



Abstract
Huntington's disease is a devastating neurodegenerative condition associated with the formation of intraneuronal aggregates by mutant huntingtin. Aggregate formation is a property shared by the nine related diseases caused by polyglutamine codon expansion mutations and also by other neurodegenerative conditions like Parkinsons's disease. The roles of aggregates and aggregation in these diseases have been a subject of heated controversy. Here, we have addressed the question in vivo by generating a new transgenic mouse overexpressing the yeast chaperone hsp104, as hsp104 overexpression reduced mutant huntingtin aggregation and toxicity in cell models. Hsp104 has no close mammalian orthologues and does not appear to have effects on mammalian cell death pathways. We crossed hsp104 transgenic mice with mice expressing the first 171 residues of mutant huntingtin. Hsp104 reduced aggregate formation and prolonged the lifespan of the HD mice by 20%. This protection may be mediated at the level of changing the conformation of a putative toxic monomer, reducing oligomerization or aggregation, reducing the levels of oligomeric species or aggregates or combinations of these non-mutually exclusive possibilities.
INTRODUCTION
Huntington's disease (HD) is a devastating autosomal dominant neurodegenerative condition that typically strikes in middle age and is characterized by movement disorders, cognitive decline and psychiatric symptoms (1). HD is caused by the abnormal expansion of a CAG trinucleotide repeat tract to more than 35 repeats. The CAG trinucleotides in the gene are translated into a polyglutamine stretch. HD is the most prevalent of nine known diseases caused by CAG/polyglutamine repeat expansions. These diseases are believed to be caused predominantly by gain-of-function mechanisms. The pathological hallmark of HD and other polyglutamine diseases are the presence of intracellular aggregates formed by the mutant proteins (2,3).
The formation of intraneuronal aggregates is a feature of many other neurodegenerative disorders such as Alzheimer's disease and Parkinson's disease. The proteins causing these diseases are unrelated in size, sequence or function, but share a common propensity to misfold and accumulate into amyloid-like structures. When first discovered, aggregates were believed to be responsible for cell death but new data have argued that they may be inert or even protective (4–6). While the roles of aggregates are controversial, it has also been postulated that the intermediate oligomeric states that occur during the aggregation process may be pathogenic (7). Furthermore, the aggregation process itself, rather than the aggregates themselves, may be toxic.
Aggregates recruit molecular chaperones in the human diseases and in cell (8) and mouse disease models (9–11). These molecular chaperones, like hsp70 and hsp40 family members, play key roles by preventing aggregation or by refolding misfolded intracellular proteins. Overexpression of different molecular chaperones, especially heat shock proteins (hsp), has been used to rescue cell death caused by proteins with abnormal polyglutamine expansions in tissue culture models. For instance, the chaperones hsp40 (HDJ-1) and hsp70 suppress polyglutamine aggregation and reduce nuclear fragmentation in SK-N-SH cells transfected with the first exon of huntingtin containing 74 glutamines (74Q) (12). Hsp40 and hsp70 have also been used in a cell model of spinal bulbar muscular atrophy, causing a decrease in aggregate load and apoptosis in Neuro2a cells transiently transfected with a truncated and expanded (97Q) androgen receptor (13).
Hsp70, in particular, has been studied in different animal models. This chaperone reduces polyglutamine toxicity in Drosophila (14) where it acts in synergy with hsp40 (15) but had no obvious effect on aggregate content. However, in mouse models of polyglutamine diseases, the results of hsp70 overexpression are not consistent. High expression levels of hsp70, at 50–100 times the endogenous levels, have a protective effect on neurodegeneration and rescue behavioural phenotypes in a mouse model of spinocerebellar ataxia 1 (16). However, Hay et al. (17) showed that hsp70 overexpression resulted in a transient delay in aggregate formation but failed to attenuate the behavioural phenotype in R6/2 HD mice. In this study, the increase in hsp70 expression was not quantified. In another study (18), a 5- to 15-fold increase in hsp70 expression slowed weight loss in double transgenics compared with HD animals but did not rescue aggregate load, neurodegeneration or the clasping phenotype. Likewise, overexpression of hsp70 with hsp40 had no effect on aggregate load or neurotoxicity in the retinas of mice with another polyglutamine disease, spinocerebellar ataxia type 7 (19). One possible reason for the failure of certain studies to observe effects may be due to the need for chaperones to operate with co-chaperones and the observation that levels of hsp70 and co-chaperones like Hdj1and Hdj2 progressively decrease over time in HD mice (17).
Hsp70 can act as a molecular chaperone aiding protein folding, but also has direct anti-apoptotic roles [e.g. by inhibiting apoptosome formation (20)]. Thus, when beneficial effects of overexpression of this chaperone are seen in polyglutamine disease, it is difficult to infer that hsp70 is protecting simply by reducing aggregation. We aimed to test whether overexpression of a molecular chaperone (that does not have anti-apoptotic effects) could reduce polyglutamine aggregation and toxicity. For these experiments, we used hsp104, an essential chaperone involved in thermotolerance in Saccharomyces cerevisiae. Hsp104, which has no orthologue in the animal genome, forms a large hexameric ring structure (21,22) and can recycle previously aggregated proteins (23). Hsp104 has been shown to rescue aggregate formation in yeast (24), Caenorhabditis elegans (25) and in human leukemic T cell lines (26). Hsp104 overexpression reduced aggregation and toxicity caused by the first exon of huntingtin containing 74Q or polyalanine-expanded proteins in cell models (27,28). However, hsp104 does not appear to have anti-apoptotic properties, in the context of H2O2 or staurosporine insults in mammalian cells (28). As these cell model experiments suggested a correlation between reducing aggregation and lower toxicity of mutant huntingtin fragments, we tested whether the same occurred in vivo using the N171-82Q mice, which express the first 171 amino acids of mutant huntingtin under the control of the mouse prion protein promoter (MoPrP) (29). This promoter restricts expression of the protein mainly to the brain. N171-82Q mice exhibit a clear behavioural phenotype, including a loss of motor coordination and tremors. N171-82Q mice are also characterized by the presence of intranuclear aggregates in the brain and premature death. We have generated the first transgenic mouse expressing hsp104 under the control of the MoPrP promoter and crossed them with N171-82Q mice. Hsp104 expression in this HD mouse model decreased aggregate load in the brain and increased survival of the animals.
RESULTS
Generation of hsp104 transgenic mice
We generated hsp104 transgenic mice by standard pronuclear injections of DNA in FvB embryos using hsp104 constructs driven by the MoPrP, the same promoter that drives transgene expression in the N171-82Q HD mice. Prior to injection, the constructs were validated by DNA sequencing and western blotting after expression in cell lines (data not shown). Mice were genotyped by PCR and Southern blot (data not shown). To confirm expression of the transgene, western blot analyses were performed on different tissues using an anti-hsp104 antibody. As expected from patterns of expression obtained from previous transgenes driven by the MoPrP in mice (30), hsp104 protein was strongly expressed in brain (including the cerebellum), heart, kidney and testis (Fig. 1A, H, B and D). The expression of the transgene in the other tested tissues, muscle, spleen, liver and lung (Fig. 1C, E, F and G) was below the limits of detection. As hsp104 acts in conjunction with hsp70 in yeast and transfected human cells (26), we examined whether the expression of hsp104 had any effect on the endogenous levels of hsp70 in the mouse brain. Western blots of whole brain lysates of 14-week-old transgenic mice and non-transgenic littermates confirmed that endogenous levels of hsp70 were not different between hsp104 transgenic animals and their non-transgenic littermates (Fig. 1I).
Hsp104 expression does not affect the levels of soluble mutant huntingtin in mouse brain
To generate N171-82Q mice expressing hsp104, we mated either Bl6/C3Hf1 heterozygous males transgenic for mutant huntingtin and FvB females expressing hsp104 or the converse. The offspring consisted of non-transgenic (NT), HD only (HD), HDxhsp104 double transgenic (Db Tg) or hsp104-only. Western blots of whole brain lysates from 13-week-old mice were probed using an antibody directed against the expanded polyglutamine sequence (1C2). The levels of soluble mutant huntingtin showed some mouse-to-mouse variability (possibly due to aggregation/oligomerization), but we did not detect any difference in huntingtin transgene expression between double transgenic animals and mice expressing only the mutant huntingtin (Fig. 2A and B). We then examined whether hsp104 expression had any effect on the endogenous levels of hsp in the mutant huntingtin-expressing mice. In both HD only and double transgenic animals, hsp25 levels showed a non-significant tendency to be reduced, compared with the hsp25 levels in non-transgenic animals. The levels of hsp70 were not different between double transgenic and mice expressing only the mutant huntingtin but were significantly lower than in non-transgenic animals. This is not surprising as lower levels of hsp70 and hsp40 have been previously reported in another mouse model of HD (17). Hsp104 expression in HD mice did not modify the levels of either endogenous hsp70 or hsp25 (Fig. 2C–F).
Hsp104 expression reduces the number of aggregates in the piriform cortex of HD mice
One of the neurological hallmarks of HD is the presence of aggregates in the striatum and deeper layers of the cortex, and at later stages in the hippocampus and hypothalamus. Immunohistochemistry was performed on coronal sections from HD mice, double transgenic animals and non-transgenic littermates. On coronal sections, the piriform cortex represents a well-defined region, and its size allows one to count aggregates in the whole area. At 14 weeks, the piriform cortex of double transgenic mice contained fewer aggregates than in the HD-only animals (Fig. 3A). We did not detect any obvious differences of aggregate size or shape between HD and double transgenic animals (Fig. 3B). At 14 weeks, we could not detect any aggregates in the striatum. At 22 weeks, the number of aggregates in the piriform cortex was too high for accurate quantification. However, we still saw a trend towards fewer aggregates in double transgenic mice compared to HD mice (Supplementary Material, Fig. S1A). At 22 weeks, there were very few aggregates in the striatum and again the double transgenic animals showed a non-significant trend towards having fewer aggregates than their HD counterparts (Supplementary Material, Fig. S1B). As the anti-hsp104 antibody gives a high non-specific background with immunohistochemistry, it was not possible to examine whether the hsp104 protein was recruited into the aggregates.
Hsp104 does not improve the motor phenotype in HD mice
A decrease in motor performances occurring during the evolution of the disease has been well characterized in N171-82Q mice (29). We tested mice at 4 weeks of age to estimate baseline motor performances. Mice were then tested every other week to assess the evolution of their motor impairment, from 12 weeks old, when N171-82Q mice show a clear difference in motor performances compared with non-transgenic littermates. Grip strength was evaluated by two methods, a qualitative SHIRPA test from 12 to 24 weeks old (Table 1) (Supplementary Material, Fig. S2A–H) and a quantitative determination of the strength using a grip strength meter from 14 to 24 weeks old (Table 1) (Supplementary Material, Fig. S4A–F). Both methods suggested that grip strength was similar in HD mice with or without hsp104 expression (Table 1) (Supplementary Material, Figs S2 and S4). The mice were also tested for their capacity to climb back on a horizontal wire when hung on the wire by their forelimbs, the ‘wire manoeuvre’. HD and double transgenic showed worsening performances with aging but the expression of hsp104 did not rescue the wire manoeuvre phenotype in HD mice (Table 1) (Supplementary Material, Fig. S6A–H). We also evaluated the motor functions using an accelerating Rotarod apparatus. Double transgenic mice and HD mice performances decreased between 4 and 12 weeks old (Table 1) (Supplementary Material, Fig. S8). However, hsp104 expression did not rescue the loss in Rotarod performances (Table 1) (Supplementary Material, Fig. S8). Non-transgenic and hsp104-only mice were tested at the same time points as the double transgenic and HD mice. There were no differences between non-transgenic and hsp104-only animals on grip strength (Supplementary Material, Figs S3 and S5), ‘wire manoeuvre’ (Supplementary Material, Fig. S7) or Rotarod (Supplementary Material, Fig. S9).
Hsp104 expression does not prevent weight loss in HD animals
N171-82Q mice typically start to lose weight by 4 months of age (29). We started weighing the mice immediately after weaning, at 4 weeks old, then three times per week until the death of the animal. Interestingly, the HD and double transgenic animals gained weight faster than their hsp104 and non-transgenic littermates (Fig. 4). (This contrasts with what is normally observed with these HD mice on the Bl6/C3Hf1 background, which show a slower weight gain and lower plateaus compared with their wild-type littermates and may be an effect of FvB alleles). At about 26 weeks old, the weight of the HD and double transgenic mice reached a ‘plateau’ and then started to decrease. The weights of HD and double transgenic were never significantly different, neither during the increasing phase nor when they were losing weight.
Hsp104 increases survival in HD mice
Double transgenic and HD animals showed a shortened lifespan compared with hsp104 and non-transgenic littermates. However, hsp104 expression increased survival in mutant huntingtin-expressing mice (P<0.0001). HD mice lived on an average for 172 days (±6.8) whereas the double transgenic animals lived on an average for 206 days (±6.5), which represent an increase of 20% of survival. While our survival analyses included mice that either died spontaneously or need to be culled when their disease severities exceeded defined moderate humane endpoints, this is unlikely to confound our interpretations, as all but two of the HD mice died spontaneously in the experiment (Fig. 5).
DISCUSSION
Hsp104 is a yeast molecular chaperone that reduces aggregate formation by various proteins in S. cerevisiae and can even disaggregate existing aggregates (20,22). We have previously shown that hsp104 reduces aggregation and toxicity of three different codon-expanded proteins: mutant huntingtin fragments (26), GFP-tagged polyalanine expansions and polyalanine-expanded mutant poly-(A) binding protein nuclear 1 that causes oculopharyngeal muscular dystrophy (27). Our cell model data suggested that hsp104 acts specifically at the level of aggregation in these models as it does not appear to protect against other pro-apoptotic insults such as staurosporine or H2O2 (27). As hsp70 overexpression protects against polyglutamine toxicity in HD cell models (12) but has not shown efficacy in rodent models of HD (16,17), it is crucial to test whether chaperones are protective in vivo. Accordingly, we have tested whether hsp104, a molecular chaperone that appears to act primarily at the level of protein folding, could alleviate the toxicity of the HD mutation in vivo. To this end, we have generated hsp104 transgenic mice driven by the same promoter as the N171-82Q HD mice. Overexpression of hsp104 reduced aggregation of mutant huntingtin fragments in vivo and significantly extended survival by ∼20%. It is interesting to note that the most effective extension of lifespan reported in an HD mouse model (by drug or genetic approaches) is 29% (31).
Interestingly, hsp104 did not affect motor signs in these mice. The phenomenon of a protective strategy improving survival but having no effects on motor function or weight loss has been previously noted in the N171-82Q mice treated post-symptomatically with phenylbutyrate, which increased survival by 23% (32). It is likely that the survival of HD transgenic mice and HD human patients have different biological determinants. In this mouse model, there are low levels of neurodegeneration and a significant proportion of mice die unpredictably, without obvious previous signs of severe disease. Schilling et al. (29), who described this model, noted that the transgenic mice started to die from about 14 weeks. However, the cause of death is still unknown, despite autopsies performed by our lab and others. We have used the terms ‘unpredictable death/died spontaneously’, to differentiate between the animals clearly moribund (no interaction with peers, no righting reflexes, very sudden and major loss of weight) needing to be culled immediately for humane reasons and the animals losing weight more progressively, showing some signs of disease (poor Rotarod performance, tremors and reduced grip strength) but not manifesting moribund phenotypes prior to being found dead in the cage. Indeed, it is worth stressing that the cause of death of all the HD mouse models is unclear, and this phenomenon is certainly not confined to the Ross/Borchelt model. Thus, death in these mice may be due to neuronal dysfunction in a critical domain, rather than what is seen in the human diseases where death follows typically 15 years after disease onset and in the context of major neurodegeneration and functional disability. So death in this mouse model may be more like a sign of neurological dysfunction than an endpoint after major neurodegeneration.
We were surprised that the reduction in aggregation did not lead to a more substantial rescue of phenotype. There are a number of possible reasons that may contribute to this. First, if hsp70 levels decrease over time in N171-82Q mice as in R6/2 mice (17) (Fig. 2), then the efficacy of hsp104 in reducing aggregation may be lessened in older mice, as hsp104 function requires interactions with hsp70 (21,23,33). Secondly, it is possible that aggregation may contribute to disease in particular cell types or cellular sites, more than in others. It is interesting to note that the similarities of our data with those of Orr and Zogbi's studies of an ataxin-1 mouse model where the self-association domain of ataxin-1 was deleted. Mice expressing polyglutamine-expanded ataxin-1 with this deleted domain had reduced aggregation but disease initiated at similar rates, compared with mice with the transgene without the deletion (4). However, subsequent analyses suggested that the mice with less aggregation had a slower progression of disease (34). It is difficult to draw mechanistic conclusions at the cellular level in relation to progression of disease in the whole organism. Progression of disease in the whole animal may not be simply due to a worsening of cellular dysfunction and progression along the path to cell death in a defined set of affected neurons—it may also be due to newer sets of neurons being affected as the animal ages.
In summary, hsp104 overexpression reduces aggregation and prolongs lifespan in an HD mouse model. As hsp104 is a yeast molecular chaperone that has no close mammalian orthologues and does not appear to have effects on cell death pathways (28), it is likely that the effects are mediated as a chaperone for mutant huntingtin folding. This may be at the level of changing the conformation of a putative toxic monomer, reducing oligomerization or aggregation or reducing the levels of oligomeric species or aggregates or combinations of these non-mutually exclusive possibilities. As hsp104 reduces aggregation and toxicity of a variety of codon-expanded proteins in cell cultures, it would be interesting in the future to examine the effects of this chaperone in mouse models of other polyglutamine diseases and related conditions associated with intracellular aggregate formation.
MATERIALS AND METHODS
Plasmid construction and generation of founder
The hsp104 sequence was excised from the vector previously described in Carmichael et al. (27) with the restriction enzymes AflII and NotI. The DNA fragment was then subcloned into the XhoI site of the MoPrP.Xho vector as described in Schilling et al. (29). In contrast to the Schilling et al. (29) paper, the hsp104 sequence contains its own stop codon. Plasmids were digested with NotI overnight at 37°C. Products of digestion were separated on TAE-agarose gel, without ethidium bromide. The piece of gel containing the band of interest was dialysed in TAE buffer. Products of dialysis were purified using the Elutip-D microcolumn protocol (Schleicher and Schuell), precipitated overnight in 100% ethanol at −20°C. The pellet was washed with 70% ethanol, dissolved in injection buffer (Sigma ultrapure water, 10 mm Tris pH 7.4, 0.1 mm NaEDTA) and microinjected into the pronucleus of fertilized oocytes of FvB mice.
Hsp104 transgenic mice were identified by PCR of genomic DNA extracted from tail tips. Two primers, complementary to the Hsp104 sequence, were used: Fwd-Hsp104-218 (5′-ATTGAAACGCCAGAAGATGG-3′) and Rvs-Hsp104-388 (5′-AAGGACTTTCCCCAAAGCAT-3′) giving an amplified product of 170 bp. One male founder was generated and bred to FvB females to generate a colony on pure FvB background.
Generation of HD×hsp104 mice
HD-N171-N82Q mice (B6C3F1/J-Tg(HD82Gln)81Dbo/J, Jackson Laboratory, Bar Harbour, ME) expressing the first 171 amino acids of mutant human huntingtin under the expression of a mouse PrP promoter were mated with FvB-hsp104 transgenic animals to generate double transgenic mice. All studies and procedures were performed under the jurisdiction of appropriate Home Office Project and Personal animal licences and with local Ethical Committee approval. The litters produced by the mating of HD-N171-N82Q mice and FvB-hsp104 mice were genotyped between 3 and 5 weeks of age by PCR and Southern blot.
HD×hsp104 mouse genotyping
The presence of HD transgene was identified by PCR, according to the protocol recommended by the Jackson laboratory. Two sets of primers were used IL-2wt-fwd (5′-CTAGGCCACAGAATTGAAAGATCT-3′), IL-2wt-Rvs (5′-GTAGGTGGAAATTCTAGCATCATCC-3′), HD82Gln (5′-GTGGATACCCCCTCCCCCAGCCTAGACC-3′) and HD-591-5′ (5′-GAACTTTCAGCTACCGAAGAAAGACCGTGT-3′). Hsp104 transgenic animals were identified by PCR, as described earlier, and Southern blot. For Southern blot, 371 bp probes were made by PCR, the hsp104-MoPrP vector, using internal primers for Hsp104: Fwd-Hsp104-729 (5′-AATTGGTGAGCCAGGTATCG-3′) and Rvs-Hsp104-1080 (5′-TTCTTTCAAAGGCACCATCC-3′). PCR products were separated on TBE agarose gel, purified using QIAquick gel extraction kits (Qiagen) and labelled according to the Prime-a-gene® labelling system (Promega). Products of labelling were purified using MicroSpin G25 columns (Amersham).
Genomic DNA extracted from tail tips were digested with the restriction enzyme DraI. Products of digestion were separated on TAE-agarose gel and transferred onto nitrocellulose membrane. Membranes were incubated at 65°C for 6–8 h in hybridization buffer [0.5 m phosphate-sodium buffer (0.4 m NaH2PO4; 0.6 m Na2HPO4, pH 7.2), 0.01 m EDTA, 1% herring sperm DNA, 7% SDS] without the probe. Membranes were hybridized overnight at 65°C in hybridization buffer and washed twice in 1% SDS–0.04 m phosphate-sodium buffer and once in 0.1% SDS–1×SSC. Membranes were exposed to X-ray films (Kodak).
Western blot analysis
Western blot analysis was done using standard techniques with ECL detection kit (Amersham). The primary antibodies used were anti-polyglutamine (1C2, Chemicon), anti-hsp104 (Stressgen) and anti-actin (Sigma). Brains were homogenized in 2.5 volumes of buffer B (50 mm Tris pH 7.5, 10% glycerol, 5 mm magnesium acetate, 0.2 mm EDTA, 0.5 mm DTT and protease inhibitor) at 4°C. The homogenate was centrifuged at 13 400 g at 4°C. The supernatant was removed and used for western blot analysis.
Immunohistochemistry
Thirty micrometer frozen sections of non-transgenic, HD and double transgenic brains were analyzed for neuronal inclusions according to the protocol by Davies et al. (35). Sections were labelled with anti-huntingtin antibody (EM48, Chemicon). Staining was performed by peroxidase labelling using Vectastain Avidin: Biotinylated enzyme Complex (ABC) kit. Inclusions (both intranuclear and neuritic) were counted in various brain areas on six sections per animal.
Mice and behavioural tests
Transgenic mice and non-transgenic littermates were compared in the trial. Mice were coded with alpha-numeric identities which provided no clues to their genotype. Thus, observers were blind to their treatment and genetic status during testing. Mice were monitored daily and weighed three times a week. Motor performances were assessed at 4, 12, 14, 16, 18, 20, 22 and 24 weeks of age with a Rotarod apparatus (Accelerating Model, Ugo Basile, Biological Research Apparatus, Varese, Italy). The mice were given two training sessions per day, for two consecutive days, to acclimatize them to the apparatus. On the third day, the mice were given six separate trials. On the training and testing days, the speed was set to increase from 4 to 40 rpm in 250 s; the animals were put on the Rotarod for a maximum of 300 s. Grip strength and wire manoeuvre monitoring are part of the SHIRPA battery of behavioural tests. These tests were assessed at 4, 12, 14, 16, 18, 20, 22 and 24 weeks of age. To assess grip strength, mice were allowed to hold a metal grid with their forelimbs, lifted by the tail so that the hind limbs were not in contact with the grid and gently pulled backwards by the tail until they could no longer hold the grid. Grip strength was scored as follows: 0=none; 1=slight grip, semi effective; 2=moderate grip, effective; 3=active grip, effective. For the wire manoeuvre, the animals were held above a horizontal wire by tail suspension and lowered to allow the forelimbs to grip the wire. The mice were held in extension, rotated around to the horizontal and released. They received a mark as follows: 0=active grip with hind legs; 1=difficulty grasping with hind legs; 2=unable to grasp with hind legs; 3=unable to lift hind legs, falls within 10 s; 4=falls immediately.
Grip strength was monitored quantitatively by using a grip strength meter (Bioseb, France) at 14, 16, 18, 20, 22, 24 weeks of age. The mice were held above the apparatus grid with their front paws (2 leg test) or all four paws (4 leg test) grasping the transverse bar on the grid, then pulled back by the tail following the axle of the sensor, horizontally and steadily, until they released the grid. The apparatus was used in peak mode, the recorded value corresponding to the maximum force developed by the animal.
Statistics
Significance levels for comparisons between groups were determined with t-tests, repeated measure or factorial ANOVA, where appropriate, for parametric data and with Mann–Whitney U-tests for non-parametric data, using the STATVIEW software, version 4.53 (Abacus Concepts, Berkeley, CA).
Survival analysis was determined by Kaplan–Meier test, followed by a logrank (Mantel–Cox) test. When the animals were showing any of the following clinical signs, they had to undergo euthanasia: marked loss of appetite and fluid intake or staring coat, hunched posture and subdue behaviour or >20% weight loss over a period of more than 3 days.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
D.C.R. is grateful for funding from the MRC (Programme Grant with Prof Steve Brown) and a Wellcome Trust Senior Clinical Fellowship. We are grateful to Lauren Gilroy and Oana Sadiq for technical assistance.
Conflict of Interest statement. None declared.
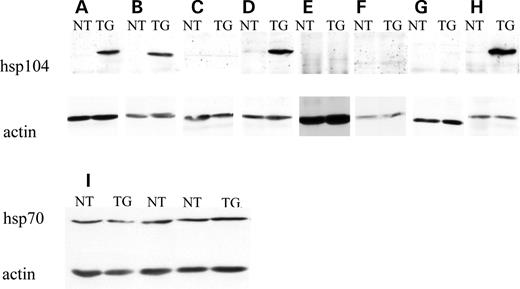
Figure 1. Detection of the hsp104 protein by western blot analysis. (A–H) Whole brain homogenates from a 14-week-old transgenic mouse (TG) and non-transgenic littermate (NT) were probed with anti-hsp104 antibody. The pattern of expression is typical of the MoPrP promoter: the hsp104 protein (upper panels) is specifically expressed in the brain (A), kidney (B), testis (D) and heart (H) of the transgenic mouse but not in the muscle (C), spleen (E), liver (F) or lung (G). (I) The expression of hsp104 in the whole brain of otherwise normal mouse does not modify the levels of endogenous hsp70. Lanes 2 and 5, Hsp104 mice (Tg); lanes 1, 3 and 4, non-transgenic littermates (NT). Actin (lower bands) was used as a loading control.
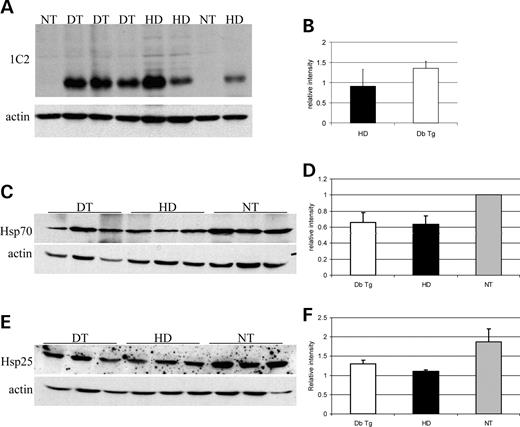
Figure 2. The expression of hsp104 protein does not modify the levels of soluble mutant huntingtin or endogenous heat shock proteins in HD mouse brains. (A) Upper panel: whole brain homogenates from 13-week-old HD mice (HD, lanes 5, 6 and 8), double transgenic mice (DT, lanes 2, 3, 4) and non-transgenic littermates (NT, lanes 1 and 7) probed with anti-polyglutamine antiboby (1C2, Chemicon). (B) The levels of huntingtin, relative to actin, are not different between HD and the double transgenic animals (nHD=3, nDb Tg=3, P=0.3797). (C and E) Upper panels: whole brain homogenates from 12-week-old double transgenic mice (DT, lanes 1, 2, 3), HD mice (HD, lanes 4, 5, 6) and non-transgenic littermates (NT, lanes 7, 8, 9) probed with anti-hsp70 (hsp72) antibody (C) or anti-hsp25 antibody (E). (D) The levels of hsp70 expression are reduced in mutant huntingtin-expressing animals, HD only and double transgenic animals, compared with non-transgenic mice. The means were obtained by pooling data from four different experiments. The values for HD (n=10) and Db Tg (n=11) are relative to the values for NT (n=11) animals (PHD,Db Tg=0.8789, P=Db Tg,NT=0.0105, PHD,NT=0.0014). (F) The levels of hsp25 expression do not differ between the three groups (PHD,Db Tg=0.1551, P=Db Tg,NT=0.1696, PHD,NT=0.0804, nHD=3, nDb Tg=3, nNT=3). Actin was used as a loading control (A, C, E—lower panels). NT, non-transgenic; DT, double transgenic; HD, HD mice.
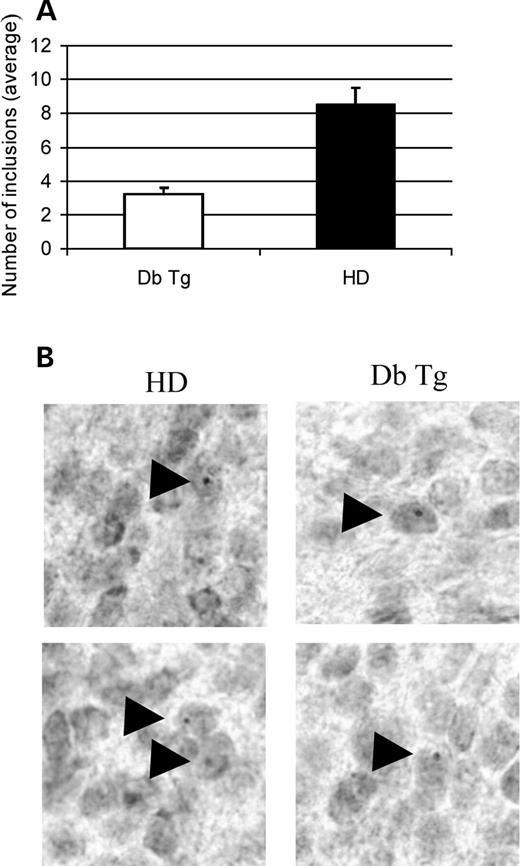
Figure 3. Hsp104 reduces aggregate load in HD mice. (A) We counted aggregates in the whole piriform cortex on both sides of the brain, on two mice for each transgenic status and six brain sections per animal (×60 objective). The piriform cortex of 14-week-old double transgenic mice showed 38% fewer aggregates than their HD littermates (P<0.0001). Error bars show SEM. (B) Peroxidase immunohistochemistry of double transgenic (Db Tg, right panels) and HD (HD, left panels) transgenic mouse brain at 14 weeks of age. The aggregates in the double transgenic and HD animals did not show any difference in shape or size. Arrowheads point out the aggregates.
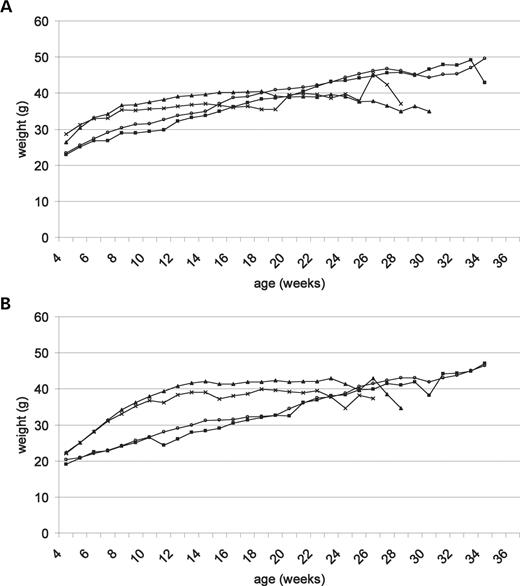
Figure 4. Weight of the mice expressing hsp104 only (squares), mutant huntingtin only (cross), double transgenic (triangles) and non-transgenic littermates (open circles). (A) Weight of the males. There is no significant overall effect between 4 and 28 weeks of age on the weights of the males of the four different genotypes (P=0.1138). Between 4 and 15 weeks of age, the overall effect on the weights of the four different genotypes is significant (nNT=7, nHsp104=12, nHD=7, nDb Tg=9, P<0.0001). The double transgenic animals are significantly bigger than the animals expressing hsp104 only (P<0.0001) and their non-transgenic littermates (P=0.0087) but not the HD animals (P=0.5117). The HD animals are also significantly bigger than the hsp104 animals (P=0.0004) and marginally heavier than their non-transgenic littermates (P=0.0604). (B) Weights of the females. Between 4 and 25 weeks of age, the overall effect on the weights of the four different genotypes is highly significant (nNT=11, nHsp104=3, nHD=9, nDb Tg=9, P<0.0001). The double transgenic animals are significantly bigger than the animals expressing hsp104 only (P=0.0024) and their non-transgenic littermates (P<0.0001) but not than the HD animals (P=0.38). The HD animals are also marginally bigger than their non-transgenic littermates (P=0.02) but not the animals expressing only hsp104 (P=0.18).
![Figure 5. Kaplan–Meier survival curve for the four different genotypes. The expression of hsp104 has a protective effect on the survival of mice expressing the mutant huntingtin [P=0.0005, logrank (Mantel–Cox) test]. HD, black circles; Db Tg, open circles; NT and hsp104,+sign. No non-transgenic or hsp104 only animals died during the time of the experiment. Asterisks represent the animals that required euthanasia when the disease exceeded defined, moderately severe, humane endpoint. The other animals died spontaneously.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/22/10.1093_hmg_ddi372/1/m_ddi37205.jpeg?Expires=1716415072&Signature=xKkb4AQkZKx9LFv1jgjQYEu9oRWwAJI2rSJJ688FMbYaslIZwyT4inw-Wvl6i0I1JZEzIjNQdLP20GEUvI1652nZ-ottCSDax4rTDZ-0sRrBMKtn0SaNjZSNdtFPgEzvCGfFShmgA5~I9twDqAUWLLrqJMUdW8cOAODn2z-8y1DAyvs1Hk88tVM5~iIq2Yn2gKr9Y3esVJs~63TrTnsy9IY5x8jgKHHvMtip5fNxwALaoQYdCBx3Vfp0pnacLmF1QBoMgF5kOfVUBBdbXtqKLDPNj48hq2e9bLEhEwYtgr3pOUkfCLBBgQca8kwlHmlWqzMvzvkDO03Dl3slvMv7Fg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 5. Kaplan–Meier survival curve for the four different genotypes. The expression of hsp104 has a protective effect on the survival of mice expressing the mutant huntingtin [P=0.0005, logrank (Mantel–Cox) test]. HD, black circles; Db Tg, open circles; NT and hsp104,+sign. No non-transgenic or hsp104 only animals died during the time of the experiment. Asterisks represent the animals that required euthanasia when the disease exceeded defined, moderately severe, humane endpoint. The other animals died spontaneously.
Summary of motor phenotyps of HD and HD/hsp104 double transgenic mice (Db Tg) at various ages
| Age (weeks) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 4 | 12 | 14 | 16 | 18 | 20 | 22 | 24 | ||
| Rotarod | Db Tg | 118.6±6.6 | 62.6±5.2 | 73.0±5.5 | 72.9±5.4 | 61.2±4.3 | 67.6±4.9 | 69.5±5.5 | 71.1±6.4 |
| HD | 119.1±8.0 | 75.1±7.3 | 67.6±6.7 | 62.8±5.8 | 73.2±6.4 | 76.5±7.5 | 82.1±14.7 | 82.9±10.5 | |
| P-value | 0.9813 | 0.5082 | 0.7712 | 0.5535 | 0.4439 | 0.6250 | 0.6008 | 0.6695 | |
| Grip strength (2 legs) | Db Tg | 75.4±4.8 | 71.7±4.0 | 70.9±2.7 | 77.3±7.1 | 65.2±1.4 | 66.7±5.8 | ||
| HD | 83.0±5.0 | 86.3±3.8 | 69.5±2.5 | 68.2±3.4 | 59.1±3.0 | 58.2±4.9 | |||
| P-value | 0.4024 | 0.0919 | 0.7673 | 0.3794 | 0.4942 | 0.5134 | |||
| Grip strength (4 legs) | Db Tg | 198.3±5.0 | 177.5±5.4 | 180.2±6.3 | 172.6±7.5 | 158.2±11.2 | 170.6±9.2 | ||
| HD | 189.0±5.6 | 183.7±5.9 | 187.4±6.4 | 177.6±7.1 | 146.8±4.2 | 155.2±7.0 | |||
| P-value | 0.4203 | 0.5345 | 0.4422 | 0.8195 | 0.4629 | 0.4508 | |||
| Grip SHIRPA | P= | 0.3870 | 0.6538 | 0.2142 | 0.2695 | 0.3006 | 0.0827 | 0.2981 | 0.3095 |
| Wire manoeuvre | P-value | 0.2017 | 0.1425 | 0.9587 | 0.5011 | 0.3340 | 0.6908 | 0.6902 | 0.3912 |
| Age (weeks) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 4 | 12 | 14 | 16 | 18 | 20 | 22 | 24 | ||
| Rotarod | Db Tg | 118.6±6.6 | 62.6±5.2 | 73.0±5.5 | 72.9±5.4 | 61.2±4.3 | 67.6±4.9 | 69.5±5.5 | 71.1±6.4 |
| HD | 119.1±8.0 | 75.1±7.3 | 67.6±6.7 | 62.8±5.8 | 73.2±6.4 | 76.5±7.5 | 82.1±14.7 | 82.9±10.5 | |
| P-value | 0.9813 | 0.5082 | 0.7712 | 0.5535 | 0.4439 | 0.6250 | 0.6008 | 0.6695 | |
| Grip strength (2 legs) | Db Tg | 75.4±4.8 | 71.7±4.0 | 70.9±2.7 | 77.3±7.1 | 65.2±1.4 | 66.7±5.8 | ||
| HD | 83.0±5.0 | 86.3±3.8 | 69.5±2.5 | 68.2±3.4 | 59.1±3.0 | 58.2±4.9 | |||
| P-value | 0.4024 | 0.0919 | 0.7673 | 0.3794 | 0.4942 | 0.5134 | |||
| Grip strength (4 legs) | Db Tg | 198.3±5.0 | 177.5±5.4 | 180.2±6.3 | 172.6±7.5 | 158.2±11.2 | 170.6±9.2 | ||
| HD | 189.0±5.6 | 183.7±5.9 | 187.4±6.4 | 177.6±7.1 | 146.8±4.2 | 155.2±7.0 | |||
| P-value | 0.4203 | 0.5345 | 0.4422 | 0.8195 | 0.4629 | 0.4508 | |||
| Grip SHIRPA | P= | 0.3870 | 0.6538 | 0.2142 | 0.2695 | 0.3006 | 0.0827 | 0.2981 | 0.3095 |
| Wire manoeuvre | P-value | 0.2017 | 0.1425 | 0.9587 | 0.5011 | 0.3340 | 0.6908 | 0.6902 | 0.3912 |
Full details in Supplementary Material Figs 2–9. Rotarod values represent the latency to fall (in s, mean±SEM), grip strength values represent the strength of the forelegs (2 legs) or all legs (4 legs, in g, means±SEM). P-values compare HD to double transgenic (Db Tg).
Summary of motor phenotyps of HD and HD/hsp104 double transgenic mice (Db Tg) at various ages
| Age (weeks) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 4 | 12 | 14 | 16 | 18 | 20 | 22 | 24 | ||
| Rotarod | Db Tg | 118.6±6.6 | 62.6±5.2 | 73.0±5.5 | 72.9±5.4 | 61.2±4.3 | 67.6±4.9 | 69.5±5.5 | 71.1±6.4 |
| HD | 119.1±8.0 | 75.1±7.3 | 67.6±6.7 | 62.8±5.8 | 73.2±6.4 | 76.5±7.5 | 82.1±14.7 | 82.9±10.5 | |
| P-value | 0.9813 | 0.5082 | 0.7712 | 0.5535 | 0.4439 | 0.6250 | 0.6008 | 0.6695 | |
| Grip strength (2 legs) | Db Tg | 75.4±4.8 | 71.7±4.0 | 70.9±2.7 | 77.3±7.1 | 65.2±1.4 | 66.7±5.8 | ||
| HD | 83.0±5.0 | 86.3±3.8 | 69.5±2.5 | 68.2±3.4 | 59.1±3.0 | 58.2±4.9 | |||
| P-value | 0.4024 | 0.0919 | 0.7673 | 0.3794 | 0.4942 | 0.5134 | |||
| Grip strength (4 legs) | Db Tg | 198.3±5.0 | 177.5±5.4 | 180.2±6.3 | 172.6±7.5 | 158.2±11.2 | 170.6±9.2 | ||
| HD | 189.0±5.6 | 183.7±5.9 | 187.4±6.4 | 177.6±7.1 | 146.8±4.2 | 155.2±7.0 | |||
| P-value | 0.4203 | 0.5345 | 0.4422 | 0.8195 | 0.4629 | 0.4508 | |||
| Grip SHIRPA | P= | 0.3870 | 0.6538 | 0.2142 | 0.2695 | 0.3006 | 0.0827 | 0.2981 | 0.3095 |
| Wire manoeuvre | P-value | 0.2017 | 0.1425 | 0.9587 | 0.5011 | 0.3340 | 0.6908 | 0.6902 | 0.3912 |
| Age (weeks) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 4 | 12 | 14 | 16 | 18 | 20 | 22 | 24 | ||
| Rotarod | Db Tg | 118.6±6.6 | 62.6±5.2 | 73.0±5.5 | 72.9±5.4 | 61.2±4.3 | 67.6±4.9 | 69.5±5.5 | 71.1±6.4 |
| HD | 119.1±8.0 | 75.1±7.3 | 67.6±6.7 | 62.8±5.8 | 73.2±6.4 | 76.5±7.5 | 82.1±14.7 | 82.9±10.5 | |
| P-value | 0.9813 | 0.5082 | 0.7712 | 0.5535 | 0.4439 | 0.6250 | 0.6008 | 0.6695 | |
| Grip strength (2 legs) | Db Tg | 75.4±4.8 | 71.7±4.0 | 70.9±2.7 | 77.3±7.1 | 65.2±1.4 | 66.7±5.8 | ||
| HD | 83.0±5.0 | 86.3±3.8 | 69.5±2.5 | 68.2±3.4 | 59.1±3.0 | 58.2±4.9 | |||
| P-value | 0.4024 | 0.0919 | 0.7673 | 0.3794 | 0.4942 | 0.5134 | |||
| Grip strength (4 legs) | Db Tg | 198.3±5.0 | 177.5±5.4 | 180.2±6.3 | 172.6±7.5 | 158.2±11.2 | 170.6±9.2 | ||
| HD | 189.0±5.6 | 183.7±5.9 | 187.4±6.4 | 177.6±7.1 | 146.8±4.2 | 155.2±7.0 | |||
| P-value | 0.4203 | 0.5345 | 0.4422 | 0.8195 | 0.4629 | 0.4508 | |||
| Grip SHIRPA | P= | 0.3870 | 0.6538 | 0.2142 | 0.2695 | 0.3006 | 0.0827 | 0.2981 | 0.3095 |
| Wire manoeuvre | P-value | 0.2017 | 0.1425 | 0.9587 | 0.5011 | 0.3340 | 0.6908 | 0.6902 | 0.3912 |
Full details in Supplementary Material Figs 2–9. Rotarod values represent the latency to fall (in s, mean±SEM), grip strength values represent the strength of the forelegs (2 legs) or all legs (4 legs, in g, means±SEM). P-values compare HD to double transgenic (Db Tg).
References
Rubinsztein, D.C. (
DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsattel, J.P. and Aronin, N. (
Taylor, J.P., Hardy, J. and Fischbeck, K.H. (
Klement, I.A., Skinner, P.J., Kaytor, M.D., Yi, H., Hersch, S.M., Clark, H.B., Zoghbi, H.Y. and Orr, H.T. (
Tompkins, M.M., Basgall, E.J., Zamrini, E. and Hill, W.D. (
Arrasate, M., Mitra, S., Schweitzer, E.S., Segal, M.R. and Finkbeiner, S. (
Caughey, B. and Lansbury, P.T. (
Wyttenbach, A., Carmichael, J., Swartz, J., Furlong, R.A., Narain, Y., Rankin, J. and Rubinsztein, D.C. (
Cummings, C.J., Mancini, M.A., Antalffy, B., DeFranco, D.B., Orr, H.T. and Zoghbi, H.Y. (
Jana, N.R., Tanaka, M., Wang, G. and Nukina, N. (
Auluck, P.K., Chan, H.Y., Trojanowski, J.Q., Lee, V.M. and Bonini, N.M. (
Wyttenbach, A., Sauvageot, O., Carmichael, J., Diaz-Latoud, C., Arrigo, A.P. and Rubinsztein, D.C. (
Kobayashi, Y., Kume, A., Li, M., Doyu, M., Hata, M., Ohtsuka, K. and Sobue, G. (
Warrick, J.M., Chan, H.Y., Gray-Board, G.L., Chai, Y., Paulson, H.L. and Bonini, N.M. (
Chan, H.Y., Warrick, J.M., Gray-Board, G.L., Paulson, H.L. and Bonini, N.M. (
Cummings, C.J., Sun, Y., Opal, P., Antalffy, B., Mestril, R., Orr, H.T., Dillmann, W.H. and Zoghbi, H.Y. (
Hay, D.G., Sathasivam, K., Tobaben, S., Stahl, B., Marber, M., Mestril, R., Mahal, A., Smith, D.L., Woodman, B. and Bates, G.P. (
Hansson, O., Nylandsted, J., Castilho, R.F., Leist, M., Jaattela, M. and Brundin, P. (
Helmlinger, D., Bonnet, J., Mandel, J.L., Trottier, Y. and Devys, D. (
Saleh, A., Srinivasula, S.M., Balkir, L., Robbins, P.D. and Alnemri, E.S. (
Lee, S., Sowa, M.E., Choi, J.M. and Tsai, F.T. (
Mogk, A. and Bukau, B. (
Glover, J.R. and Lindquist, S. (
Krobitsch, S. and Lindquist, S. (
Satyal, S.H., Schmidt, E., Kitagawa, K., Sondheimer, N., Lindquist, S., Kramer, J.M. and Morimoto, R.I. (
Mosser, D.D., Ho, S. and Glover, J.R. (
Carmichael, J., Chatellier, J., Woolfson, A., Milstein, C., Fersht, A.R. and Rubinsztein, D.C. (
Bao, Y.P., Cook, L.J., O'Donovan, D., Uyama, E. and Rubinsztein, D.C. (
Schilling, G., Becher, M.W., Sharp, A.H., Jinnah, H.A., Duan, K., Kotzuk, J.A., Slunt, H.H., Ratovitski, T., Cooper, J.K., Jenkins, N.A. et al. (
Borchelt, D.R., Davis, J., Fischer, M., Lee, M.K., Slunt, H.H., Ratovitsky, T., Regard, J., Copeland, N.G., Jenkins, N.A., Sisodia, S.S. et al. (
Ferrante, R.J., Ryu, H., Kubilus, J.K., D'Mello, S., Sugars, K.L., Lee, J., Lu, P., Smith, K., Browne, S., Beal, M.F. et al. (
Gardian, G., Browne, S.E., Choi, D.K., Klivenyi, P., Gregorio, J., Kubilus, J.K., Ryu, H., Langley, B., Ratan, R.R., Ferrante, R.J. et al. (
Krzewska, J., Konopa, G. and Liberek, K. (
Skinner, P.J., Vierra-Green, C.A., Emamian, E., Zoghbi, H.Y. and Orr, H.T. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}