




Abstract
Mutations in the dysferlin (DYSF) and caveolin-3 (CAV3) genes are associated with muscle disease. Dysferlin is mislocalized, by an unknown mechanism, in muscle from patients with mutations in caveolin-3 (Cav-3). To examine the link between Cav-3 mutations and dysferlin mistargeting, we studied their localization at high resolution in muscle fibers, in a model muscle cell line, and upon heterologous expression of dysferlin in muscle cell lines and in wild-type or caveolin-null fibroblasts. Dysferlin shows only partial overlap with Cav-3 on the surface of isolated muscle fibers but co-localizes with Cav-3 in developing transverse (T)-tubules in muscle cell lines. Heterologously expressed dystrophy-associated mutant Cav3R26Q accumulates in the Golgi complex of muscle cell lines or fibroblasts. Cav3R26Q and other Golgi-associated mutants of both Cav-3 (Cav3P104L) and Cav-1 (Cav1P132L) caused a dramatic redistribution of dysferlin to the Golgi complex. Heterologously expressed epitope-tagged dysferlin associates with the plasma membrane in primary fibroblasts and muscle cells. Transport to the cell surface is impaired in the absence of Cav-1 or Cav-3 showing that caveolins are essential for dysferlin association with the PM. These results suggest a functional role for caveolins in a novel post-Golgi trafficking pathway followed by dysferlin.
INTRODUCTION
Muscular dystrophy (MD) comprises a group of inherited disorders characterized by progressive weakness of skeletal muscle. Several muscle membrane proteins have been linked to a variety of MDs. The best characterized are those that make-up the dystrophin–glycoprotein complex, such as dystrophin, sarcoglycans and dystroglycan, which link components of the extracellular matrix to the intracellular cytoskeleton providing stability to the muscle fibers (1). Dysferlin and caveolin-3 (muscle-specific caveolin, Cav-3), although not part of the dystrophin complex, have also been linked to different muscle disorders. Dysferlin is a 230 kDa transmembrane protein homologous to the Caenorhabditis elegans sperm vesicle-fusion protein, fer-1 (2). The ferlin family of proteins includes dysferlin, myoferlin and otoferlin, of which dysferlin and otoferlin have been associated with genetic diseases (3–5). Mutations in the dysferlin gene can cause limb girdle muscular dystrophy (LGMD) type 2B, Miyoshi myopathy and distal anterior compartment myopathy (2,6–8). Dysferlin has been implicated in membrane fusion events (2), and more recently suggested to play a role in membrane repair processes, as the ability to reseal the sarcolemma upon injury is impaired in dysferlin-null mice (9).
Caveolins are 21–24 kDa integral membrane proteins and the crucial structural component of caveolae, flask-shaped 55–65 nm diameter plasma membrane (PM) pits. Three mammalian caveolins have been described. They are linked to membrane trafficking (10–14) and signal transduction events (15–17). Caveolin-1 (Cav-1) and caveolin-2 form hetero-oligomeric complexes mainly found in endothelial cells, smooth muscle cells, skeletal myoblasts, fibroblasts and adipocytes (18), whereas Cav-3 is largely restricted to striated muscle cells (skeletal and heart muscle) (19,20) although it has been reported in smooth muscle (21,22). The role of caveolae and Cav-3 in muscle has recently gained clinical attention as defects in the CAV3 gene are associated with a number of muscle pathologies including LGMD type 1C, distal myopathy, hyperCKemia and rippling muscle disease (RMD) (23–32). In addition, Cav-3 has been implicated in transverse (T)-tubule system biogenesis (33), co-purifies with dysferlin (34,35) and when Cav-3 expression is defective dysferlin localization is abnormal (29–31,34,36).
In spite of these findings, the cell biology underlying the Cav-3 and dysferlin interaction and its association with MD pathogenesis is not well understood. At present 15 different point mutations and a 9 bp deletion in the Cav-3 gene have been described in patients with muscle disease (23–29,31,32,37). Of these, a missense mutation within the membrane-spanning domain of Cav-3 (Pro104→Leu, Cav3P104L) is one of the best characterized. Cav3P104L is associated with autosomal dominant LGMD type 1C in humans (24), and dysferlin shows abnormal sarcolemmal labeling in muscle tissue derived from patients bearing this mutation (30,34). In addition, Cav3P104L expressed in C2C12 myotubes (38) and non-muscle cells (39,40) is strongly retained in the Golgi complex. This mutant causes accumulation of wild-type (WT) Cav-3 in the Golgi apparatus resulting in decreased PM targeting (39), or increased ubiquitin-dependent degradation of WT protein through the proteasome pathway (41), indicating that it acts in a dominant-negative manner. Another missense autosomal dominant mutation in the Cav-3 gene, Cav3R26Q (Arg26→Gln; assuming a single starting methionine), has been previously associated with RMD (26,27,36,42), as well as with MD including LGMD type 1C (30), idiopathic hyperCKemia (25) and distal myopathy (29).
To investigate the potential role of caveolins in dysferlin function, we have examined the distribution of endogenous and heterologously expressed forms of these proteins in muscle and non-muscle cells. We have found that dysferlin and Cav-3 show partial co-localization in muscle (isolated muscle fibers and differentiated C2C12 myotubes). Co-expression of epitope-tagged dysferlin and muscle disease-associated Cav-3 mutants, Cav3R26Q or Cav3P104L, cause a dramatic redistribution of dysferlin to the Golgi complex. Furthermore, dysferlin traffic to the cell surface is impaired in the absence of Cav-1 or Cav-3. Our results show that caveolins are required for correct localization of dysferlin to the PM. We suggest that the reduced sarcolemmal expression of dysferlin seen in patients with mutant forms of Cav-3 may be because of disruption of trafficking of dysferlin to the sarcolemma and T-tubules via a novel caveolin-dependent trafficking pathway from the Golgi complex.
RESULTS
Dysferlin co-localizes with Cav-3 in mature muscle fibers and C2C12 myotubes
We first studied the expression of dysferlin and Cav-3 in muscle from an RMD patient with a missense mutation in the CAV3 gene, which involves an amino acid substitution (Arg→Glu) at position 26 of the Cav-3 protein (Cav3R26Q, from here on RMD-R26Q) (30,34,42). In accordance with previous studies (30,34), in normal human skeletal muscle, dysferlin and Cav-3 are localized at the sarcolemma of muscle fibers (Fig. 1A). In contrast, Cav-3 and dysferlin labeling is severely reduced at the sarcolemma of muscle fibers from the RMD-R26Q patient (Fig. 1A). In some fibers dysferlin appeared to be concentrated in patches underlying the muscle cell membrane (Fig. 1A). Western blot revealed a marked reduction in Cav-3 in the RMD-R26Q patient when compared with control muscle (Fig. 1B), whereas dysferlin expression levels were normal despite an observed reduction in sarcolemmal immunolabeling (Fig. 1B). Thus, the RMD-R26Q patient shows aberrant intracellular (IC) distribution of dysferlin.
To gain further insights into the perturbation of dysferlin distribution associated with mutations in the CAV3 gene, we examined the localization of dysferlin with respect to Cav-3 in mature muscle and in model cell systems. Dysferlin has been reported to localize to the sarcolemma (43) and to cytoplasmic vesicles in mouse muscle (44,45) but the precise distribution of dysferlin with respect to caveolae and Cav-3 has not been resolved. To gain high-resolution views of the surface distribution of dysferlin with respect to Cav-3, we cultured isolated mice skeletal muscle fibers (46). The distribution of dysferlin and Cav-3 was then examined over the surface of the muscle fiber at high resolution by confocal microscopy. Dysferlin and Cav-3 showed partial co-localization in striations over the surface of isolated adult muscle fibers. As previously described Cav-3 showed a highly organized lattice-like surface labeling along the length of isolated myofibers with low labeling within the fiber (46,47) (Fig. 1C). Interestingly, dysferlin showed a similar discontinuous pattern of staining at the sarcolemma (Fig. 1C); labeling was mainly localized to transversely oriented regions of the sarcolemma (Fig. 1C) that overlapped only partially with Cav-3 domains. Confocal imaging inside the fiber showed higher labeling for dysferlin than for Cav-3 in transverse bands suggestive of labeling of the T-tubules (Fig. 1D). This was supported by labeling of these transverse elements with antibodies to triadin, a marker of the T-tubule/SR triad junction (Fig. 1E), or to the alpha subunit of the dihydropyridine receptor (DHPR; not shown).
These results show that dysferlin and Cav-3 are targeted to the sarcolemma in adult skeletal muscle but are not completely co-associated. In addition dysferlin localizes to the region of the T-tubules. We further investigated this potential association in a model muscle cell line, C2C12. Both dysferlin and Cav-3 have been shown to be upregulated in C2C12 cells following differentiation (19,35,48,49). In differentiated C2C12 cells, endogenous dysferlin localized to a tubular network within the cytoplasm of myotubes (Fig. 1F) and showed extensive, although incomplete, co-localization with Cav-3 (Fig. 1F). This is consistent with co-localization of dysferlin with Cav-3 in the developing T-tubule system (33).
Heterologously expressed epitope-tagged dysferlin is targeted to the PM in fibroblasts
To gain a better understanding of the association between dysferlin and caveolin, and the potential pathogenic mechanisms underlying Cav-3-associated muscle disease, we sought to develop a model system in which the trafficking of dysferlin could be examined with respect to caveolin and caveolin mutants. To facilitate these studies we constructed a dysferlin cDNA with an N-terminal GFP tag and a C-terminal (lumenal/extracellular) myc tag (GFP–dysf). GFP–dysf expressed in BHK cells was evident as a single polypeptide of the predicted molecular weight of approximately 260 kDa with no evidence of degradation as judged by western blot analysis with an anti-GFP antibody (Fig. 2A). We first examined whether the transiently expressed construct trafficked normally in C2C12 cells. As shown in Figure 2B, GFP–dysf co-localized with Cav-3 and showed an identical distribution to endogenous dysferlin in C2C12 cells. We then characterized the subcellular distribution of the epitope-tagged dysferlin in non-muscle cells. GFP–dysf expressed in primary mouse embryonic fibroblasts (MEFs) and BHK cells associated with the PM as well as to intracellular puncta (Fig. 2C). Immunoelectron microscopy of transfected BHK cells revealed GFP–dysf localized to the PM and to vesicular profiles (Fig. 2D) but no labeling could be detected in the Golgi complex (results not shown). At the PM, GFP–dysf was evident along planar PM with no apparent enrichment within caveolae (Fig. 2D). Thus in muscle and non-muscle cell lines dysferlin is mainly targeted to the PM but also shows some IC staining.
Disease-associated mutants of Cav-3 and Cav-1 cause retention of dysferlin in the Golgi complex of muscle and non-muscle cells
We next used the epitope-tagged form of dysferlin to examine whether trafficking of dysferlin is affected by mutated epitope-tagged forms of Cav-3 when co-expressed in muscle cell lines as well as in a non-muscle context. Epitope-tagged Cav3R26Q (Cav3R26Q-HA) and Cav3P104L [Cav3P104L-HA; (38,39)], accumulated in the Golgi complex of C2C12 cells and MEFs as judged by co-localization with the Golgi marker, GM130 (Fig. 3A and B). The detailed subcellular distribution of WT and mutant forms of Cav-3 was then examined by immunoelectron microscopy in transfected BHK cells (which gave higher transfection efficiency than the other cell types tested). Epitope-tagged Cav-3 (Cav3-HA) associated predominantly with PM caveolae (Fig. 4A) but also was detected within the Golgi complex (Fig. 4B). Labeling for Cav-3 was observed throughout the Golgi stacks and associated vesicles (Fig. 4B). GM130, a medial/cis Golgi cisternae marker, showed a more restricted distribution (Fig. 4B). In contrast to the WT Cav3-HA, the Cav3P104L-HA mutant was predominantly associated with the Golgi complex with a striking lack of PM caveolae labeling even in cells with high levels of expression (Fig. 4C). Thus, the P to L mutation causes a dramatic block in caveolin trafficking from the Golgi to the PM. We therefore examined whether the mutant protein was associated with a particular subcompartment of the Golgi complex, which might indicate the site of the block. As shown in Figure 4, Cav3P104L-HA was observed throughout the Golgi complex (Fig. 4C and D), with no particular association with a subdomain of the Golgi under a range of different expression levels. Both cisternal and vesicular profiles were labeled with limited overlap of labeling with GM130. Like Cav3P104L-HA, Cav3R26Q-HA localized to IC vesicular elements with little labeling of the PM in low expressing cells (Fig. 4E and F). However, Golgi cisternae were not as readily recognizable in Cav3R26Q-HA cells expressing higher levels suggesting that expression might disrupt Golgi morphology visualized at the ultrastructural level (unpublished data).
As dysferlin shows an abnormal distribution in the RMD-R26Q patient (Fig. 1) we examined whether accumulation of Cav3R26Q in the Golgi complex affects traffic of dysferlin to the PM. We first looked at the effect of expression of epitope-tagged Cav3R26Q on the distribution of endogenous dysferlin in C2C12 myotubes. As demonstrated in Figure 5, in C2C12 myotubes expressing Cav3R26Q-HA, endogenous dysferlin redistributed to a perinuclear region and co-localized with Cav3R26Q (Fig. 5A). Endogenous dysferlin localized to intracellular tubular structures upon expression of WT epitope-tagged Cav-3 (Fig. 5A). We further examined the subcellular distribution of epitope-tagged dysferlin upon expression of disease-associated Cav-3 mutants in muscle and non-muscle cells. Upon co-expression in C2C12 cells (Fig. 5B) or MEFs (Fig. 5C), Cav3R26Q-HA caused a dramatic accumulation of GFP–dysf in the Golgi complex as determined by triple labeling with an antibody against the Golgi cisternae protein, GM130. Identical results were obtained upon expression of Cav3P104L-HA (results not shown) but not Cav3-HA (Fig. 5B and C). We further confirmed the effect of Cav-3 mutants upon dysferlin subcellular distribution by carrying out a quantitative analysis of the amount of dysferlin at the PM with respect to IC in WT-MEF cells (Fig. 5C). Non-permeabilized cells expressing GFP–dysf or co-expressing GFP–dysf and Cav3R26Q-HA or Cav3P104L-HA were immunolabeled with antibodies to the C-terminal extracellular myc tag of dysferlin at 4°C, fixed, permeabilized and immunolabeled for the HA tag. In cells expressing GFP–dysf, dysferlin was mainly localized at the PM as demonstrated by a PM/IC ratio value of 2.4. However, expression of either Cav3R26Q-HA (PM/IC 0.33±0.12) or Cav3P104L-HA (PM/IC 0.4±0.19) caused a significant reduction in surface dysferlin (Fig. 5C). Ultrastructural analysis of the distribution of GFP–dysf and Cav3R26Q-HA co-expressed in BHK cells, revealed dysferlin and Cav3R26Q-HA co-localizing in IC membranes, with virtually no labeling at the PM (Fig. 5D and E). Identical results were obtained with the Cav-1 mutant, Cav1P132L (a mutation described in breast cancer) (50,51), with a missense mutation at the analogous position to Cav3P104L (results not shown).
We next examined the specificity of the effect of Cav-3 mutants on post-Golgi trafficking. As caveolins have been implicated in glycosyl phosphatidylinositol (GPI)-anchored protein trafficking to the PM (52), we examined whether expression of Cav-3 dystrophy mutants would sequester GPI-anchored proteins in the Golgi complex by utilizing the MEFs as a model system to study this effect. Transient expression of GPI-GFP in MEFs showed GPI labeling at the PM with no localization to the Golgi complex (Fig. 5G). Expression of Cav3R26Q-HA had no detectable effect upon GPI-GFP targeting to the PM in MEF cells as judged qualitatively and using the above quantitative assay (Fig. 5H). These results suggest that retention of dysferlin in the Golgi complex is a specific effect of Cav-3 mutants.
Taken together, these data suggest that mutant forms of caveolin specifically affect the trafficking of dysferlin through the biosynthetic pathway. The effect is a feature of both Cav-1 and -3 and is not a muscle-specific phenomenon.
Caveolin deficiency perturbs PM targeting of dysferlin
We made use of MEF cells derived from WT and Cav-1 null (KO) mice. These cells have no detectable caveolae as they lack Cav-1 (as well as the muscle-specific isoform Cav-3). This provides a powerful system to analyze the dependence of dysferlin on caveolin for trafficking. Expressed GFP–dysf was localized to the PM and to punctate IC structures in both WT- and KO-MEFs (Fig. 6A and B). In WT-MEFs, some GFP–dysf positive, apparently intracellular, structures also labeled for endogenous Cav-1 (Fig. 6A). Although dysferlin reached the surface in KO-MEFs, as demonstrated by labeling non-permeabilized cells expressing GFP–dysf with antibodies against its C-terminal extracellular myc-tag (refer to Materials and Methods), we noticed a consistent increase in dysferlin-positive structures inside the cell (Fig. 6B). This internal staining appeared as puncta mainly concentrated in the perinuclear region but distinct from the evident Golgi staining observed with expression of mutant caveolins. Quantitation of surface to IC (refer to Materials and Methods) dysferlin revealed a highly significant difference in GFP–dysf ability to traffic to the PM between WT- (PM to IC ratio 2.4±1.04) versus KO-MEF cells (PM to IC ratio 0.47±0.22) (Fig. 6C). Although dispersed punctate labeling in the cell periphery was also evident, this pool might have been underestimated by the quantitation method employed. We predicted that if we re-expressed Cav1-HA in KO-MEFs we would restore dysferlin trafficking to the PM. We co-expressed GFP–dysf and HA-tagged Cav-1 (Cav1-HA) in KO-MEFs (Fig. 6D) and quantified the amount of dysferlin at the PM with respect to IC (refer to Materials and Methods) (Fig. 6E). KO-MEFs co-expressing GFP–dysf and Cav1-HA showed a distribution of dysferlin similar to that seen in WT-MEFs (refer to Fig. 6A) demonstrating that the ability of dysferlin to traffic to the PM has been rescued by expression of Cav-1 (Fig. 6D). Quantitative analysis revealed that the dysferlin was efficiently re-localized to the PM (PM to IC ratio 2.04±0.94; Fig. 6D and E). We conclude that caveolin is essential for efficient retention at, or delivery of dysferlin to the PM.
DISCUSSION
Mutations in Cav-3 or in dysferlin have been associated with muscle disease. In addition, dystrophy-associated mutations of Cav-3 result in down-regulation of Cav-3 from the PM and in an abnormal distribution of dysferlin (7,30). However, little is known about the cell biology underlying dysferlin mislocalization and the precise role of Cav-3 mutants in MD. In this work, we have described the subcellular distribution of dysferlin with respect to Cav-3 and show for the first time that muscle disease-associated Cav-3 mutants affect dysferlin traffic to the PM. We demonstrate that co-expression of epitope-tagged dysferlin and mutated Cav-3 or Cav-1 cause a dramatic accumulation of dysferlin in the Golgi complex in both muscle and non-muscle cells. Furthermore dysferlin traffic to the cell surface is impaired in the absence of Cav-1 and Cav-3. Our results suggest that caveolins are necessary for PM localization of dysferlin. The reduced sarcolemmal expression of dysferlin seen in an RMD patient with a missense mutation in the CAV3 gene, Cav3R26Q, may be because of retention of dysferlin in the Golgi complex by mutated Cav-3. In view of the postulated role of dysferlin in muscle repair (9,45), our results provide new insights into the molecular mechanisms involved in Cav-3-associated muscle disease.
Dysferlin and Cav-3 are localized to sarcolemmal domains and to the T-tubule system
We have examined the subcellular distribution of dysferlin with respect to Cav-3 in mature muscle fibers, the C2C12 model system and in a heterologous system in which an epitope-tagged dysferlin was expressed in non-muscle cells. In isolated mature muscle fibers, both Cav-3 and dysferlin localize to the sarcolemma, labeling specific domains of the cell surface. Cav-3 and dysferlin show a similar labeling pattern at the sarcolemma but only show limited overlap as judged by confocal microscopy. This suggests that dysferlin is not exclusively localized to caveolae. This is consistent with our electron microscopic observations of epitope-tagged dysferlin expressed in fibroblasts; expressed dysferlin localized to the cell surface but was not enriched in caveolae. However, limited co-localization was observed in intracellular caveolin-positive structures.
In addition to surface labeling of mature muscle, dysferlin was also evident within transverse tubular elements consistent with significant dysferlin levels within the T-tubule system. Cav-3 is also found within the T-tubules of mature muscle but at a lower density than at the sarcolemma (33,47) and it appears that the ratio of T-tubule labeling to sarcolemmal labeling is much higher for dysferlin than for Cav-3 (Fig. 1). However, during development, and in C2C12 myotubes, Cav-3 is concentrated in the developing T-tubule system (33) and we have shown that it extensively co-localizes with dysferlin in this membrane system. Interestingly, in MEFs, expressed dysferlin also shows some limited co-localization in apparently internal caveolin-rich structures, which are yet to be defined.
Dysferlin trafficking; role of caveolins in a novel Golgi exit pathway
Using a model system to study dysferlin trafficking, we have shown that dysferlin is efficiently transported to the cell surface when expressed in muscle or non-muscle cells. Caveolin plays an important role in dysferlin localization to the PM as shown by two independent sets of experiments. First, co-expression of Cav-3 or Cav-1 mutants causes accumulation of dysferlin in the Golgi complex. Ultrastructural studies showed that the mutant caveolin proteins accumulated throughout the Golgi complex, with no particular enrichment in a specific subdomain, but were not efficiently transported to the PM. A patient with the Cav3R26Q mutation showed reduced expression of dysferlin at the sarcolemma consistent with mistargeting of dysferlin. Secondly, we have shown that in cells lacking Cav-1 and Cav-3 (and caveolae), PM association of dysferlin is greatly reduced. Interestingly, these cells showed no accumulation of dysferlin in the Golgi complex, but dysferlin accumulated in punctate structures within the cell. Furthermore re-expressing Cav-1 in these cells restored dysferlin traffic to the PM. The identification of the site of dysferlin accumulation should provide insights into the mechanism by which dysferlin trafficking is disrupted in the caveolae-null cells. The dramatic effect of caveolin mutants on trafficking of dysferlin is not because of a general perturbation of post-Golgi traffic. We have shown that GFP-GPI trafficking is unaffected by the expression of Cav-3 mutants. From analysis of patients' muscle samples it appears that only a subset of proteins are affected by mutations in the CAV3 gene; for example surface levels of β-dystroglycan, dystrophin and sarcoglycans generally appear to be unaffected (24,25,35,37,53), whereas α-dystroglycan levels have been shown to be reduced (27,53). A detailed analysis of the subset of proteins whose trafficking is disrupted by Cav-3 mutants is now required.
These results strongly suggest that dysferlin trafficking is closely linked to trafficking of caveolin. Does the perturbation of dysferlin trafficking by caveolin mutants reflect a direct interaction between the two proteins? Cav-3 and dysferlin have been shown to co-immunoprecipitate in muscle (34,35) but this was evident only under mild detergent conditions, which may not disrupt all membrane domains. Our own studies with co-expressed WT Cav-3/Cav-3 mutants with dysferlin in BHK cells suggest a limited co-immunoprecipitation of the Cav-3 proteins and dysferlin (results not shown). The fact that Cav-3 and dysferlin only show limited co-localization at the sarcolemma in mature muscle fibers and expressed dysferlin is not greatly enriched in caveolae argues that an association between caveolins and dysferlin may occur during the secretory pathway but not at the cell surface. A similar scenario has been suggested for the angiotensin-II type 1 receptor [AT1R; (14)]. Trafficking of the AT1R to the cell surface was inhibited by caveolin mutants and was perturbed in Cav-1 KO cells but AT1R did not localize to caveolae at the cell surface. For both dysferlin and AT1R, an association of the caveolin scaffolding domain with putative caveolin binding domains has been proposed (14,34). However, other studies have suggested that the scaffolding domain of caveolin is partially inserted into the lipid bilayer (54). An interesting possibility is that this region is exposed and available for association with cargo proteins only during transport through the Golgi complex when caveolin is in a detergent-soluble monomeric form (55). In contrast, this region would be inserted into the membrane in surface caveolae. This may coordinate dissociation of cargo proteins with formation of caveolae or caveolar carriers. This is consistent with the exposure of specific caveolin epitopes close to the caveolin scaffolding domain, within the Golgi complex but not at the PM (55).
How then does dysferlin reach the cell surface in cells with and without caveolin? Possible clues come from real-time studies of caveolin trafficking (56). This study showed that ‘quanta’ of caveolin-GFP formed within the Golgi and fused directly with the cell surface to generate caveolae. These carriers appear to be distinct from those mediating transport of other known cargo proteins to the cell surface. Together with other studies these results suggest a model in which caveolae are formed in the Golgi complex as caveolin oligomerizes and acquires the detergent-insolubility properties characteristic of a ‘lipid raft’ protein (40,55). Caveolin may therefore be required for the formation of distinct exocytic carriers with a specialized set of transported cargo proteins. We postulate that dysferlin and the AT1R may be examples of such cargo proteins. Mutant caveolins would ‘freeze’ this exit pathway by binding to cargo proteins but not forming caveolar carriers in the Golgi. This would cause accumulation of trapped cargo in the Golgi complex. This is not seen in caveolin-KO cells. Assuming that caveolin is required for generation of post-Golgi caveolar carriers, as it is for biogenesis of caveolae at the cell surface (57), in caveolin-KO cells this pathway would be absent. Cargo proteins would then be randomly incorporated into other exocytic carriers rather than remaining trapped in the Golgi complex. Alternatively, such proteins might be transported within the same carriers that lack caveolin. This raises the question of why in the absence of caveolin dysferlin accumulation at the cell surface is perturbed. One possible explanation is that the caveolin-enriched carriers are targeted to specific domains of the surface where dysferlin dissociates away from caveolin and is retained and stabilized. This would be particularly important in muscle fibers which have distinct surface domains, accessed by different exocytic trafficking pathways (46) and would be consistent with the high level of co-localization of Cav-3 and dysferlin during development in the T-tubule system.
In conclusion, these studies show an essential role for caveolin in dysferlin trafficking. The effect of caveolin mutants and caveolin loss can be reconstituted in a simple non-muscle cell culture system and is a feature of both Cav-1 and Cav-3. The effect of caveolin-associated mistargeting or Golgi retention of dysferlin is likely to have functional consequences relevant to caveolin-related muscle disease. The localization of dysferlin to both the sarcolemma and T-tubules suggests that dysferlin may play a role in both membrane systems. In view of the essential role of dysferlin in muscle repair after sarcolemmal damage, elucidation of the cellular pathways and molecular mechanisms involved in dysferlin trafficking and function will be crucial to understanding muscle function in health and disease.
MATERIALS AND METHODS
DNA constructs, reagents and antibodies
Cell culture reagents were purchased from Gibco-BRL, Gaithersburg, MD. The following antibodies were used: mouse anti-dysferlin (NCL-Hamlet), mouse anti-spectrin (NCL-SPEC1) (Novocastra Laboratories, Newcastle-Upon-Tyne, UK), mouse anti-Cav-3, mouse anti-GM130 (BD Biosciences, Lexington, KY, USA), mouse anti-triadin (Affinity Bioreagents, CO, USA), rabbit polyclonal antibody generated against the conserved region of Cav3 [characterized previously (58)], rabbit anti-HA (gift from Dr T. Nilsson, Gothenburg University, Gothenburg, Sweden), and rabbit anti-dysferlin (Abcam, Cambridge, UK). Secondary antibodies conjugated to Alexa Fluor 488, 350 (Molecular Probes, OR, USA) and CY3 (Jackson Immunoresearch Laboratories, PA, USA) were used. HRP-conjugated secondary antibodies were from Zymed Laboratories. Supersignal substrate was obtained from Pierce Chemical Company, Rockford, IL, USA. All other chemicals and reagents were purchased from Sigma–Aldrich Chemical Company, St Louis, MO, USA.
HA-tagged Cav3 point mutation R26→Q (Cav3R26Q-HA) was generated by PCR using mouse HA-tagged Cav3 cDNA as a template. The following forward 5′ GATAGACTTGGTGAACCAGGATCCCAAGAACATC 3′, and reverse 5′ GATGTTCTTGGGATCCTGGTTCACCAAGTCTATC 3′ primers were used. This final construct was sequenced using ABI-PRISM BigDye Terminator v3.1 (Applied Biosystems, Foster City, CA, USA) in the Australian Genome Research Facility, University of Queensland (Brisbane). Cav3-HA, Cav3P104L-HA and Cav1P132L-HA were generated as described previously (19,38,58). The complete human dysferlin cDNA sequence was isolated in a series of clones using RT-PCR from human skeletal muscle cDNA (BD-Clontech) and ligated together to form a single clone. The cDNA sequence was verified and found to be identical to the published sequence (Accession AF075575) with the exception of three silent changes (T2200C, C2803T and T3172C). This clone was inserted adjacent to the EGFP coding sequence isolated from pEGFP1-C1 (BD-Clontech) to form an N-terminal fusion between GFP and dysferlin in the expression vector pcDNA4/TO/MycHis. Expression of the complete fusion protein has been confirmed in multiple cell lines. The GPI-GFP and Cav1-HA constructs were gifts from C. Zurzolo (Institut Pasteur, France) and D. Brown (State University of New York at Stony Brook, USA), respectively.
Cell culture and transfection
Muscle fibers were isolated from the flexor digitorum brevis muscle from adult female mice using the method of Bekoff and Betz (1977) (59), and described previously by Rahkila et al. (1996) (60). Muscle fibers were maintained MEM with 20% (v/v) FCS, 1% (v/v) horse serum 2 mml-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin was added. Fibers were used for experiments after 12 h. C2C12 cells were cultured as described by Way and Parton (1995) (19). Primary MEFs were obtained from 13.5-day-old embryos from either Cav1 knockout or WT mice (61). To immortalize MEFs, cells were propagated according to the 3T3-passaging protocol (i.e. 3×105 cells were plated per 60 mm dish every 3 days) as previously described and characterized (52,61).
Cells were grown on glass coverslips or on 35 mm Petri dishes (C2C12 cells) and cDNAs were expressed by transient transfection using Lipofectamine 2000 (Gibco-BRL) according to manufacturer's directions.
Immunofluorescence and microscopy
Frozen 8 µm muscle cryosections were fixed with methanol:acetone (1:1) for 4 min at room temperature. After fixation, cells were blocked in 2% (v/v) BSA in PBS, and incubated with primary (overnight at 4°C) and secondary antibodies (1 h at room temperature) diluted in block solution. Images were taken on a Leica SP2 laser scanning confocal microscope. Isolated muscle fibers were labeled as previously described by (46). Immunolabeling of C2C12, MEFs and BHK cells were carried out as described previously (62,63). For dysferlin labeling, cells were fixed with methanol:acetone for 4 min at room temperature, for all other labelings cells were fixed with 4% PFA.
Fluorescent images were obtained using a Zeiss LSM 510 META confocal microscope system (Carl Zeiss, Germany) or an Olympus Provis AX-70 epifluorescence microscope. For confocal images, single optical sections were captured using Plan apochromatic 63X 1.4 NA oil immersion objective. Epifluorescent images were captured using a charge-coupled device 300ET-RCX camera (DAGE-MTI, Michigan City, IN, USA) with Plan Apo 40X (NA 1.35) or 60X (NA 1.4) oil objectives. Images were processed and merged using Adobe Photoshop 7.0 software (Adobe Systems, Mountain View, CA, USA). To facilitate comparison, identical imaging and processing parameters were used for all figures.
Surface labeling and single cell fluorescence quantification
3T3-immortalized MEF cells (52,61) transiently expressing GFP-dysferlin-myc or GPI-GFP alone, or co-expressing GFP-dysferlin-myc or GPI-GFP and Cav3R26Q-HA or Cav3P104L-HA were incubated at 4°C for 30 min with mouse anti-myc or anti-GFP antibody (1:100, in CO2 independent media/0.1% BSA), respectively. Cells were next fixed in 4% PFA, permeabilized with 0.1% saponin and non-specific sites of antibody adsorption were blocked with 0.2% BSA/0.2% FSG/PBS. Cells were incubated with polyclonal anti-HA antibody followed with Cy3-labeled goat anti-mouse and Alexa Fluor 350-labeled anti-rabbit secondary antibodies.
To quantify the fluorescence intensity of dysferlin or GPI pool at the PM, one region of interest was traced around the cell and for intracellular perinuclear/Golgi pools one region of interest was drawn around the perinuclear region. The average pixel intensity for PM and intracellular/Golgi pools were measured using Adobe Photoshop 7.0 software after subtraction of background outside the cell or cytoplasm overlap. Images used for quantification corresponding to the different treatments were each done on 15–20 cells; performed in parallel and captured at the same contrast and intensity. Results were expressed as the ratio of fluorescent intensity at the PM versus intracellular pool.
Electron microscopy
Immunoelectron microscopy of ultrathin cryosections was performed as described previously (64). BHK cells were transfected with GFP–dysf, Cav3-HA or Cav3P104L-HA or co-transfected with GFP–dysf and Cav3R26Q-HA were washed once in 0.1 M PHEM buffer (pH 6.9) and fixed either in 4% PFA overnight at 4°C, or in 2% PFA/0.2% glutaraldehyde in 0.1 M PHEM buffer (pH 6.9) for 1 h at room temperature and then replaced with 2% PFA in 0.1 M PHEM buffer (pH 6.9) and fixed overnight at 4°C.
Immunoblot analysis
Western blot analysis of human skeletal muscle was performed as described previously (65) using NCL-Hamlet (1:1000) and anti-Cav3 (1:1000) antibodies. Samples were obtained with approval from the Ethics Committee at the Children's Hospital at Westmead.
ACKNOWLEDGEMENTS
The authors would like to thank R. Luetterforst for technical assistance, Dr Susan Nixon, Annika Stark and Matthew Kirkham for preparation of primary MEF cells, Dr Michelle Hill for generating the immortalized MEF cell line, Matthew Kirkham and Dr Piers Walser for assistance with quantitation and statistical analysis. We also thank members of the Parton group for critical reading of the manuscript. Confocal microscopy was performed at the ACRF/IMB Dynamic Imaging Facility for Cancer Biology, established with funding from the Australian Cancer Research Foundation. The Institute for Molecular Bioscience is a Special Research Centre of the Australian Research Council. This research was supported by grants from the National Health and Medical Research Council of Australia (to R.G.P. and K.N.N.), from the Muscular Dystrophy Campaign and Association Francais contre les Myopathies (AFM) (to K.B. and S.H.L.), and from the Muscular Dystrophy Association Development (to S.T.C).
Conflict of Interest statement. None declared.
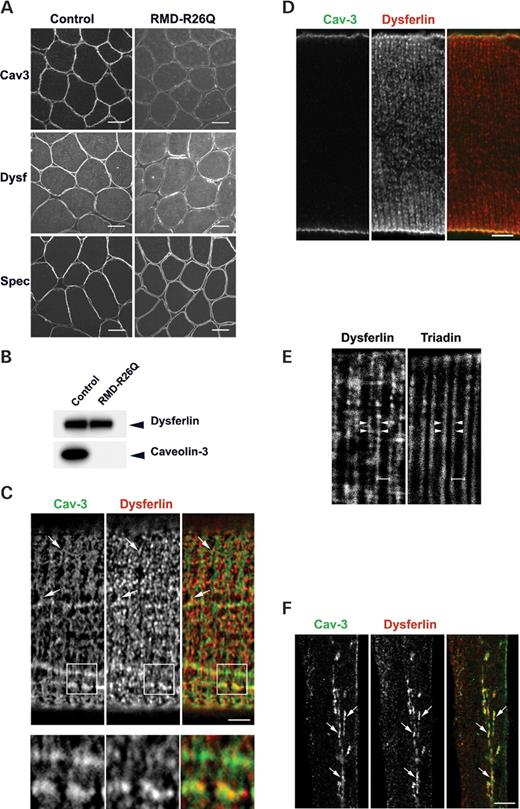
Figure 1. Localization of endogenous dysferlin and Cav-3 in human skeletal muscle, isolated muscle fibers and C2C12 myotubes. (A) Dysferlin and Cav-3 localization in control and RMD-R26Q patient. In patient muscle, both dysferlin and Cav-3 show significantly reduced immunolabeling of the sarcolemma. Spectrin is shown as a control of membrane integrity. (B) Western blot analysis of Cav-3 and dysferlin expression in control and RMD-R26Q patient human skeletal muscle. Cav-3 expression is undetectable in the patient, whereas expression of dysferlin is normal. (C–F) Dysferlin and Cav-3 are targeted to similar sarcolemmal domains. (C–E) Isolated mice muscle fibers in culture were co-labeled with anti-dysferlin (NCL-Hamlet) and anti-Cav-3 (C and D), or with anti-dysferlin and anti-triadin (E) antibodies. (C) Confocal image over the surface of the muscle fiber shows Cav-3 labeling as a highly organized lattice along the length of the muscle fiber. Dysferlin has a similar staining pattern, mainly localized to transversally oriented striations that show partial co-localization with Cav-3 (arrows, inset). (D) Confocal image taken within the interior of the muscle fiber shows dysferlin labeling localized to transversally oriented strands in the region of the T-tubule system. Cav-3 staining is predominantly sarcolemmal. (E) Dysferlin and triadin are localized to the region of the T-tubule system. Confocal image taken within the interior of the muscle fiber shows dysferlin and triadin labeling co-localized (arrowheads) to transversally oriented strands. (F) C2C12 myotubes in culture were co-labeled with anti-dysferlin (NCL-Hamlet) and anti-Cav-3 antibodies. Fluorescent images of C2C12 myotubes showing similar labeling pattern of endogenous Cav-3 and dysferlin. Labeling is localized predominantly in tubular structures within the cytoplasm (arrows) where there is extensive, but incomplete co-localization. Bar: A, 40 µm; C and D, 200 µm; E, 1 µm; F, 20 µm.
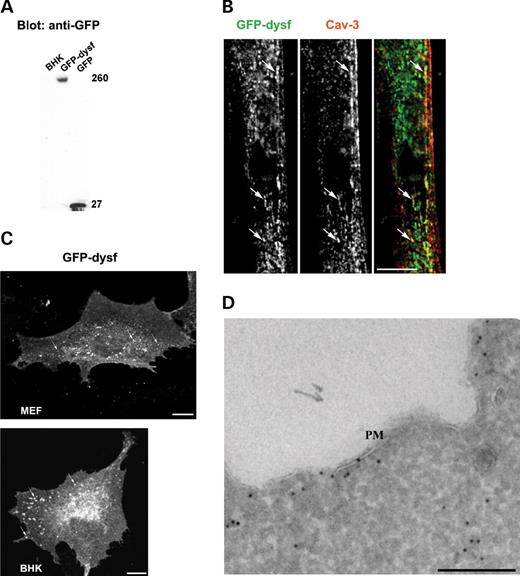
Figure 2. Expression and localization of a GFP-tagged dysferlin construct in muscle and non-muscle cell lines. (A) BHK cells were transiently transfected with either GFP or GFP–dysf and immunoblotted using anti-GFP antibody. GFP–dysf appears as a single polypeptide of 260 kDa. (B and C) GFP–dysf is targeted to the PM and to IC structures in both muscle and non-muscle cell lines. (B) C2C12 myotubes were transiently transfected with GFP-tagged dysferlin and labeled with anti-Cav-3 antibody. GFP–dysf and endogenous Cav-3 labeling are localized to the PM and to IC tubular network (arrows) showing extensive co-localization mainly in the reticular network within the cytoplasm. (C) In MEF and BHK cells expressing GFP-tagged dysferlin, GFP–dysf is targeted to the PM and punctate structures throughout the cytoplasm (arrows). (D) Electron micrograph of BHK cells expressing GFP–dysf. GFP–dysf is localized to the PM and to vesicular structures near the PM as revealed by immunogold labeling. Bars: A–C, 20 µm; D, 200 nm.
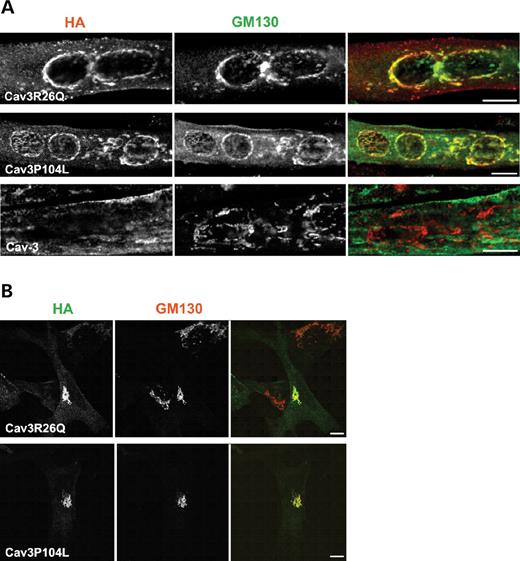
Figure 3. Disease-associated mutants of Cav-3 are retained in the Golgi complex in muscle and non-muscle cell lines. (A) C2C12 were transfected with Cav3R26Q-HA, Cav3P104L-HA or Cav3-HA and co-labeled with anti-HA and anti-GM130 antibodies. In C2C12 myotubes, either epitope-tagged Cav3R26Q or Cav3P104L, but not epitope-tagged Cav3, accumulate in the Golgi complex as demonstrated by co-localization with the Golgi marker, GM130. (B) Similarly, in MEF cells expressing either Cav3R26Q-HA or Cav3P104L-HA and co-labeled with anti-HA and anti-GM130 antibodies, Cav3R26Q and Cav3P104L are retained in the Golgi complex. Bars: 10 µm.
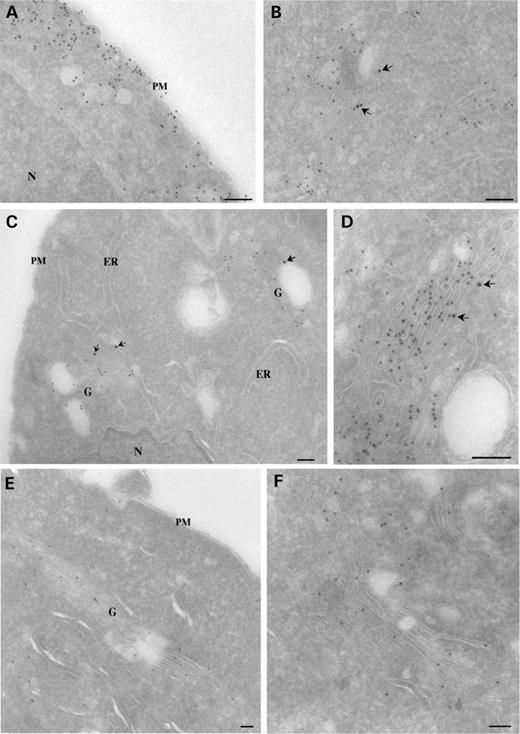
Figure 4. Immunoelectron microscopic detection of Cav-3, Cav3P104L and Cav3R26Q. Cav3-HA (A and B), Cav3P104L-HA (C and D) and Cav3R26Q-HA (E and F) were transfected into BHK cells. After allowing expression overnight, cells were incubated with cycloheximide for 30 min before fixation. The cells were then processed for cryosectioning and immunoEM. Cryosections were double-labeled for HA (A–D), detected using goat anti-rabbit 10 nm gold and GM130 detected using goat anti-mouse 15 nm gold (arrows), or were single-labeled for HA (E and F). Cav3-HA associates with surface caveolae (A) and with the Golgi complex, labeled by GM130 (B). In contrast, Cav3P104L-HA is not detectable on the PM or the endoplasmic reticulum (ER) but is concentrated in the Golgi complex (G) as shown in the low magnification overview in (C) and at higher magnification in (D). A similar distribution is seen for the Cav3R26Q-HA mutant as shown in a low magnification overview of a low expressing cell (E) and with a higher magnification view of the Golgi complex (F). Labeling for Cav3P104L-HA and Cav3R26Q-HA is evident throughout the Golgi complex and is associated with both cisternae and tubular/vesicular profiles (D and F). Bars: 100 nm.
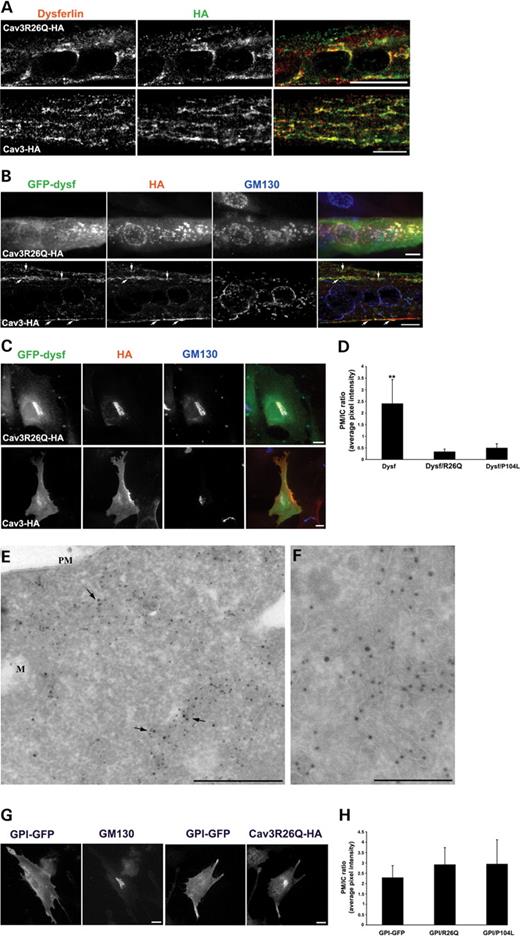
Figure 5. Disease-associated mutants of Cav-3 cause retention of dysferlin in the Golgi complex in muscle and non-muscle cell lines. (A) Differentiated C2C12 myotubes were transfected with HA-tagged Cav3R26Q or WT Cav-3 and labeled with anti-dysferlin (NCL-Hamlet) and anti-HA antibodies. In C2C12 myotubes, expression of Cav3R26Q-HA, but not of WT Cav3-HA, causes endogenous dysferlin to redistribute and co-localize with Cav3R26Q in a perinuclear region. Differentiated C2C12 myotubes (B) and WT-MEFs (C) were co-transfected with GFP–dysf and HA-tagged Cav3R26Q or WT Cav-3 and labeled with anti-HA and anti-GM130 antibodies. (B) In differentiated C2C12 myotubes, GFP–dysf is dramatically redistributed to the Golgi apparatus upon co-expression of Cav3R26Q-HA, as judged by triple labeling with anti-GM130 antibody. WT Cav-3 expression does not affect GFP–dysf localization; dysferlin co-localizes with Cav-3 at the PM and in IC tubules (arrows). (C) WT-MEFs co-expressing GFP–dysf and Cav3R26Q-HA resulted in a similar retention in the Golgi complex as in C2C12 myotubes. Control cell expressing WT-Cav3 and GFP–dysf shows no change in dysferlin localization. (D) Expression of Cav3R26Q-HA or Cav3P104L-HA causes a significant depletion of dysferlin from the PM. Surface labeling of dysferlin and quantification of PM and IC pools of dysferlin were performed as described in Materials and Methods. Results are expressed as PM/IC ratio (average pixel intensity). (E and F) GFP–dysf (15 nm gold particle) and Cav3R26Q-HA (10 nm gold particle) were co-expressed in BHK cells and processed for immunoelectron microscopy. Dysferlin and Cav3R26Q co-localize to internal tubular/vesicular profiles (arrows), with no detectable labeling at the PM. (G) Expression of Cav3R26Q-HA does not affect GPI-GFP targeting to the PM. GPI-GFP is localized at the PM in MEF cells and this localization remains unaffected upon expression of Cav3R26Q-HA. Quantification of PM and intracellular pools of GPI-GFP were performed as described in Materials and Methods. The mean fluorescence intensity of GPI associated with the PM (anti-GFP and Cy3 labeling) and intracellular structures (GFP labeling) was measured and expressed as PM/IC ratio (H). Error bars are mean±SD (n=15–20); **P<0.0001. Bars: A–C, G, 10 µm; E and F, 500 nm.
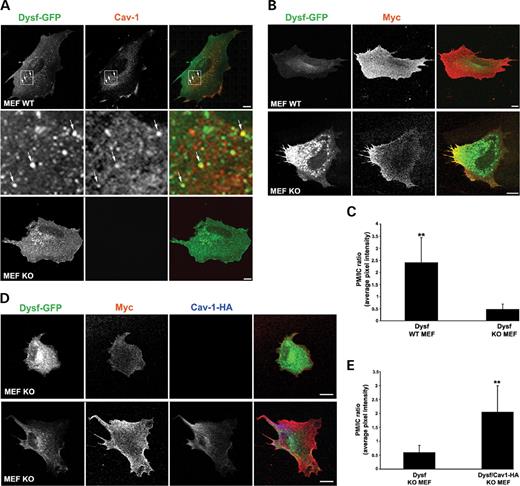
Figure 6. Dysferlin transport to the cell surface is dependent on caveolin. WT- and KO-MEFs were transfected with GFP–dysf and labeled with either rabbit anti-caveolin (A) or mouse anti-myc (B) antibodies followed by incubation with Cy3-conjugated anti-rabbit or -mouse IgG antibody, and visualized by confocal microscopy. (A) GFP–dysf localizes to the PM and to punctate structures (arrows, inset) throughout the cytoplasm of WT- and KO-MEFs with limited co-localization with endogenous caveolin in WT cells. (B and C) Lack of caveolin causes a significant depletion of dysferlin from the PM. Surface labeling of dysferlin and quantification of PM and IC pools of dysferlin were performed as described in Materials and Methods. Both WT- and KO-MEFs show uniform myc labeling at the PM, however KO-MEFs show an increase in dysferlin-positive IC puncta with respect to WT cells (B and C). (D and E) Dysferlin traffic to the PM is rescued by expression of epitope-tagged Cav-1 in KO-MEFs. KO-MEFs were transfected with GFP–dysf or co-transfected with GFP–dysf and Cav1-HA and labeled with rabbit anti-HA or mouse anti-myc antibodies. Surface labeling of dysferlin and quantification of PM and IC pools of dysferlin show a significant increase in dysferlin at the PM upon expression of Cav1-HA (D and E). The mean fluorescence intensity of dysferlin associated with the PM (myc labeling) and IC structures (GFP labeling) was measured and expressed as PM/IC ratio (C and E). Error bars are mean±SD (n=15–25); **P<0.0001. Bars: A and B, 10 µm.
References
Durbeej, M. and Campbell, K.P. (
Bashir, R., Britton, S., Strachan, T., Keers, S., Vafiadaki, E., Lako, M., Richard, I., Marchand, S., Bourg, N., Argov, Z. et al. (
Yasunaga, S., Grati, M., Cohen-Salmon, M., El-Amraoui, A., Mustapha, M., Salem, N., El-Zir, E., Loiselet, J. and Petit, C. (
Britton, S., Freeman, T., Vafiadaki, E., Keers, S., Harrison, R., Bushby, K. and Bashir, R. (
Davis, D.B., Delmonte, A.J., Ly, C.T. and McNally, E.M. (
Liu, J., Aoki, M., Illa, I., Wu, C., Fardeau, M., Angelini, C., Serrano, C., Urtizberea, J.A., Hentati, F., Hamida, M.B. et al. (
Matsuda, C., Aoki, M., Hayashi, Y.K., Ho, M.F., Arahata, K. and Brown, R.H., Jr (
Illa, I., Serrano-Munuera, C., Gallardo, E., Lasa, A., Rojas-Garcia, R., Palmer, J., Gallano, P., Baiget, M., Matsuda, C. and Brown, R.H. (
Bansal, D. and Campbell, K.P. (
Montesano, R., Roth, J., Robert, A. and Orci, L. (
Parton, R.G. (
Scheiffele, P., Verkade, P., Fra, A.M., Virta, H., Simons, K. and Ikonen, E. (
Verkade, P., Harder, T., Lafont, F. and Simons, K. (
Wyse, B.D., Prior, I.A., Qian, H., Morrow, I.C., Nixon, S., Muncke, C., Kurzchalia, T.V., Thomas, W.G., Parton, R.G. and Hancock, J.F. (
Roy, S., Luetterforst, R., Harding, A., Apolloni, A., Etheridge, M., Stang, E., Rolls, B., Hancock, J.F. and Parton, R.G. (
Kurzchalia, T.V. and Parton, R.G. (
Razani, B., Schlegel, A. and Lisanti, M.P. (
Scherer, P.E., Lewis, R.Y., Volonte, D., Engelman, J.A., Galbiati, F., Couet, J., Kohtz, D.S., van Donselaar, E., Peters, P. and Lisanti, M.P. (
Way, M. and Parton, R.G. (
Segal, S.S., Brett, S.E. and Sessa, W.C. (
Song, K.S., Scherer, P.E., Tang, Z., Okamoto, T., Li, S., Chafel, M., Chu, C., Kohtz, D.S. and Lisanti, M.P. (
Doyle, D.D., Upshaw-Earley, J., Bell, E. and Palfrey, H.C. (
McNally, E.M., de Sa Moreira, E., Duggan, D.J., Bonnemann, C.G., Lisanti, M.P., Lidov, H.G., Vainzof, M., Passos-Bueno, M.R., Hoffman, E.P., Zatz, M. et al. (
Minetti, C., Sotgia, F., Bruno, C., Scartezzini, P., Broda, P., Bado, M., Masetti, E., Mazzocco, M., Egeo, A., Donati, M.A. et al. (
Carbone, I., Bruno, C., Sotgia, F., Bado, M., Broda, P., Masetti, E., Panella, A., Zara, F., Bricarelli, F.D., Cordone, G. et al. (
Betz, R.C., Schoser, B.G., Kasper, D., Ricker, K., Ramirez, A., Stein, V., Torbergsen, T., Lee, Y.A., Nothen, M.M., Wienker, T.F. et al. (
Vorgerd, M., Ricker, K., Ziemssen, F., Kress, W., Goebel, H.H., Nix, W.A., Kubisch, C., Schoser, B.G. and Mortier, W. (
Merlini, L., Carbone, I., Capanni, C., Sabatelli, P., Tortorelli, S., Sotgia, F., Lisanti, M.P., Bruno, C. and Minetti, C. (
Tateyama, M., Aoki, M., Nishino, I., Hayashi, Y.K., Sekiguchi, S., Shiga, Y., Takahashi, T., Onodera, Y., Haginoya, K., Kobayashi, K. et al. (
Figarella-Branger, D., Pouget, J., Bernard, R., Krahn, M., Fernandez, C., Levy, N. and Pellissier, J.F. (
Fischer, D., Schroers, A., Blumcke, I., Urbach, H., Zerres, K., Mortier, W., Vorgerd, M. and Schroder, R. (
Kubisch, C., Schoser, B.G., von During, M., Betz, R.C., Goebel, H.H., Zahn, S., Ehrbrecht, A., Aasly, J., Schroers, A., Popovic, N. et al. (
Parton, R.G., Way, M., Zorzi, N. and Stang, E. (
Matsuda, C., Hayashi, Y.K., Ogawa, M., Aoki, M., Murayama, K., Nishino, I., Nonaka, I., Arahata, K. and Brown, R.H., Jr (
Capanni, C., Sabatelli, P., Mattioli, E., Ognibene, A., Columbaro, M., Lattanzi, G., Merlini, L., Minetti, C., Maraldi, N.M. and Squarzoni, S. (
Yabe, I., Kawashima, A., Kikuchi, S., Higashi, T., Fukazawa, T., Hamada, T., Sasaki, H. and Tashiro, K. (
Sugie, K., Murayama, K., Noguchi, S., Murakami, N., Mochizuki, M., Hayashi, Y.K., Nonaka, I. and Nishino, I. (
Carozzi, A.J., Roy, S., Morrow, I.C., Pol, A., Wyse, B., Clyde-Smith, J., Prior, I.A., Nixon, S.J., Hancock, J.F. and Parton, R.G. (
Galbiati, F., Volonte, D., Minetti, C., Chu, J.B. and Lisanti, M.P. (
Ren, X., Ostermeyer, A.G., Ramcharan, L.T., Zeng, Y., Lublin, D.M. and Brown, D.A. (
Galbiati, F., Razani, B. and Lisanti, M.P. (
Roberts HL, L.H., Day B, North KN (
Anderson, L.V., Davison, K., Moss, J.A., Young, C., Cullen, M.J., Walsh, J., Johnson, M.A., Bashir, R., Britton, S., Keers, S. et al. (
Piccolo, F., Moore, S.A., Ford, G.C. and Campbell, K.P. (
Bansal, D., Miyake, K., Vogel, S.S., Groh, S., Chen, C.C., Williamson, R., McNeil, P.L. and Campbell, K.P. (
Rahkila, P., Takala, T.E., Parton, R.G. and Metsikko, K. (
Ralston, E. and Ploug, T. (
Davis, D.B., Doherty, K.R., Delmonte, A.J. and McNally, E.M. (
Tang, Z., Scherer, P.E., Okamoto, T., Song, K., Chu, C., Kohtz, D.S., Nishimoto, I., Lodish, H.F. and Lisanti, M.P. (
Lee, H., Park, D.S., Razani, B., Russell, R.G., Pestell, R.G. and Lisanti, M.P. (
Hayashi, K., Matsuda, S., Machida, K., Yamamoto, T., Fukuda, Y., Nimura, Y., Hayakawa, T. and Hamaguchi, M. (
Sotgia, F., Razani, B., Bonuccelli, G., Schubert, W., Battista, M., Lee, H., Capozza, F., Schubert, A.L., Minetti, C., Buckley, J.T. et al. (
Herrmann, R., Straub, V., Blank, M., Kutzick, C., Franke, N., Jacob, E.N., Lenard, H.G., Kroger, S. and Voit, T. (
Arbuzova, A., Wang, L., Wang, J., Hangyas-Mihalyne, G., Murray, D., Honig, B. and McLaughlin, S. (
Pol, A., Martin, S., Fernandez, M.A., Ingelmo-Torres, M., Ferguson, C., Enrich, C. and Parton, R.G. (
Tagawa, A., Mezzacasa, A., Hayer, A., Longatti, A., Pelkmans, L. and Helenius, A. (
Fra, A.M., Williamson, E., Simons, K. and Parton, R.G. (
Luetterforst, R., Stang, E., Zorzi, N., Carozzi, A., Way, M. and Parton, R.G. (
Bekoff, A. and Betz, W. (
Rahkila, P., Alakangas, A., Vaananen, K. and Metsikko, K. (
Razani, B., Engelman, J.A., Wang, X.B., Schubert, W., Zhang, X.L., Marks, C.B., Macaluso, F., Russell, R.G., Li, M., Pestell, R.G. et al. (
Carozzi, A.J., Ikonen, E., Lindsay, M.R. and Parton, R.G. (
Richards, A.A., Stang, E., Pepperkok, R. and Parton, R.G. (
Martin, S., Millar, C.A., Lyttle, C.T., Meerloo, T., Marsh, B.J., Gould, G.W. and James, D.E. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}