote had 420 units of activity. Cbs−/− animals expressing wild-type human CBS from the mMT-I promoter had 260 units of activity. In contrast, mice expressing I278T and I278T/T424N had 25 and 18 units of activity, respectively. We saw an inverse correlation between levels of CBS activity in the liver and tHcy in the serum. These results show that CBS activity was significantly reduced by the presence of the I278T and I278T/T424N mutations. In addition, these results show that the addition of the T424N mutation does not enhance enzyme function of the I278T enzyme in mice, contrasting with its behavior in S. cerevisiae.
DISCUSSION
In the original mouse model for CBS deficiency developed by Watanabe et al. (11) ∼80% of the Cbs homozygous animals died by 4 weeks of age. The few survivors that were characterized had extremely elevated tHcy and steatosis in the liver. In our earlier work, we demonstrated that expression of the wild-type human CBS enzyme from a metallotheinein-driven transgene could rescue this lethality (16). We had assumed that this was due to the fact that Cbs homozygous mice expressing the wild-type human CBS enzyme had significantly lower tHcy levels. However, in the work presented here, we show that the expression of mutant and largely inactive CBS enzymes can also rescue viability, despite the fact that the surviving animals have extremely elevated tHcy levels. The average tHcy level was actually slightly higher in the transgene containing Cbs−/− animals than that observed for the surviving homozygous animals lacking the transgene. We also found that the surviving Cbs homozygous animals had moderate focal steatosis of the liver and a loss of facial hair. The fact that two different mutant transgene lines had identical phenotypes argues against the suppression of neonatal lethality being due to an insertion effect of the transgene. These findings show that elevated tHcy per se is not what is responsible for the poor survival of the CBS-deficient animals.
How might the mutant transgene rescue the neonatal lethality, but not affect the tHcy or steatosis phenotypes? One possibility is that even though the mutant transgenes are largely inactive, they may have enough enzyme activity to rescue the phenotype. Our measurement of CBS activity in mouse liver extracts indicates that both transgenes provide ∼2% of the enzyme activity of that found in wild-type animals. However, we found no difference in the tHcy levels between Tg+ and Tg−Cbs−/− animals. It may be that the surviving minority of Tg−Cbs−/− are actually those that are selected for tHcy levels compatible with life, i.e. the ones that die have even higher levels. Alternatively, it is possible that the neonatal lethality is actually due to the production of cystathionine or some downstream metabolite as opposed to excessive elevations of tHcy. We were unable to detect any cystathionine in the serum of our Tg-278 and Tg-278/424 Cbs−/− animals using an amino acid analyzer, but our sensitivity is such that we would be unable to detect cystathionine at levels <10–20% observed in wild-type animals.
Another possibility would be that CBS has a function in addition to its enzymatic function that may be important during development. This idea is not without precedent. Phosphoglucose isomerase (PGI), a glycolytic enzyme, is also a secreted neurotrophic growth factor called neuroleukin (20). Dimeric PGI is active in the isomerase reaction, while the monomeric form has neurotrophic activity (21). Thus the enzyme activity is separable from the growth activity. Glutathione transferase Pi (GST-Pi), a phase II detoxification enzyme, doubles as a regulator of Jun N-terminal kinase (JNK), a key enzyme in stress response (22). Like PGI, the monomer affects JNK function, but GST activity requires dimerization.
A surprising finding is that there was essentially no difference between mice expressing I278T and mice expressing I278T+T424N human CBS. Our earlier work in S. cerevisiae showed that the human I278T enzyme had very little enzyme activity (2.4% of wild-type) and did not complement the cysteine auxotrophy of a cys4Δ yeast strain (4,19). T424N was identified in a random mutagenesis screen designed to identify functional suppressors of I278T. Cys4Δ yeast expressing double mutant I278T/T424N could grow on cysteine free media and that extracts derived from these yeast had ∼30% of the CBS activity as extracts from strains expressing wild-type human CBS. In contrast, liver extracts from Tg-278 and Tg-278/424 Cbs−/− animals had essentially identical low levels of CBS activity. One possible explanation was that the presence of the N-terminal HA tag might be acting to inhibit T424N suppression. However, expression of the HA-tagged I278T/T424N protein in S. cerevisiae also complemented the cysteine auxotrophy, suggesting this was not the case (L. Wang, unpublished data). Our current working hypothesis is that there may be subtle differences in the protein folding environments between yeast and mouse cells such that in yeast the T424N mutation allows proper folding of the I278T enzyme, but in mouse cells it does not.
A second unexpected finding was that the transgene rescued Cbs−/− mice were relatively healthy despite extremely elevated tHcy. We did not observe any obvious excess mortality in these animals and we have several animals that are over a year in age. The most obvious phenotypes are the slightly smaller size, distinctive facial hair loss and steatosis in the liver. Recently, another group reported very similar phenotypes in their surviving Cbs−/− animals (13–15). These animals were obtained by feeding the animals diets supplemented with betaine. Betaine is an alternative methyl-group donor that can be used to catalyze the conversion of homocysteine to methionine via the enzyme betaine-dependent homocysteine methyltransferase. Interestingly, the betaine rescued animals had tHcy levels >200 µm, supporting the view that high tHcy may not be responsible for the neonatal lethality. It is unclear from these reports how efficiently betaine rescues Cbs−/− neonates.
A distinctive difference between the mouse and human CBS deficiency concerns plasma methionine levels. In humans with CBS deficiency plasma methionine levels are extremely elevated. It is not uncommon to see plasma methionine levels in CBS-deficient patients as high as 1 or 2 mm. In contrast, the Cbs−/− mice had very modest plasma methionine levels, despite having tHcy levels quite similar to that observed in human patients. This suggests the possibility that some of the pathological consequences of excess tHcy in CBS-deficient patients not observed in mice, such as increased thrombosis, may be due to the consequences of an interaction between excess tHcy and excess methionine. The cause of the lack of methionemia in our Tg Cbs−/− animals is unknown.
Our ability to generate animals with excessive tHcy should be useful in the study of the pathology of elevated tHcy. Many studies have compared phenotypes of Cbs+/+ with Cbs−/+ animals, but few have examined phenotypes in Cbs−/− animals because of the difficulty of obtaining the required number of animals. The Tg-278 and Tg278/424 mice can be produced efficiently and should be useful to confirm the effects of tHcy on a variety of pathological processes.
MATERIALS AND METHODS
Constructs
Site-directed mutagenesis was used to introduce a 833T>C (I278T) change into pLW2, which contains a hemagglutinin epitope tagged version of the human CBS cDNA (16) using the Quick Change XL site-directed mutagenesis kit from Stratagene (La Jolla, CA). The entire open reading frame of the resulting clone, pLW2:278, was sequenced to verify no additional changes occurred. Plasmid pLW2:278 was subsequently digested with MfeI and cloned into MT-LCR expression vector 2999 (18). The resulting plasmid was designated pLW3:278. For the double mutant construct pLW2:278 was used as a template for site-directed mutagenesis in which the 1271A>C change was introduced and the resulting construct was subcloned into 2999 to generate pLW3:278/424.
Mouse generation
Transgenic mice containing either pLW3:278 or pLW3:278/424 were created as previously described (16). In brief, DNA was digested with SalI, purified and microinjected into the pronuclei of day 0.5 C57B6/C3H F2 embryos. Injected embryos were then transferred to pseudopregnant mice. Tails from the resulting pups were then screened for the presence of the transgene by PCR as described (16). DNA positive animals were then bred to C57B6 animals and the offspring was then tested for germline transmission. Five of seven of the pLW3:278 founders had germline transmission, as well as one out of one of the pLW3:278/424 founders.
Transgene positive offspring were then bred to C57B6 Cbs− heterozygous animals originally obtained from Jackson Laboratories (Bar Harbor, ME) to obtain Tg+Cbs−/+ animals. These animals were then backcrossed a second time to C57B6 Cbs− heterozygous animals on water containing 25 mol/l ZnSO4 to induce transgene expression. In some cases Tg+Cbs−/+ siblings were intercrossed in the presence of 25 mol/l ZnSO4.
Genotyping of offspring was generally done at the time of weaning as previously described (16). Animals were fed standard rodent chow (LabDiet 5013).
Immunoblot and CBS enzyme analysis
Mouse liver and tissue extracts were prepared as previously described (16). In brief, mice were sacrificed and tissues were immediately put on ice and homogenized with a dounce homogenizer in extract buffer. Extracts were then centrifuged at 12 000g at 4oC and the supernatant was retained. Protein concentration was determined by the Coomassie blue protein assay reagent (Pierce) using BSA as a standard. For western blot analysis, 25 µg of extract were run on precast 7% NuPage Tris-acetate SDS gel (Invitrogen) and transferred to PVDF membrane as described (23). Samples were probed with a 1/15 000 dilution of rabbit anti-hCBS serum as previously described (10).
CBS enzyme activity was measured using a Biochrom 30 amino acid analyzer as previously described (16).
Homocysteine and methionine analysis
Serum homocysteine and methionine were measured using a BioChrom 30 amino acid analyzer. Mice were bled from the eye socket in a standard Eppendorf tube and immediately put on ice. The samples were then centrifuged at 4oC for 10 min and the serum was collected. Fifty microliters of serum was then treated with 5 µl of 12% dithiothreitol at room temperature for 5 min to reduce the samples. Fifty-five microliters of 10% sulfosalicylic acid was added followed by 1 h at 4oC and then followed by centrifugation. The supernatant was then loaded on the amino acid analyzer. Peaks were identified and quantitated by comparing to a known standard.
Histology
The liver, kidney and brain were excised, fixed overnight in 10% buffered formaldehyde, embedded in paraffin and sectioned. Three representative longitudinal sections 6 µm thick from each tissue were collected. All sections were stained with hematoxylin and eosin and evaluated for abnormalities by a trained mouse pathologist (A.K.S.).
ACKNOWLEDGEMENTS
We thank Dr. Elizabeth Henske and Dr. Cynthia Kelcher for the critical reading of the manuscript. We also wish to thank the Sequencing, Experimental Histopathology, Transgenic Mouse, and Laboratory Animal Facilities of the Fox Chase Cancer Center for their assistance. This work was supported by grants HL57299 and Core grant CA06927 from the National Institutes of Health and in part, by a grant from the Pennsylvania Department of Health. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
Conflict of Interest statement. None declared.
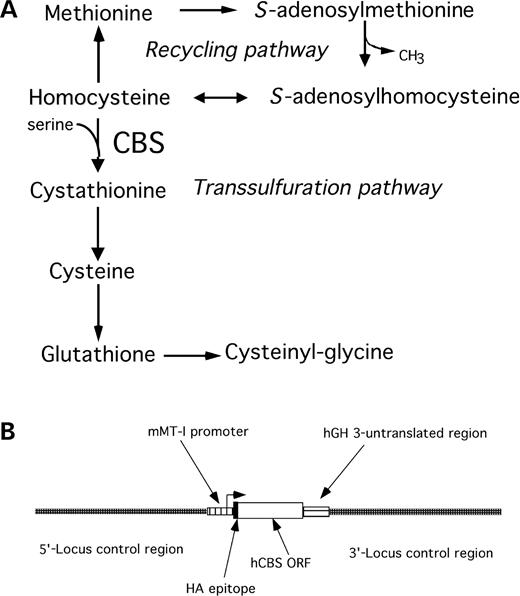
Figure 1. (A) Methionine metabolism in mammals. The methionine recycling and transsulfuration pathways are shown. (B) Schematic of the Tg-CBS construct used in this study. The regulatory and coding sequences are indicated by the arrows.
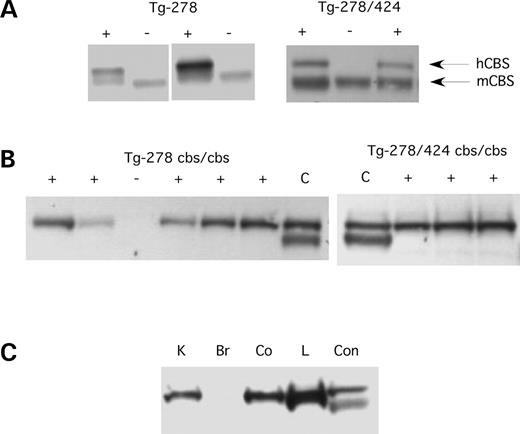
Figure 2. Immunoblot analysis of CBS. (A) Livers were harvested from Tg-278 and Tg-278/424 animals on zinc water and total protein extracts were examined for CBS levels using polyclonal anti-serum against human CBS. This serum detects both human and mouse CBS protein. The left panel shows two offspring from two different Tg-278 founder mice, while the right panel shows three offspring from the Tg-278/424 founder. The transgene status of the offspring is indicated at the top. The arrows indicate the human and the mouse CBS proteins. (B) CBS expression in Tg-278 and Tg-278/424 mice that lack mouse CBS (homozygous for the Cbs− allele) on zinc water. Lane C is a control extract from Tg-hCBS mouse on zinc water (16). (C) Tissue expression of transgene. A Tg-278/424 mouse lacking endogenous CBS on zinc water was sacrificed and extracts were prepared from the kidney (K), brain (Br), liver (L) and colon (Co).
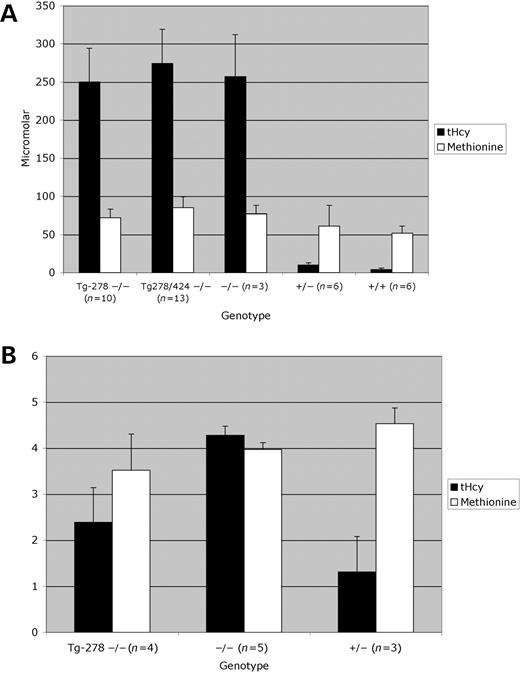
Figure 3. Measurement of tHcy and methionine. (A) Serum measurement. Animals of the indicated genotypes on zinc water had serum collected and analyzed using an amino acid analyzer for methionine and homocysteine. The number of animals in each group is indicated at the bottom. The error bar indicates the standard deviation of the measures. (B) Measurement in liver extracts. The mice of the indicated genotype on zinc water were sacrificed and had their livers harvested and processed as described in materials and methods. The extracts were then analyzed using an amino acid analyzer. The units are nanomoles per milligram of protein.
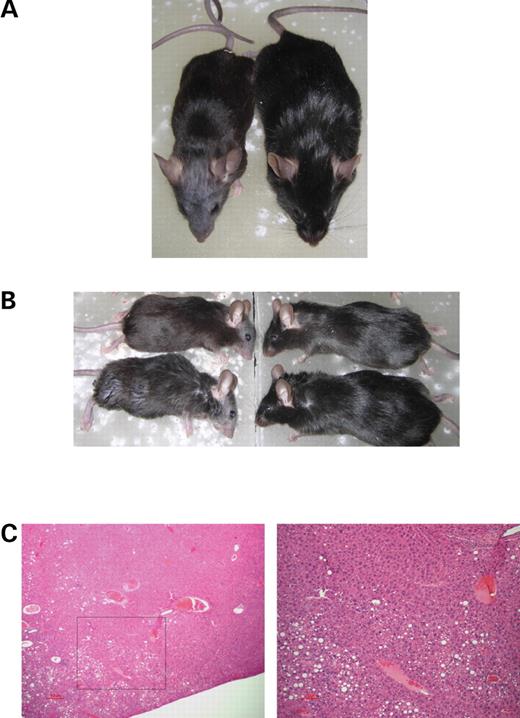
Figure 4. Tg-278/424 Cbs−/− and Tg-278/424 Cbs−/+ comparisons. (A) Overhead view of sibling male Tg-278/424 Cbs−/− and Tg-278/424 Cbs−/+ animals. Note the smaller size and loss of facial hair. (B) Pair of Tg-278/424 Cbs−/− and Tg-278/424 Cbs−/+ animals from the same litter. On the left are the knockout animals, while on the right are heterozygous animals. The top pair are females, while the bottom pair are males. (C) Example of liver sections from a Tg-278/424 Cbs−/− animal. On the left is a hematoxylin and eosin stained section at 40× magnification. On the right is a 100× magnification of the area shown in the square.
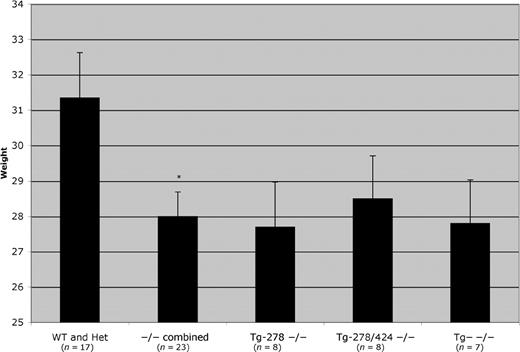
Figure 5. Weight comparison of Cbs−/− animals. The animals ranged in age from 4 months to 1 year. Control animals were matched by age and sex. The column labeled −/− combined is a grouping of all three of the Cbs−/− genotypes. The star indicates a significant difference (P=0.018) between the Cbs−/− animals and the control siblings.
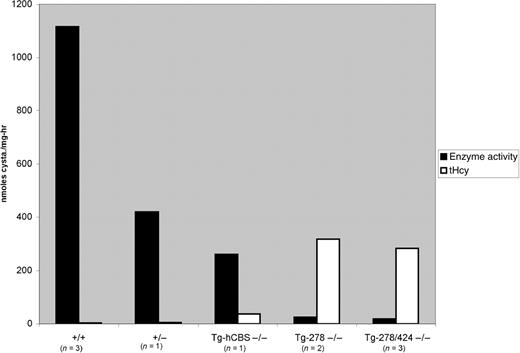
Figure 6. Liver CBS enzyme activity. Extracts from mice of the indicated genotype were assessed for CBS enzyme activity, shown on the left Y-axis. The right axis shows the same animals tHcy levels.
References
Mudd, S.H., Levy, H.L. and Kraus, J.P. (
Kraus, J.P., Janosik, M., Kozich, V., Mandell, R., Shih, V., Sperandeo, M.P., Sebastio, G., de Franchis, R., Andria, G., Kluijtmans, L.A. et al. (
Gallagher, P.M., Ward, P., Tan, S., Naughten, E., Kraus, J.P., Sellar, G.C., McConnell, D.J., Graham, I. and Whitehead, A.S. (
Kim, C.E., Gallagher, P.M., Guttormsen, A.B., Refsum, H., Ueland, P.M., Ose, L., Folling, I., Whitehead, A.S., Tsai, M.Y. and Kruger, W.D. (
Sperandeo, M.P., de Franchis, R., Andria, G. and Sebastio, G. (
De Stefano, V., Dekou, V., Nicaud, V., Chasse, J.F., London, J., Stansbie, D., Humphries, S.E. and Gudnason, V. (
Kruger, W.D. and Cox, D.R. (
Shan, X., Dunbrack, R.L., Jr., Christopher, S.A. and Kruger, W.D. (
Scott, J.W., Hawley, S.A., Green, K.A., Anis, M., Stewart, G., Scullion, G.A., Norman, D.G. and Hardie, D.G. (
Shan, X. and Kruger, W.D. (
Watanabe, M., Osada, J., Aratani, Y., Kluckman, K., Reddick, R., Malinow, M.R. and Maeda, N. (
Caudill, M.A., Wang, J.C., Melnyk, S., Pogribny, I.P., Jernigan, S., Collins, M.D., Santos-Guzman, J., Swendseid, M.E., Cogger, E.A. and James, S.J. (
Robert, K., Maurin, N., Ledru, A., Delabar, J. and Janel, N. (
Robert, K., Nehme, J., Bourdon, E., Pivert, G., Friguet, B., Delcayre, C., Delabar, J.M. and Janel, N. (
Robert, K., Maurin, N., Vayssettes, C., Siauve, N. and Janel, N. (
Wang, L., Jhee, K.H., Hua, X., DiBello, P.M., Jacobsen, D.W. and Kruger, W.D. (
Palmiter, R.D., Norstedt, G., Gelinas, R.E., Hammer, R.E. and Brinster, R.L. (
Palmiter, R.D., Sandgren, E.P., Koeller, D.M. and Brinster, R.L. (
Kruger, W.D., Wang, L., Jhee, K.H., Singh, R.H. and Elsas, L.J., II (
Sun, Y.J., Chou, C.C., Chen, W.S., Wu, R.T., Meng, M. and Hsiao, C.D. (
Mizrachi, Y. (
Adler, V., Yin, Z., Fuchs, S.Y., Benezra, M., Rosario, L., Tew, K.D., Pincus, M.R., Sardana, M., Henderson, C.J., Wolf, C.R. et al. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}