




Abstract
The study of common fragile sites has its roots in the early cytogenetic investigations of the fragile X syndrome. Long considered an interesting component of chromosome structure, common fragile sites have taken on novel significance as regions of the genome that are particularly sensitive to certain forms of replication stress, which are frequently rearranged in cancer cells. In recent years, much has been learned about the genomic structure at fragile sites and the cellular checkpoint functions that monitor their stability. Recent findings suggest that common fragile sites may serve as markers of chromosome damage caused by replication stress during early stages of tumorigenesis. Thus, the study of common fragile sites can provide insight not only into the nature of fragile sites, but also into the broader consequences of replication stress on DNA damage and cancer. However, despite recent advances, many questions remain regarding the normal functional significance of these conserved regions and the basis of their fragility.
INTRODUCTION
Common fragile sites are chromosomal regions that are particularly sensitive to forming gaps or breaks on metaphase chromosomes after partial inhibition of DNA synthesis (Fig. 1). These lesions were first seen in cells grown under conditions of folate stress used to induce cytogenetic expression of the fragile X site (FRAXA) in individuals with fragile X-linked mental retardation. It was later found that these fragile sites are also specifically induced by aphidicolin, an inhibitor of DNA polymerase α and other polymerases. Fragile sites are now generally categorized into two main classes. ‘Rare’ fragile sites, such as the fragile X site, are seen in few individuals, are associated with CGG or AT repeat expansion mutations and segregate in a Mendelian manner (1,2). ‘Common’ fragile sites are seen in all individuals and, therefore, represent a component of normal chromosome structure.
At present, 84 common fragile sites are listed in the genome database (GDB). The exact number of common fragile sites that exist is a matter of interpretation based on the criteria for inclusion and statistical analyses of data. The greater the stress placed on DNA replication, the more fragile sites are observed, until replication ceases. However, not all fragile sites form breaks at the same frequency, and a small number of sites in the genome are most prone to form lesions (3,4). Gaps and breaks at just 20 fragile sites represent >80% of all lesions observed in lymphocytes, following treatment with low doses of aphidicolin (3). FRA3B at 3p14.2 stands out as the most ‘fragile’ site in the genome and can be induced to break or form gaps in the majority of treated cells. Other highly expressed fragile sites in lymphocytes include those at 16q23 (FRA16D), 6q26 (FRA6E), 7q32.3 (FRA7H) and Xp22.3 (FRAXB) (3). We have observed some variation in the relative sensitivity of individual fragile sites in different cell types, but the basis for this has not been fully explored.
FRAGILE SITES ARE UNSTABLE IN VITRO
Common fragile site regions are normally stable in cultured human cells. However, when cells are cultured under conditions of folate or thymidylate stress or with low doses of aphidicolin that only partially inhibit DNA synthesis, common fragile site regions form chromosome gaps and breaks. Other inhibitors of replication, such as hydroxyurea, are less specific in inducing lesions primarily at fragile sites, probably due to differences in mechanisms of inhibition.
In addition to gaps and breaks, common fragile sites display a number of characteristics of unstable DNA in cultured cells. Following induction, they are ‘hotspots’ for increased sister chromatid exchange (SCE) (5) and show a high rate of translocations and deletions in somatic cell hybrid systems (6,7). Rassool et al. (8) demonstrated that fragile sites are preferred sites of recombination, or integration, with pSV2neo-plasmid DNA transfected into cells pretreated with aphidicolin. Perhaps related to this characteristic are reports of the coincidence of viral integration sites in tumors or tumor cell lines and fragile sites (9–12). Fragile sites have also been implicated in intrachromosomal gene amplification events in cultured Chinese hamster ovary (CHO) cells and in cancer cells by leading to DNA strand breaks that trigger breakage–fusion–bridge cycles (13).
GENES AT COMMON FRAGILE SITES
Eleven common fragile sites, FRA2G, FRA3B, FRA4F, FRA6E, FRA6F, FRA7E, FRA7G, FRA7H, FRA9E, FRA16D and FRAXB, have been cloned and characterized. All have been found to extend over hundreds of kilobases as defined by the induction of gaps or breaks on metaphase chromosomes occurring throughout these regions. As might be predicted given their size, most sites either lie within or span known genes (Table 1). FRA3B was the first fragile site to be mapped and cloned and it was found to lie within the FHIT gene (14–16). FHIT spans ∼900 kb and includes two large introns where FRA3B is centrally located, but encodes only a small 1.1 kb transcript. There are no large di- or trinucleotide repeats within FRA3B, but the region was found to contain human cervical cancer HPV-16 integration sites (9), which led to other findings of HPV integrations at fragile sites in cervical tumors (17).
Two other of the most frequently expressed fragile sites also lie within large genes. FRA16D lies within the large WWOX gene, and like FHIT, WWOX encodes a small 2.2 kb transcript but extends >1 Mb due to the presence of two very large introns (18,19). Similarly, FRA6E lies within the large PARK2 gene, which extends >500 kb (20). Recently, the GRID2 gene, a hotspot for mutation and translocations in mouse and humans, has been associated with fragile site FRA4F (21). GRID2, like FHIT, WWOX and PARK2, extends over more than a megabase, but has a small ∼3 kilobase transcript. The significance of the relationship of fragile sites to large genes is intriguing and could be a reflection of the base composition of large introns found in these genes.
FRAGILE SITES ARE EVOLUTIONARILY CONSERVED
Common fragile sites have been observed in several other mammalian species, including other primates, cat, dog, pig, horse, cow, Indian mole rat, deer mouse and laboratory mouse (22–29), thus suggesting a conserved function. Of these species, common fragile sites are currently best characterized in the laboratory mouse. Two murine sites, Fra14a2 and Fra8e1, are orthologs of human FRA3B and FRA16D, respectively, and are associated with the murine Fhit and Wwox genes (24,30–33). Like human fragile sites, these sites are highly AT-rich. There are several large intronic regions within these fragile sites with high similarity to their human counterpart, suggesting functional conservation. However, despite these comparisons between mouse and human fragile site sequences, it still has been difficult to identify particular sequence elements that make these sites fragile.
Studies from two laboratories suggest that there may also be fragile sites in Saccharomyces cerevisiae. Cha and Kleckner (34) reported that yeast Mec1-deficiency results in global replication fork stalling, followed by non-random double-strand breaks (DSBs) that occur late in the S/G2-phase. These sites of breakage occur in regions that replicate slowly, called ‘replication slow zones’. Lemoine et al. (35) created yeast mutants with reduced DNA polymerase α and found increased chromosome translocations and loss. The translocation breakage sites were non-random, with a preferred site located at a pair of Ty elements on chromosome III. Although their size and distribution indicate a more simple structure, these two reported classes of unstable regions in yeast may be functionally analogous to mammalian common fragile sites and could provide unique models for understanding fragile site instability and function.
SEQUENCES AND FLEXIBILITY PEAKS AT FRAGILE SITES
Sequence analyses of cloned fragile sites did not readily reveal why these sites are unstable. All fragile sites cloned to date are relatively AT-rich (18,31,36–38) and have no expanded di- or trinucleotide repeats. Mishmar et al. (37) designed a program named FlexStab to measure local variation in the twist angle between bases and found that the FRA7H region contained more areas of high flexibility, termed ‘flexibility peaks’, than the non-fragile region. FRA2G, FRA3B, FRAXB, FRA7E and FRA16D have now been analyzed in this manner, and like FRA7H, they all contain a high number of flexibility peaks relative to non-fragile regions (18,38–40). Zlotorynski et al. (40) have shown that the flexible sequences are composed of interrupted runs of AT-dinucleotides and these sequences show similarity to the AT-rich minisatellite repeats that underlie the fragility of the rare fragile sites, FRA16B and FRA10B. Such sequences have the potential to form secondary structures and hence may affect replication at fragile sites; however, functional studies have not been performed to test this prediction.
FRAGILE SITES ARE LATE REPLICATING
Late replication is a feature of the rare fragile sites, as first shown for the fragile X site in FMR1 and later for FRAXE, FRA16B and FRA10B (41–46). A likely explanation for the delayed replication at rare fragile sites is that the expansions found at these sites, CGG and AT repeats, can form secondary structures that inhibit progression of replication forks (47,48).
Late replication is also a feature of the common fragile sites. Le Beau et al. (49) were the first to show that sequences at FRA3B replicate very late and that the addition of aphidicolin resulted in a further significant delay in replication. Perhaps even more noteworthy is the finding that ∼16.5% of FRA3B sites remained unreplicated in G2 after the addition of aphidicolin. Replication timing studies of common fragile sites, FRA16D and FRA7H, indicate that these fragile sites also may experience difficulty in replication fork progression (50,51). These studies support a model in which common fragile site regions can initiate replication in early-mid S-phase but are slow to complete replication, and the chromosomal breaks and gaps observed in metaphase cells result from unreplicated DNA (50). A high-resolution investigation of replication is needed to more clearly define the timing of origin firing and replication fork progression in fragile site regions. As other regions in the genome also replicate late, this feature alone does not define a fragile site. However, it is clear that late-replicating DNA in fragile site regions is particularly sensitive to further delay in response to replication inhibitors and as such play a role in fragile site instability.
CELL-CYCLE CHECKPOINTS ACTIVELY MONITOR FRAGILE SITES
Cells have evolved cell-cycle checkpoints to delay cell division in response to DNA lesions to allow for their repair. We have been interested in elucidating those mechanisms that recognize the fragile site lesions as a means of understanding the molecular nature of fragile sites and the cellular processes that affect their stability.
Based on the appearance of chromosome gaps and breaks at fragile sites and the methods used for their induction, the ATM and ATR kinases were primary candidates. ATM and ATR act in parallel, partially overlapping, checkpoint pathways in response to replication stress and DNA damage (52,53). ATM is activated primarily by DNA DSBs, such as those induced by ionizing irradiation (54), whereas ATR responds primarily to replication stalling such as those caused by ultraviolet light, hydroxyurea, aphidicolin and hypoxia (55–58). ATM signaling can activate the G1 and later checkpoints (52). ATR functions during the intra-S and G2/M checkpoints to stabilize, and later restart, stalled replication forks (59,60), inhibit late origin firing in the presence of stresses (60) and block cell-cycle progression into mitosis before replication is complete (57).
Casper et al. (61) have studied the effects of ATM and ATR on fragile site stability. No significant difference was found between ATM-deficient cells and normal control cells in spontaneous or aphidicolin-induced chromosome gaps or breaks, or breaks at specific fragile sites. However, ATR-deficient cells were very sensitive to aphidicolin and showed a highly significant increase in gaps and breaks at fragile sites. Importantly, fragile sites were expressed in ATR-deficient cells even without the addition of replication inhibitors. Casper et al. (62) also found that cells from individuals with Seckel syndrome that have hypomorphic mutations in ATR are sensitive to aphidicolin and show increased instability at common fragile sites. Thus, ATR is necessary for the maintenance of stability at fragile sites during normal cell divisions, as well as in cells with replication stress.
These results suggest that perturbed replication at fragile sites leads to ATR-dependent activation of cell-cycle checkpoints. Although the lack of increased fragile site expression in ATM-deficient cells suggests that DSBs are not the initial or primary cause of fragile site expression, the ATM pathway may be important in subsequent events at fragile sites, particularly in the resolution of DSBs that must sometimes occur at fragile sites in giving rise to chromosomal rearrangements. Given the overlapping functions of these two proteins, it is also possible that ATR can compensate for ATM deficiency with regard to fragile site stability, but not vice versa.
The ATR checkpoint was the first major pathway found to be associated with control of fragile site expression and this discovery was important in linking cell-cycle checkpoint function with fragile site stability. Subsequent investigations have focused on further delineation of these mechanisms, and a number of targets or modifiers of the ATR pathway have now been shown to influence fragile site stability, including BRCA1, the Fanconi anemia (FA) pathway proteins and SMC1.
BRCA1 has a number of important functions in the DNA damage response, including roles in checkpoint and repair processes. Arlt et al. (63) showed that BRCA1 is important for the maintenance of fragile site stability, specifically via its G2/M checkpoint. This result is consistent with the late replication seen at fragile sites and points to a specific checkpoint involved in fragile site stability. These data do not rule out other functions of BRCA1, such as homologous repair (HR), in common fragile site stability, but it is clear that BRCA1 helps to activate the G2/M checkpoint in response to stalled replication via ATR-dependent phosphorylation of BRCA1 serine 1423 and other residues.
Howlett et al. (64) have recently examined the role of the FA pathway in the maintenance of fragile site stability. Several lines of evidence had suggested a role for the FA pathway in the response to DNA replication stress and in the maintenance of fragile site stability. The FANCD2 protein is activated via mono-ubiquitination (mono-Ub) during S-phase, signaling its translocation to BRCA1- and RAD51-positive nuclear foci (65). It had also been shown that ATR phosphorylates FANCD2 and is required for its efficient mono-Ub (66). The FA pathway was found to be strongly activated by aphidicolin and disruption of the FA pathway resulted in a 3–5-fold increase in fragile site breaks. These results further support an important role for the FA pathway in the ATR-dependent replication stress response and also demonstrated a role in the maintenance of fragile site stability.
More recently, Musio et al. (67) reported that SMC1 deficiency increases breakage at fragile sites. SMC1 is a member of the structural maintenance of chromosomes protein family, whose members play important roles in chromosome condensation, sister chromatid cohesion and DNA repair (68). SMC1 forms a heterodimer with SMC3 to form the core of the cohesin complex. It is also a direct target of ATM phosphorylation after IR (69) and was shown by Musio et al. (67) to be phosphorylated in response to aphidicolin. This phosphorylation does not affect binding to chromatin or to SMC3 (70), but is required for S-phase checkpoint activity (71). Further studies on the relationship of SMC proteins to fragile site instability and repair should better define this relationship.
Replication is routinely arrested by a variety of stresses, and checkpoint recognition and the associated repair of these sites are essential for genome maintenance. Currently, much more is known about the checkpoint and repair responses to DNA DSBs than to stalled replication due to replication inhibition. Most studies of these checkpoint and repair pathways have used high concentrations of replication inhibitors, such as aphidicolin and hydroxyurea, which would normally completely block replication. Fragile sites are induced at much lower concentrations that only partially inhibit replication and that may be more relevant to natural genotoxic exposure levels. Thus, fragile sites provide a novel cytological assay to help further delineate these checkpoints and associated DNA repair pathways in mammalian cells.
DNA REPAIR AT FRAGILE SITES
Little is known about how lesions at fragile sites are repaired. Most studies of repair responses have focused on DNA DSBs, whereas little is known about the repair of stalled replication forks or lesions resulting from replication stress that occur at fragile sites. HR plays the major role in responding to DSBs and stalled or collapsed replication forks during S and G2, when the sister chromatid is present. Interestingly, Glover and Stein (5) reported that, on average, 70% of all gaps and breaks at FRA3B after aphidicolin treatment had an SCE at that site. SCEs were also observed at FRA3B sites that were not broken, and the percentage of unbroken FRA3B sites with an SCE doubled after aphidicolin treatment. The presence of the SCEs at fragile sites is an indication that some type of repair has occurred at the site. This result is not surprising as the checkpoint response to cellular stress during replication is tightly linked to DNA repair. The molecular basis for formation of SCEs in mammalian cells is not well-understood, but it has been hypothesized that SCEs are formed by the action of HR during replication repair. Cells with mutations in RAD51 and other members of the HR pathway have been reported to have reduced spontaneous and MMC-induced SCE formation, although the reduction is not severe and may even be species specific (72–78). Although these findings suggest a role for HR in repair at fragile sites, there is no direct evidence for any protein involved in HR being involved in the maintenance of fragile site stability.
FRAGILE SITE INSTABILITY IN TUMOR CELLS
A number of early fragile site studies attempted to correlate the location of fragile sites with the sites of frequent chromosome breaks and rearrangements in cancers. These attempts were hampered by the large number of both fragile sites and chromosome breakpoints in tumors and the relatively low resolution of banded chromosomes. However, with the cloning of fragile sites, numerous studies have shown that common fragile sites are unstable in tumor cells, albeit not necessarily in the ways originally proposed.
Most such studies have focused on FRA3B and FRA16D due to the fact that they are the two most frequently expressed and best-characterized fragile sites, and both lie within large tumor suppressor genes. FRA3B frequently shows allelic loss or homozygous deletions in many tumor types including lung, digestive tract, kidney and breast (79–81). The rearrangements are usually one or more large deletions of tens to hundreds of kilobases directly within the fragile site region. Investigation of chromosome 3 homologs from tumor cell lines showed multiple variable deletions within FRA3B, thus suggesting ongoing instability in the region (82). Only a few translocations with breakpoints in FRA3B have been reported (83–85). Whether this represents a form of ascertainment bias or is a true reflection of the frequency of translocations at fragile sites is not known. The deletions at FRA3B inactivate the FHIT gene, and partly based on these deletions, FHIT was suggested to be a tumor suppressor gene. More recent functional studies strengthen this hypothesis. FHIT-deficient mice have increased NMBA-induced gastric tumors (86) and this susceptibility can be rescued by the introduction of a functional FHIT allele. In addition, overexpression of FHIT has been shown to suppress the growth of different cancer cell lines both in vitro and in vivo (87). Thus, loss of FHIT appears to play a role in mutagen sensitivity and cancer susceptibility.
FRA16D maps within regions of frequent loss-of-heterozygosity (LOH) in breast and prostate cancers and is associated with homozygous deletions in various adenocarcinomas and with chromosomal translocations in multiple myeloma. These rearrangements inactivate the WWOX gene, which is defined as a tumor suppressor gene by the virtue of these large, intragenic, homozygous deletions and point mutations in cancers and its ability to suppress the growth of mammary tumors in mice, possibly via a role in apoptosis pathways (18,88–91). Similar patterns of deletions in cancer cells have also been shown for other common fragile sites including FRA6E (20), FRA9E (92) and FRA7G (19,93).
It is clear from numerous studies that FRA3B, FRA16D and other common fragile sites contain large, intralocus deletions or translocations that frequently inactivate associated genes, such as FHIT and WWOX, in a variety of tumor cells (94). The type of mutations primarily seen in these genes in tumors, large deletions and translocations, as opposed to point mutations, supports the involvement of the associated fragile sites in the mutations. This raises the important question of whether the deletions are simply a result of chromosome instability at fragile sites or the selection for cells with loss of gene function or a combination of both. A number of tumors have been identified with deletions in more than one fragile site, suggesting a common mechanism (38,95). In addition, fragile site instability in tumors appears not to be driven solely by associated genes, because FRAXB at Xp23.3 also shows frequent deletions in tumor cells, yet none of the genes identified at FRAXB is involved in tumor progression (38). However, given the function of FHIT and WWOX in tumorigenesis, it seems likely that at least in some tumors, selection for loss of function driven by fragile site instability plays a role in tumor progression.
In addition to deletions, instability at fragile sites has also been implicated in gene amplification. Coquelle et al. (13) have shown that breakage at fragile sites can initiate breakage– fusion–bridge cycles, a mechanism responsible for the accumulation of intrachromosomal amplicons, in drug-resistant mutant CHO cells. More recently, FRA7I and FRA7G have been identified at one boundary of the amplicons found in two tumor cell lines (96). We recently characterized the boundaries of amplicons involving the MET oncogene in six primary esophageal adenocarcinomas (97) and found that all have one boundary within FRA7G, consistent with the mechanisms proposed by Coquelle and co-workers.
Why are fragile site regions unstable in these tumor cells but not in normal cells? Casper et al. (61) suggested that breaks at fragile sites may serve as ‘signatures’ of stalled replication in tumor cells caused by mutations or perturbations of the S-phase and G2/M checkpoints or associated repair genes during tumor progression. Two recent reports support this concept and experimentally tie together what is known about the mechanisms leading to fragile site expression and chromosomal rearrangements at fragile sites in tumors. Gorgoulis et al. (98) and Bartkova et al. (99) examined bladder, breast, colorectal and lung tumors at various stages of progression for signs of a DNA damage response. Both found that early stages of cancer development, before malignancy, are associated with an active DNA damage response, including phosphorylated ATR, ATM, CHK1 and CHK2 kinases, and phosphorylated histone H2AX and p53. Concomitant with these events was a high frequency of LOH at known fragile site regions, including FRA3B. These findings suggest that in precancerous lesions, replication stress leads to stalled or collapsed replication forks, activation of the ATR and with subsequent DNA DSBs, the ATM checkpoints. In cells that do not undergo apoptosis or cell-cycle arrest, allelic imbalances will preferentially target fragile sites as they are most sensitive to replication stress. Further mutation in p53 or other genes will release additional checkpoints and lead to progression. These findings suggest that lesions at common fragile sites are indicators of replication stress during early stages of tumorigenesis. They are also consistent with a role for some fragile site-associated genes in cancer.
A MODEL FOR COMMON FRAGILE SITES AND FUTURE DIRECTIONS
Data from a number of laboratories support a model for common fragile sites. This model (Fig. 2) is based on late or delayed replication, unusual sequence composition and the role of checkpoint proteins in fragile site stability. It suggests that sequences at fragile sites present difficulties to replication, which are further exacerbated by aphidicolin and certain other forms of replication stress. Given that aphidicolin, an inhibitor of pol α, is the most specific inducer of common fragile sites, it is likely that lagging strand synthesis is centrally involved. A perturbation of lagging strand synthesis could result in the formation of unreplicated regions, and perhaps sequence-specific secondary structures (40), behind an advancing helicase (100,101). Alternatively, inhibition of leading strand synthesis could lead to stalled or collapsed replication forks following replication stress. The single-stranded, unreplicated regions are coated with the ssDNA binding protein RPA and stimulate the activation of the S-phase and/or G2/M checkpoints, in which ATR plays a key role. However, the appearance of fragile sites on metaphase chromosomes suggests that many of these lesions can escape these checkpoint controls.
Thus, fragile site gaps are believed to represent unreplicated regions or single-stranded gaps on metaphase chromosomes. Deletions or translocations at fragile sites could result from DNA DSBs caused by the breakage of already weakened single-stranded regions or aberrant processing of Holliday junctions at damaged forks. Thus, the deletions seen in tumor cells are proposed to arise from unequal or faulty HR of stalled forks and would be enhanced by mutations in the replication checkpoint or associated repair pathways. In this way, deletions at fragile sites in tumor cells are ‘signatures’ of replication stress.
Largely because of the identification of frequent fragile site instability in tumor cells and the link to important cell-cycle checkpoint pathways, the study of common fragile sites has become increasingly significant over the past few years, and a great deal has been learned about their genomic structure and mechanisms of instability. However, many questions remain. These include: Why are fragile site regions ‘fragile?’ What are the dynamics of fragile site replication and repair? What forms of replication stress in vivo lead to frequent fragile site deletions in tumor cells? Do fragile sites have a conserved function, possibly one tied to cell-cycle progression? The answers to these and other questions are important to fully understand the normal functional significance of these conserved regions and the nature of their fragility in normal and cancer cells.
ACKNOWLEDGEMENTS
We thank Niall Howlett and Ryan Ragland for helpful discussions and Cynthia Gaffney for assistance. T.W.G. is supported by NIH grant CA43222.
Conflict of Interest statement. None declared.
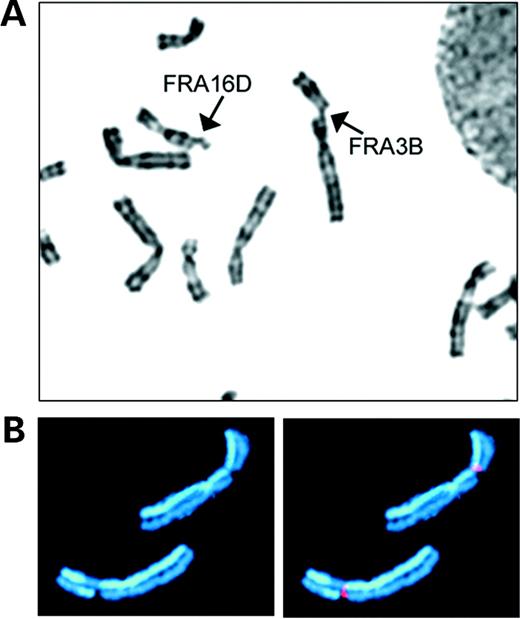
Figure 1. Examples of common fragile sites. (A) Human G-banded metaphase chromosomes with breaks at fragile sites FRA3B and FRA16D (arrows). (B) An example of an SCE at a fragile site. Both homologs of chromosome 3 with breaks at FRA3B were stained to show SCEs. The chromosome at the bottom has an SCE at the fragile site. A yeast artificial chromosome mapping to FRA3B was used as a fluorescence in situ hybridization probe (red) to identify breakage at the fragile site.
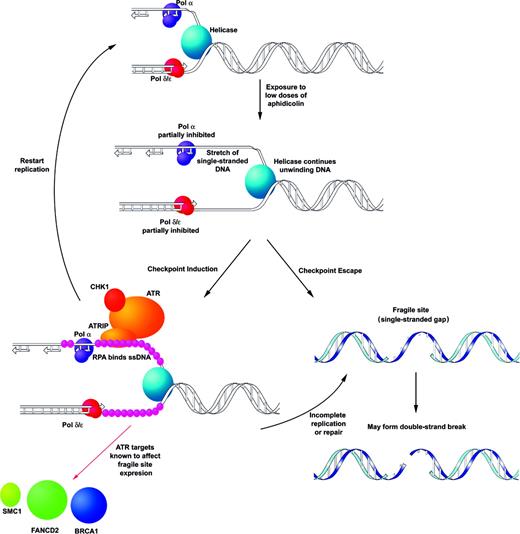
Figure 2. Model for instability at common fragile sites. This model predicts that fragile sites are regions of unreplicated (single stranded) DNA that arise from delay or stalling of replication in these regions, in this case, owing to low doses of aphidicolin. The single-stranded, unreplicated regions are coated with the ssDNA binding protein RPA leading to the activation of the S-phase and/or G2/M checkpoints, in which ATR plays a key role. The ATR/ATRIP complex responding to replication delay on the lagging strand and target proteins, BRCA1, FANCD2 and SMC1, which have been shown to affect fragile site stability are shown. The appearance of fragile sites on metaphase chromosomes implies that these lesions can occasionally escape checkpoint control. The result is an unreplicated region that, if not properly repaired, can secondarily lead to DSBs.
Genes associated with characterized, common fragile sites
| Fragile site | Location | Associated genes | References |
|---|---|---|---|
| FRA2G | 2q31 | IGRP, RDHL, LRP2 and others | (39) |
| FRA3B | 3p14.2 | FHIT | (16) |
| FRA4F | 4q22 | GRID2 | (21) |
| FRA6E | 6q26 | PARKIN, MAP3K4, LPA and others | (20) |
| FRA6F | 6q21 | REV3L, DIF13, FKHRL1 and others | (102) |
| FRA7E | 7q21.11 | LEP | (40) |
| FRA7G | 7q31.2 | CAV1, CAV2, TESTIN and MET | (19,93) |
| FRA7H | 7q32.3 | None identified | (37) |
| FRA9E | 9q32-33.1 | PAPPA, ROD1, KLF4 and others | (92) |
| FRA16D | 16q23.3 | WWOX | (18,19) |
| FRAXB | Xp22.3 | STS | (38) |
| Fragile site | Location | Associated genes | References |
|---|---|---|---|
| FRA2G | 2q31 | IGRP, RDHL, LRP2 and others | (39) |
| FRA3B | 3p14.2 | FHIT | (16) |
| FRA4F | 4q22 | GRID2 | (21) |
| FRA6E | 6q26 | PARKIN, MAP3K4, LPA and others | (20) |
| FRA6F | 6q21 | REV3L, DIF13, FKHRL1 and others | (102) |
| FRA7E | 7q21.11 | LEP | (40) |
| FRA7G | 7q31.2 | CAV1, CAV2, TESTIN and MET | (19,93) |
| FRA7H | 7q32.3 | None identified | (37) |
| FRA9E | 9q32-33.1 | PAPPA, ROD1, KLF4 and others | (92) |
| FRA16D | 16q23.3 | WWOX | (18,19) |
| FRAXB | Xp22.3 | STS | (38) |
Genes associated with characterized, common fragile sites
| Fragile site | Location | Associated genes | References |
|---|---|---|---|
| FRA2G | 2q31 | IGRP, RDHL, LRP2 and others | (39) |
| FRA3B | 3p14.2 | FHIT | (16) |
| FRA4F | 4q22 | GRID2 | (21) |
| FRA6E | 6q26 | PARKIN, MAP3K4, LPA and others | (20) |
| FRA6F | 6q21 | REV3L, DIF13, FKHRL1 and others | (102) |
| FRA7E | 7q21.11 | LEP | (40) |
| FRA7G | 7q31.2 | CAV1, CAV2, TESTIN and MET | (19,93) |
| FRA7H | 7q32.3 | None identified | (37) |
| FRA9E | 9q32-33.1 | PAPPA, ROD1, KLF4 and others | (92) |
| FRA16D | 16q23.3 | WWOX | (18,19) |
| FRAXB | Xp22.3 | STS | (38) |
| Fragile site | Location | Associated genes | References |
|---|---|---|---|
| FRA2G | 2q31 | IGRP, RDHL, LRP2 and others | (39) |
| FRA3B | 3p14.2 | FHIT | (16) |
| FRA4F | 4q22 | GRID2 | (21) |
| FRA6E | 6q26 | PARKIN, MAP3K4, LPA and others | (20) |
| FRA6F | 6q21 | REV3L, DIF13, FKHRL1 and others | (102) |
| FRA7E | 7q21.11 | LEP | (40) |
| FRA7G | 7q31.2 | CAV1, CAV2, TESTIN and MET | (19,93) |
| FRA7H | 7q32.3 | None identified | (37) |
| FRA9E | 9q32-33.1 | PAPPA, ROD1, KLF4 and others | (92) |
| FRA16D | 16q23.3 | WWOX | (18,19) |
| FRAXB | Xp22.3 | STS | (38) |
References
Kremer, E.J., Pritchard, M., Lynch, M., Yu, S., Holman, K., Baker, E., Warren, S.T., Schlessinger, D., Sutherland, G.R. and Richards, R.I. (
Sutherland, G.R., Baker, E. and Richards, R.I. (
Glover, T.W., Berger, C., Coyle, J. and Echo, B. (
Yunis, J.J. and Soreng, A.L. (
Glover, T.W. and Stein, C.K. (
Glover, T.W. and Stein, C.K. (
Wang, N.D., Testa, J.R. and Smith, D.I. (
Rassool, F.V., McKeithan, T.W., Neilly, M.E., van Melle, E., Espinosa, R., III and Le Beau, M.M. (
Wilke, C.M., Hall, B.K., Hoge, A., Paradee, W., Smith, D.I. and Glover, T.W. (
De Braekeleer, M., Sreekantaiah, C. and Haas, O. (
Popescu, N.C. and DiPaolo, J.A. (
Smith, P.P., Friedman, C., Bryant, E.M. and McDougall, J.K. (
Coquelle, A., Pipiras, E., Toledo, F., Buttin, G. and Debatisse, M. (
Wilke, C.M., Guo, S.W., Hall, B.K., Boldog, F., Gemmill, R.M., Chandrasekharappa, S.C., Barcroft, C.L., Drabkin, H.A. and Glover, T.W. (
Boldog, F.L., Waggoner, B., Glover, T.W., Chumakov, I., Le Paslier, D., Cohen, D., Gemmill, R.M. and Drabkin, H.A. (
Ohta, M., Inoue, H., Cotticelli, M.G., Kastury, K., Baffa, R., Palazzo, J., Siprashvili, Z., Mori, M., McCue, P., Druck, T. et al. (
Thorland, E.C., Myers, S.L., Persing, D.H., Sarkar, G., McGovern, R.M., Gostout, B.S. and Smith, D.I. (
Ried, K., Finnis, M., Hobson, L., Mangelsdorf, M., Sayan, S., Nancarrow, J.K., Woollatt, E., Kremmidiotis, G., Gardner, A., Venter, D. et al. (
Bednarek, A.K., Laflin, K.J., Daniel, R.L., Liao, Q., Hawkins, K.A. and Aldaz, C.M. (
Denison, S.R., Callahan, G., Becker, N.A., Phillips, L.A. and Smith, D.I. (
Rozier, L., El-Achkar, E., Apiou, F. and Debatisse, M. (
Stone, D.M., Stephens, K.E. and Doles, J. (
Stone, D.M., Jacky, P.B., Hancock, D.D. and Prieur, D.J. (
Elder, F.F.B. and Robinson, T.J. (
Smeets, D.F.C.M. and van de Klundert, F.A.J.M. (
Yang, M.Y. and Long, S.E. (
McAllister, B.F. and Greenbaum, I.F. (
Ruiz-Herrera, A., Garcia, F., Fronicke, L., Ponsa, M., Egozcue, J., Caldes, M.G. and Stanyon, R. (
Soulie, J. and De Grouchy, J. (
Glover, T.W., Hoge, A.W., Miller, D.E., Ascara-Wilke, J.E., Adam, A.N., Dagenais, S.L., Wilke, C.M., Dierick, H.A. and Beer, D.G. (
Shiraishi, T., Druck, T., Mimori, K., Flomenberg, J., Berk, L., Alder, H., Miller, W., Huebner, K. and Croce, C.M. (
Matsuyama, A., Shiraishi, T., Trapasso, F., Kuroki, T., Alder, H., Mori, M., Huebner, K. and Croce, C.M. (
Krummel, K.A., Denison, S.R., Calhoun, E., Phillips, L.A. and Smith, D.I. (
Cha, R.S. and Kleckner, N. (
Lemoine, F.J., Degtyareva, N.P., Lobcchev, K. and Petes, T.D. (
Boldog, F., Gemmill, R., West, J., Robinson, M. and Li, E. (
Mishmar, D., Rahat, A., Scherer, S.W., Nyakatura, G., Hinzmann, B., Kohwi, Y., Mandel-Gutfroint, Y., Lee, J.R., Drescher, B., Sas, D.E. et al. (
Arlt, M.F., Miller, D.E., Beer, D.G. and Glover, T.W. (
Limongi, M.Z., Pelliccia, F. and Rocchi, A. (
Zlotorynski, E., Rahat, A., Sakaug, J., Ben-Porat, N., Ozeri, E., Herschberg, R., Levi, A., Scherer, S.W., Margalit, H. and Kerem, B. (
Ohashi, H., Kuwano, A., Tsukahara, M., Arinami, T. and Kajii, T. (
Yu, W.D., Wenger, S.L. and Steele, M.W. (
Hansen, R.S., Canfield, T.K., Lamb, M.M., Gartler, S.M. and Laird, C.D. (
Hansen, R.S., Canfield, T.K., Fjeld, A.D., Mumm, S., Laird, C.D. and Gartler, S.M. (
Subramanian, P.S., Nelson, D.L. and Chinault, A.C. (
Handt, O., Baker, E., Dayan, S., Gartler, S.M., Woollatt, E., Richards, R.I. and Hansen, R.S. (
Chen, X., Mariappan, S.V., Catasti, P., Ratliff, R., Moyzis, R.K., Laayoun, A., Smith, S.S., Bradbury, E.M. and Gupta, G. (
Hewett, D.R., Handt, O., Hobson, L., Mangelsdorf, M., Eyre, H.J., Baker, E., Sutherland, G.R., Schuffenhauer, S., Mao, J.I. and Richards, R.I. (
Le Beau, M.M., Rassool, F.V., Neilly, M.E., Espinosa, R., III, Glover, T.W., Smith, D.I. and McKeithan, T.W. (
Palakodeti, A., Han, Y., Jiang, Y. and Le Beau, M.M. (
Hellman, A., Rahat, A., Scherer, S.W., Darvasi, A., Tsui, L.-C. and Kerem, S. (
Abraham, R.T. (
Durocher, D. and Jackson, S.P. (
Pincheira, J., Bravo, M., Navarrete, M.H., Marcelain, K., López-Sáez, J.F. and de la Torre, C. (
Cliby, W.A., Roberts, C.J., Cimprich, K.A., Stringer, C.M., Lamb, J.R., Schreiber, S.L. and Friend, S.H. (
Cortez, D., Guntuku, S., Qin, J. and Elledge, S.J. (
Nghiem, P., Park, P.K., Kim, Y.-S., Vaziri, C. and Schreiber, S.L. (
Hammond, E.M., Denko, N.C., Dorie, M.J., Abraham, R.T. and Giaccia, A.J. (
Lopes, M., Cotta-Ramusino, C., Pellicioli, A., Liberi, G., Plevani, P., Muzi-Falconi, M., Newlon, C.S. and Foiani, M. (
Tercero, J.A. and Diffley, J.F.X. (
Casper, A.M., Nghiem, P., Arlt, M.F. and Glover, T.W. (
Casper, A.M., Durkin, S.G., Arlt, M.F. and Glover, T.W. (
Arlt, M.F., Xu, B., Durkin, S.G., Casper, A.M., Kastan, M.B. and Glover, T.W. (
Howlett, N.G., Taniguchi, T., Durkin, S.G., D'Andrea, A.D. and Glover, T.W. (
Taniguchi, T. and D'Andrea, A.D. (
Andreassen, P.R., D'Andrea, A.D. and Taniguchi, T. (
Musio, A., Montagna, C., Mariani, T., Tilenni, M., Focarelli, M.L., Brait, L., Indino, E., Benedetti, P.A., Chessa, L., Albertini, A. et al. (
Losada, A. and Hirano, T. (
Kim, S.-T., Xu, B. and Kastan, M.B. (
Yazdi, P.T., Wang, Y., Zhao, S., Patel, N., Lee, E.Y.-H.P. and Qin, J. (
Kitagawa, R., Bakkenist, C.J., McKinnon, P.J. and Kastan, M.B. (
Dronkert, M.L.G., Beverloo, H.B., Johnson, R.D., Hoeijmakers, J.H.J., Jasin, M. and Kanaar, R. (
Fuller, L.F. and Painter, R.B. (
Lambert, S. and Lopez, B.S. (
Sasaki, M.S., Takata, M., Sonoda, E., Tachibana, A. and Takeda, S. (
Stark, J.M., Hu, P., Pierce, A.J., Moynahan, M.E., Ellis, N. and Jasin, M. (
Takata, M., Sasaki, M.S., Tachiiri, S., Fukushima, T., Sonoda, E., Schild, D., Thompson, L.H. and Takeda, S. (
Tucker, J.D., Jones, N.J., Allen, N.A., Minkler, J.L., Thompson, L.H. and Carrano, A.V. (
Druck, T., Hadaczek, P., Fu, T.-B., Ohta, M., Siprashvili, Z., Baffa, R., Negrini, M., Kastury, K., Veronese, M.L., Rosen, D. et al. (
Michael, D., Beer, D.G., Wilke, C.W., Miller, D.E. and Glover, T.W. (
Mimori, K., Druck, T., Inoue, H., Alder, H., Berk, L., Mori, M., Huebner, K. and Croce, C.M. (
Corbin, S., Neilly, M.E., Espinosa, R., III, Davis, E.M., McKeithan, T.W. and Le Beau, M.M. (
Geurts, J.M.W., Schoenmakers, E.F.P.M., Roijer, E., Stenman, G. and Van de Ven, W.J.M. (
Gemmill, R.M., West, J.D., Boldog, F., Tanaka, N., Robinson, L.J., Smith, D.I., Li, F. and Drabkin, H.A. (
Fang, J.M., Dagenais, S.L., Erickson, R.P., Arlt, M.F., Glynn, M.W., Gorski, J.L., Seaver, L.H. and Glover, T.W. (
Zanesi, N., Fidanza, V., Fong, L.Y., Mancini, R., Druck, T., Valtieri, M., Rudiger, T., McCue, P.A., Croce, C.M. and Huebner, K. (
Siprashvili, Z., Sozzi, G., Barnes, L.D., McCue, P., Robinson, A.K., Eryomin, V., Sard, L., Tagliabue, E., Greco, A., Fusetti, L. et al. (
Paige, A.J.W., Taylor, K.J., Taylor, C., Hillier, S.G., Farrington, S., Scott, D., Porteous, D.J., Smyth, J.F., Gabra, H. and Watson, J.E.V. (
Bednarek, A., Keck-Waggoner, C.L., Daniel, R.L., Laflin, K.J., Bergsagel, P.L., Kiguchi, K., Brenner, A.W. and Aldaz, C.M. (
Mangelsdorf, M., Ried, K., Woollatt, E., Dayan, S., Eyre, H., Finnis, M., Hobson, L., Nancarrow, J., Venter, D., Baker, E. et al. (
Paige, A.J.W., Taylor, K.J., Stewart, A., Sgouros, J.G., Gabra, H., Sellar, G.C., Smyth, J.F., Porteous, D.J. and Watson, J.E.V. (
Callahan, G., Denison, S.R., Phillips, L.A., Shridhar, V. and Smith, D.I. (
Huang, H., Qian, J., Proffit, J., Wilber, K., Jenkins, R. and Smith, D.I. (
Huebner, K. and Croce, C.M. (
Finnis, M., Dayan, S., Hobson, L., Chenevix-Trench, G., Friend, K., Ried, K., Venter, D., Woollatt, E., Baker, E. and Richards, R.I. (
Hellman, A., Zlotorynski, E., Scherer, S.W., Cheung, J., Vincent, J.B., Smith, D.I., Trakhtenbrot, L. and Kerem, B. (
Miller, C.T., Lin, L., Casper, A.M., Lim, J., Thomas, D.G., Orringer, M.B., Chang, A.C., Chambers, A.F., Giordano, T.J., Glover, T.W. et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET signaling pathways in esophageal adenocarcinoma,
Gorgoulis, V.G., Vassiliou, L.V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., Venere, M., Ditullio, R.A., Jr, Kastrinakis, N.G., Levy, B. et al. (
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., Guldberg, P., Sehested, M., Nesland, J.M., Lukas, C. et al. (
Byun, T.S., Pacek, M., Yee, M.C., Walter, J.C. and Cimprich, K.A. (
Pacek, M. and Walter, J.C. (

{kind=link}
{kind=link}