




Abstract
Bardet–Biedl syndrome (BBS) is characterized by obesity, retinopathy, polydactyly, cognitive impairment, renal and cardiac anomalies as well as hypertension and diabetes. The nine known BBS genes do not appear to belong to the same functional category; yet mutation of these genes results in a nearly identical pleiotropic phenotype. Although the precise functions of the BBS proteins have yet to be determined, current data support a role in cilia function and intraflagellar transport. To gain insight into the biological processes controlled by BBS genes, we embarked on studies of six BBS orthologues from zebrafish. Knockdown of zebrafish bbs2, bbs4, bbs5, bbs6, bbs7 or bbs8 results in disruption of Kupffer's vesicle (KV), a ciliated organ thought to play a role in left–right patterning. KV defects are due to a progressive loss of cilia within the vesicle and result in subsequent alterations to organ laterality. We also note a specific defect altering retrograde melanosome transport. These studies are the first to comprehensively compare the diverse group of BBS genes in parallel and demonstrate a common role in intracellular trafficking, indicating that BBS proteins are involved in general organelle trafficking.
INTRODUCTION
Bardet–Biedl syndrome (BBS, MIM 209 900) is a pleiotropic genetically heterogeneous disorder characterized by obesity, retinopathy, polydactyly, renal malformations and functional abnormalities, learning disabilities and hypogenitalism (1–3). BBS patients also have an increased incidence of diabetes mellitus, hypertension and congenital heart disease (1,4,5). To date, nine BBS genes have been identified (6–17). Understanding the pathophysiology of BBS syndrome is of great interest because of the fact that components of the phenotype are prevalent in the general population.
Although nine BBS genes have been identified, little is known about the molecular mechanisms of this disorder and the precise function of the BBS proteins. BBS4 and BBS8 contain tetratricopeptide repeat (TPR) domains, indicating that they interact with other proteins (6,13). BBS6 (also known as MKKS) has sequence homology to the alpha subunit of the Thermoplasma acidophilum thermosome (18), a prokaryotic chaperonin complex with similarity to a eukaryotic chaperonin called tailless complex polypeptide ring complex (19). The BBS3 protein belongs to the ARF/ARL family of small GTP binding proteins, at least some of which are involved in trafficking vesicle-associated proteins (14,16). The recently identified BBS9 protein (also known as B1) has no similarity to the other known BBS proteins and the function is unknown (17). The other known BBS proteins (BBS1, BBS2, BBS5 and BBS7) are also novel proteins (7,11,12,15).
Recent work indicates that a common feature of BBS genes is that they are involved in ciliary function and intraflagellar transport (IFT). This hypothesis is supported by the fact that highly conserved homologs of BBS proteins are found in ciliated organisms (14,15,20,21) and several BBS gene products have been shown to localize to basal bodies in Caenorhabditis elegans (13,15,16,22). BBS4 was shown to localize to the centriolar satellites of centrosomes and basal bodies of primary cilia, where it interacts with components of the dynein transport machinery (23). Furthermore, C. elegans mutants of BBS7 and BBS8 have defects in IFT (13,22). Knockout mouse models of BBS2, BBS4 and BBS6 fail to form flagella during spermatogenesis, although these animals develop other cilia. Silencing of BBS5 in Chlamydomonas leads to the absence of flagella (15). Absence of mouse Bbs2, Bbs4 or Bbs6 does not disrupt cilia formation in photoreceptors, the sensory ciliated cells of the retina. However, these proteins appear to be required for maintenance of the photoreceptor (21,24,25), and absence of mouse Bbs2 or Bbs6 results in mislocalization of rhodopsin, indicating a defect in intracellular trafficking and/or IFT (24,25).
One intriguing aspect of BBS is the fact that although each molecular form of the disease exhibits a very similar phenotype, the gene products do not have extensive sequence homology with each other, appear not to directly physically interact, nor belong to the same functional category of proteins. Numerous questions remain concerning the role of BBS gene products, including whether the function of these proteins is limited to IFT, or whether these proteins play a more general role in intracellular trafficking; whether these proteins are involved in bidirectional intracellular and IFT trafficking and whether these proteins play a role in organelle trafficking. In order to answer these questions and to gain further insight into the biological processes shared by these genes, we identified the zebrafish ortholog of six BBS genes (bbs2, bbs4, bbs5, bbs6, bbs7 and bbs8), used antisense morpholino oligonucleotides (MOs) to individually knockdown expression of each and evaluated the effect on multiple developmental and physiological processes. We initially focused this study on zebrafish bbs7 and then extended the study to five additional BBS genes. We observed strikingly overlapping phenotypes in the morphant embryos, including Kupffer's vesicle (KV) disruption leading to alterations in the left–right (l–r) asymmetry. This defect does not impact early cilia formation but rather presents itself as a progressive reduction in the number of cilia within KV over time. Of particular note, we also observe a critical role for BBS proteins in intracellular trafficking of organelles (melanosomes). The loss-of-function of each BBS gene appears to predominantly affect retrograde transport of melanosomes, indicating a functional role in vesicle transport. Our data lead to a unifying hypothesis that all BBS proteins are involved in retrograde intracellular transport. This study of multiple developmental and physiological processes in the whole animal uncovers some novel requirements for BBS function in vertebrates and can be used for common clues to the disease pathophysiology.
MATERIALS AND METHODS
Zebrafish BBS ortholog cloning
We performed BLAST analysis of BBS genes against EMBL/GenBank and genome project (Sanger Centre) databases and detected highly homologous zebrafish sequences for each of the known human BBS genes. Specific primers were designed to amplify zebrafish BBS sequences from cDNA libraries (Supplementary Material, primer sequences). Total RNA extracted from embryos collected at specific developmental stages using TRIzole® reagent (GibcoBRL) was poly(A) selected and reverse transcribed (SuperScriptTM II, Invitrogen). PCR products were cloned into the pSTBlue vector (Merk) and sequence-verified on an ABI3730 sequencer.
Whole mount in situ hybridization
Embryos were fixed with 4% paraformaldehyde/phosphate buffered saline (PBS) overnight. Embryos >24 h post-fertilization (h.p.f.) were treated with pigment inhibitor. Sense and antisense digoxigenin-UTP RNA probes (Roche) were synthesized from the linearized templates [bbs2, bbs4, bbs6 and bbs7 antisense digested with SalI, and synthesized with T7 RNA polymerase; and for the sense probes, BamHI digested and transcribed with SP6 RNA polymerase]. Other probes used include: charon (BamHI, SP6), nkx2.5, (EcoRI, T7), lft-1 (MluI, T7), lft-2 (MluI, T7) and fkd2 (ApaI, T3). Whole mount in situ hybridization (WMISH) was performed as described by Long and Rebagliati (26).
Morpholino antisense oligonucleotides
Antisense MO were designed and purchased from Gene Tools, LLC (Supplementary Material, MO sequences). MOs were microinjected into one to eight cell embryos at concentrations ranging from 200 to 500 µm.
Reverse transcriptase–polymerase chain reaction
Total RNA extracted from splice-blocking morphants (10–15 embryos/stage) at the following ages: 14–16, 24, 30, 48, 70–72, 96 and 120 h.p.f., was synthesized into cDNA and amplified with gene-specific primers flanking the targeted exon for bbs2, bbs6 and bbs7. Expression of β-actin was analyzed as an internal control (Supplementary Material, primers sequences).
Rescue of knockdown phenotypes
Morpholino-resistant wild-type bbs RNA was subcloned into pCS2+ expression vector (27). Synthetic RNA was made with Ambion mMessage mMachine high-yield Capped RNA transcription kit. mRNA (50 ng/µl bbs6 and both 50 and 100 ng/µl bbs7) was co-injected with appropriate MO or five-mismatch MOs.
Analysis of KV and cilia
Live embryos were photographed using the Zeiss Axiocam Camera. Vesicles in embryos with a diameter less than one-third that of wild-type were considered reduced and embryos in which vesicles could not be morphologically identified were scored as absent. For immunolocalization, embryos were fixed. Immunofluorescence was performed according to a standard protocol using a mouse anti-acetylated tubulin monoclonal antibody (Sigma T 6793, 1:800 dilution) and secondary antibody, Alexa 488-conjugated goat anti-mouse antibody (Molecular Probes A-11001, 1:200 dilution), followed by nuclei counterstaining TO-PRO®-3 (Molecular Probes T-3605). Embryos were imaged on the Leica SP2 AOBS scanning laser confocal microscope system with 63× magnification and 4× zoom.
Melanosome transport assay
Day 5 larvae were exposed to epinephrine (50 mg/ml, Sigma, E4375) added to embryo medium (28) for a final concentration of 500 µg/ml. Melanosome retraction was continuously monitored under the microscope and the endpoint was scored when all melanosomes in the head and the trunk were perinuclear. To evaluate anterograde movement, larvae were exposed to epinephrine to induce melanosome retraction, washed with excess embryo medium and then either returned to a dark incubator with periodic observation or exposed to caffeine (1 mg/ml, pH 7.0). Some zebrafish were pre-treated with brefeldin A (2.5 µg/ml) for 45 min.
Eye histology and ultrastructure
Five-day post-fertilization (5 d.p.f.) larvae were fixed with 4% paraformaldehyde prepared in 10 mm PBS for ∼2 h. Embryos were rinsed in PBS and were infiltrated in 30% sucrose solution in PBS for several hours. Embryos were then embedded in optimal cutting temperature compound and cryosectioned. Sections were collected at 7 µm and were stained with hematoxylin/eosin stain. For transmission electron microscopy (TEM), 5 or 6 d.p.f. larvae were fixed by immersion in one-half strength Karnovsky's fixative (29). Specimens were post-fixed in osmium tetraoxide, dehydrated and embedded in Spurr's resin. Ultrathin sections were collected through the level of the eyecup to assess photoreceptor ultrastructure. Sections were viewed on a JEOL 1230 TEM and digital images collected.
RESULTS
Identification of zebrafish BBS orthologs
In order to identify zebrafish orthologs, we performed BLAST analysis of BBS genes against the Ensembl genomic database for Danio rerio, designed gene-specific primers and cloned zebrafish BBS genes. Conceptual translation and alignment with human BBS sequences demonstrate that zebrafish BBS genes have high levels of homology to the human genes (Table 1). We performed WMISH with each of the bbs genes and demonstrated maternal expression of each of these genes with continued ubiquitous expression of these genes. Figure 1 shows this data for bbs7. Both maternal (Fig. 1A) and zygotic expression (Fig. 1C) are observed when compared with sense control embryos (Fig. 1B and D). bbs7 expression at 36 h.p.f. is enriched in an anterior domain, including the brain and eye fields and the pericardial region (Fig. 1E). At 48 h.p.f., expression in the brain and eyes persists and strong expression is also observed in the developing limb bud (Fig. 1G). Expression of zebrafish bbs7 transcripts continues throughout embryonic day 5, as determined by RT–PCR (Fig. 1I). Expression of other zebrafish bbs transcripts (bbs2, bbs4 and bbs6) appears ubiquitous at 0–24 h.p.f. (as determined by whole mount in situ) and is detectable by RT–PCR throughout embryonic day 5 (data not shown).
Knockdown of zebrafish bbs gene expression
In order to evaluate whether the loss of zebrafish BBS function results in embryonic defects, we used different antisense MO to disrupt translation targeting the initiator methionine and to disrupt RNA processing targeting splice sites for each of the bbs genes evaluated in this study (bbs2, bbs4, bbs5, bbs6, bbs7 and bbs8). The fluorescein tagged bbs MOs micro-injected into zebrafish embryos appear uniformly distributed up to and beyond 48 h.p.f. The bbs splice-donor MOs induce skipping of the targeted exons and the splice-acceptor MOs induce intron inclusion, both resulting in aberrant splice transcripts. Aberrant splice forms were cloned and sequence-verified. Aberrant splice forms were assayed by RT–PCR to confirm the efficacy of the MO-induced knockdown of endogenous expression. As an example of the success of the knockdown efficiency, complete knockdown of bbs7 at 30 h.p.f. (Fig. 1J) in embryos displaying morphological phenotypes was achieved. We note complete knockdown up to 36 h.p.f. (data not shown). MO-mediated gene knockdown persists throughout 120 h.p.f., but by 70 h.p.f., the wild-type bbs7 transcript is observed along with the aberrant transcript. The populations of affected embryos with complete loss of bbs7 were used for analyses to reveal essential roles in key developmental processes. As validation and extension of the bbs7 loss-of-function studies, knockdown phenotypes of additional bbs genes (bbs2, bbs4, bbs5, bbs6 and bbs8) were evaluated. We performed a gross morphological examination, and morphant embryos did not differ in general body size and morphology compared to controls. No differences were noted with respect to the head and eye, although some embryos displayed an enlarged pericardial cavity.
Disruption of zebrafish bbs function results in KV defects
The first morphological phenotype as a result of bbs gene knockdown is alteration of an early forming ciliated vesicle. KV is a transient structure containing monocilia and is proposed to have a function similar to the mouse node relative to the l–r axis asymmetry (30–33). The KV originates from dorsal forerunner cells during gastrulation, expands as a spherical vesicle during the somite stages and is readily observable in the tail-bud region (34–36).
bbs7 morphants were evaluated for alterations to KV formation. At the 10–13 somite stage, KV in untreated and control morphants is a readily identifiable ovoid shaped vesicle in the posterior midline, with an average diameter of 60 µm (Fig. 2A, white scale bar of 100 µm). At this stage, KV has reached an approximate maximum diameter that gradually regresses in size after the 15 somite stage. In the bbs7 morphants analyzed at the 10–13 somite stage, 28% of embryos have severely reduced KV diameter to less than one-third the size of controls (Fig. 2B) or complete morphological absence of KV (Fig. 2C). Morphant embryos injected with a lower concentration of bbs7 MO had a reduced impact on KV morphology (7%, Fig. 2D), supporting a dose-dependent response to knockdown. Embryos injected with bbs2, bbs4, bbs5, bbs6 and bbs8 MOs also displayed a KV defect, and this defect is not present in control morphants (Table 2, Supplementary Material, Fig. S1). A significance at P<0.001 was calculated for each bbs morphant on the basis of Fisher's exact test. As BBS protein function has been implicated in cilia biogenesis, we next explored the status of KV cilia in bbs morphants.
bbs morphants have KV cilia defects
KV contains a population of protruding cilia that can be decorated with anti-acetylated tubulin. Evaluation of the cilia organization in bbs7 morphants was performed by immunohistochemistry at three distinct developmental stages: 5–7 somite stage, when KV becomes morphologically visible, 8–10 somite stage, when KV is typically increasing in diameter and 11–13 somite stage, the stage we use to score the morphological KV defect. In the wild-type and control morphants (Fig. 2E–G), cilia can be observed in a spherical pattern in the region of KV. Cilia number, length and overall KV area increase with developmental age. bbs7 morphants scored as reduced KV appear similar to wild-type at the 5–7 somite stage (Fig. 2H) but have a partially reduced domain by the 8–10 somite stage (Fig. 2I) and markedly reduced cilia number and domain by the 11–13 somite stage (Fig. 2J). Interestingly, in bbs7 morphants without a morphologically visible KV, cilia are present but appear to aggregate into a small domain at the 5–7 somite stage (Fig. 2K). By the 8–10 somite stage, cilia appear shorter and dispersed (Fig. 2L), and few cilia, if any, are present at the 11–13 somite stage (Fig. 2M). Similarly, immunohistochemical analysis of bbs2, bbs4 and bbs6 morphants revealed few or no cilia at the 10–13 somite stage (Supplementary Material, Fig. S2). At the early stages of KV formation, the cilia pattern is similar in wild-type and bbs7 morphants. However, the number of cilia regresses or disappears with developmental age. These data are consistent with the mouse photoreceptor degeneration data (21,24,25) and suggest that BBS function is required for the continued maintenance of cilia and/or the survival of ciliated cells. Despite complete knockdown during these stages, we cannot rule out the possibility of maternal protein contributing to cilia formation.
Knockdown of bbs alters molecular aspects of the l–r asymmetry
Disorganized cilia have been previously described in the context of lost nodal function, resulting in a reduced or absent KV (37). A recently identified nodal antagonist, charon, has been shown to be critical for proper l–r patterning in zebrafish embryos, and the charon transcript is localized to the cells surrounding KV (38). At the 10–13 somite stage, there is typically horseshoe-shaped charon-expression domain flanking the posterior KV (Fig. 3A). charon expression is still present in bbs2, bbs4, bbs6 and bbs7 morphant embryos, although not in the prototypical shape, as shown with a bbs7 morphant with no morphologically visible KV (Fig. 3B, Supplementary Material, Fig. S1). Thus, retention of charon expression in morphants indicates that the cells enveloping the posterior region of KV are still present. The lack of the prototypical shape appears to be due to a collapsed KV, which is consistent with the observed cilia alterations in the morphants.
We then set out to determine whether molecular components of the l–r patterning are altered in bbs morphants. We monitored expression of the lefty-1(lft-1) and lefty-2(lft-2) genes, a subclass of transforming growth factor-β proteins critical for the l–r asymmetry (reviewed in Shier, 2003). In the zebrafish 21–23 somite stage embryo, lft-1 and lft-2 are expressed in the left lateral plate mesoderm, heart field and brain, and lft-1 also has expression in the midline of the embryo (Fig. 3C). In bbs7 morphants scored as absent KV, lft-1 and lft-2 expression fell into three classes: 55% showed normal left-sided expression, 36% demonstrated bilateral expression in the lateral plate and brain (Fig. 3D) and 9% had reversed right-sided expression (Fig. 3E). Bilateral and reversed lft-1 and lft-2 expression was also observed in bbs6 morphants, whereas control morphants demonstrated 100% left-sided lft-1 and lft-2 expression.
bbs morphants are predisposed to organ laterality defects
The bbs knockdown-induced KV and cilia defects as well as altered charon and lft expression support a role for BBS function in organ laterality. At 22–24 h.p.f., the heart tube elongates from the midline and migrates (termed ‘jogging’) to the left. This is one of the first morphological indications of the l–r asymmetry and embryos can be scored for left-ward (Fig. 3F), right-ward (Fig. 3H) or no laterality bias (Fig. 3G). Heart jogging was evaluated by both morphology and by WMISH using a heart-specific probe, nkx2.5 (39). When evaluating the entire population of bbs7 morphants, we observed abnormal jogging (no jog or reversed jog) in 14% of the injected embryos when compared with 1–3% abnormal jogging in uninjected or control morphants (Fig. 3I). The heart tube continues to undergo morphogenesis and between 36 and 48 h.p.f. loops into a prototypical S-shape, placing the ventricle anterior and to the right of the atrium (also referred to as D-looping). Two alterations to heart looping were observed, L-looping (opposite orientation of the ventricle relative to the atrium) and No-looping (a straight heart tube). bbs7 morphants demonstrated a 14% frequency of abnormal looping.
To determine whether KV defect was linked to altered organ laterality, a subset of bbs7 morphants that displayed altered KV were selected and analyzed for heart looping and jogging. Among bbs7 MO-injected embryos with no morphological KV, 59% displayed altered heart jogging, whereas reduced KV embryos demonstrated 34% abnormal jogging (Fig. 3J). Embryos displaying left jogging typically formed a D-loop, whereas right-ward jogging formed an L-loop and no jog displayed a random looping distribution (Fig. 3I). Altered jogging and looping is observed in bbs4, bbs5, bbs6 and bbs8 morphants with a range of penetrance (8–20% for jogging and 5–29% for looping, Table 3). These data indicate that heart laterality was compromised in bbs morphants and that jogging and looping are linked (Table 3).
Altered heart laterality observed for the bbs7 lower dose MO injections is at an equivalent frequency to that observed at the higher dose. This finding indicates that the cilia in the reduced KV class continue to degenerate and result in later-stage organ laterality defects. Gut looping scored at 48 h.p.f. is also compromised in bbs7 morphants (Supplementary Material, Fig. S3). Our data suggest that zebrafish BBS function is required for the morphological manifestation of KV and loss-of-function induces disruption of the cilia and predisposes embryos to organ laterality defects.
bbs gene knockdown results in delayed retrograde intracellular transport
In order to determine whether bbs function is required for general cellular transport processes, we monitored transport in a non-ciliated cell population. Zebrafish alter their skin pigmentation by trafficking melanosomes within melanophores in response to visual cues and hormonal stimuli. The melanosome is a lysosome-related organelle, and its bidirectional shuttling between the cell periphery and the perinuclear region is carried out by the microtubule-based molecular motors, kinesin II and dynein. Kinesin II is a plus-end-directed motor responsible for dispersion of melanosomes to the cell periphery (anterograde), whereas dynein is a minus-end-directed motor responsible for melanosome retraction to the perinuclear region (retrograde) (40–43). Taking advantage of the zebrafish pigment, a time course of organelle translocations in the whole animal can be observed. Pigment aggregation (retrograde) or dispersion (anterograde) can be stimulated within minutes upon treatment with epinephrine and caffeine, respectively (44).
In our assay, 5 d.p.f. larvae are dark-adapted to display maximum melanophore dispersion (Fig. 4A). Once exposed to epinephrine, the melanosomes rapidly contract and can be visually evaluated for reduction in pigment size (Fig. 4B). The endpoint is apparent when all the pigment cells show perinuclear accumulation of melanosomes (Fig. 4B and E). Control-injected and untreated embryos rapidly retract their melanosomes (Fig. 4C–E) (t=1.6 and 1.7 min, respectively), whereas bbs7 morphants have significantly delayed melanosome retraction (t=6.6 min, P<0.001). bbs7 morphants fall into two classes; one class initiates retraction but the melanosomes remain dispersed after 4.9 and 5.5 min of exposure (Fig. 4G and H, respectively), but eventually reach the endpoint at 7.2 min. Another class of bbs7 MO-injected embryos show a partial retraction (Fig. 4J) but do not progress beyond that point even after 20 min of exposure (Fig. 4K). All bbs genes knocked down in this study (bbs2, bbs4, bbs5, bbs6, bbs7 and bbs8) resulted in significant delays in melanosome transport (Table 4, Fig. 4L). Vision impairment had no impact on hormonally induced transport because elimination of expression of the cone–rod homeobox gene, crx, generates functionally blind larva (45,46) which responded to epinephrine treatment at the same rate as control morphants (data not shown).
Anterograde transport can be evaluated by the recovery time to full pigment dispersion after larvae are placed in epinephrine-free media or when treated with caffeine. When tested for anterograde response, bbs morphants respond at the same rate as that of wild-type and control morphants evaluated with natural recovery and caffeine-induced pigment dispersion (Table 4).
The overlapping defects observed by knockdown of multiple members of the BBS gene family suggest that BBS proteins play a role in intracellular transport. If the defect is specific to retrograde transport, we should phenocopy the bbs knockdown-induced melanosome defect by compromising transport via another reagent. Brefeldin A is a fungal metabolite that blocks cellular transport (47,48). Wild-type or control morphant embryos, pre-treated with brefeldin A and then exposed to epinephrine display delayed retraction, partial constriction but never reach the perinuclear endpoint or no response at all (Supplementary Material, Table S1). Brefeldin A treatment is reversible, and after washing out the drug, embryos respond to epinephrine treatment. In this assay, brefeldin A treatment, which also predominantly inhibits intracellular retrograde transport, mimics the bbs loss-of-function phenotype.
bbs morphants and ocular morphology and ultrastructure
Retinitis pigmentosa (RP) is a cardinal feature of BBS and results in blindness in human patients. To evaluate the ocular histology of BBS MO knockdown embryos, bbs7 MO-injected embryos sorted by KV defect were fixed at 5–6 d.p.f., an age at which photoreceptor morphogenesis is underway (49). Tissue sections through the eyes of bbs7 MO-injected embryos displayed a clearly defined ganglion cell, inner plexiform, inner nuclear, outer plexiform and outer nuclear layers. Although the inner plexiform layer did appear attenuated in some cases, the overall architecture of the retina appeared normal in bbs7 morphants (Fig. 5B) when compared with wild-type (Fig. 5A). We further explored the status of the outer segments with TEM and found that the ultrastructure of a no KV bbs7 morphant (Fig. 5D) appeared morphologically normal, with well-defined photoreceptor outer segments and membranous disks (compare to Fig. 5C).
Rescue of bbs morphant phenotypes
To confirm that the phenotypes described earlier were bbs-specific, bbs2, bbs6 and bbs7 MO-injected embryos were co-injected with MO-resistant bbs RNA. Wild-type bbs2, bbs6 and bbs7 RNA markedly reduced the incidence of KV abnormalities to levels observed in control-injected embryos (Fig. 6A). We also observed a statistically significant improvement in the epinephrine-induced melanosome retraction rate (Fig. 6B, Supplementary Material, Fig. S4).
DISCUSSION
In order to search for clues to a common biological role or function shared by BBS proteins, we used developmental studies in the zebrafish to model BBS. One intriguing aspect of BBS is the fact that mutation of each of the nine known BBS genes results in a very similar phenotype in humans, even though the gene products do not have extensive sequence homology with each other and do not belong to a common functional category of proteins (6–17). This is the first study to compare loss-of-function phenotypes of multiple BBS genes in parallel in a vertebrate model. Our loss-of-function studies with six zebrafish bbs genes (bbs2, bbs4, bbs5, bbs6, bbs7 and bbs8) show strikingly overlapping phenotypes and indicate a common biological pathway for all BBS genes. In addition, endogenous expression of the zebrafish bbs transcripts in the developing brain, eye, limb buds and cardiovascular regions is consistent with tissues that demonstrate defects in the human syndrome, including retinopathy, polydactyly, learning disabilities, hypertension and congenital heart disease. Interestingly, a defect in retrograde transport, revealed by our knockdown studies in zebrafish, is a feature shared by all six BBS genes tested.
BBS function in cilia
BBS proteins are highly conserved in ciliated organisms (14,15,20,21), and BBS4 localizes to the centriolar satellites of centrosomes and basal bodies of primary cilia (23). Previous studies support a role for BBS proteins in cilia function. In knockout mice, BBS proteins are required for flagella formation during spermatogenesis but are not required for formation of other motile or primary cilia (21,24,25). Evidence from studies in C. elegans indicates that BBS7 and BBS8 are required for normal localization of specific IFT proteins and that genetic mutations of BBS7 and BBS8 result in ciliary defects (13,22). In order to comprehensively evaluate such function of BBS proteins in vertebrates, we set out to knockdown the function of six zebrafish BBS orthologs. In this study, BBS deficiencies alter the morphological formation of a ciliated vesicle in the early embryo. We demonstrate the presence of cilia early in development, followed by the premature progressive degeneration/loss of these cilia within KV. We also note heart laterality defects and disruption of molecular markers of laterality in the presumptive organ field as well as in the brain. However, in humans, laterality defects are not a commonly observed component of BBS. Given that placental mammals require a functional heart to survive in utero, the frequency of observed laterality defects may not reflect the actual frequency of occurrence. In support of this possibility, heterozygous matings of bbs knockout mice result in ∼15% homozygous offspring instead of the expected 25% (21,24,25). Owing to their small size and external egg lay, zebrafish embryos can survive the early larval stages without a functional heart and cardiovascular system and may reveal a role for BBS proteins not commonly observed in placental mammals.
It is unlikely that the absence of individual BBS proteins results in completely non-functional cilia. Differences between the BBS phenotype and phenotypes commonly attributed to defects in motile cilia indicate that the BBS phenotype cannot be explained solely by disruption of cilia motility. For example, patients with primary ciliary dysfunction commonly have bronchiectasis and sinusitis, whereas these symptoms are not recognized as common features of BBS. Zebrafish embryos with cilia defects demonstrate a characteristic body curvature and cysts in the gut tube (50,51), defects not specifically found in bbs knockdown embryos.
BBS function and intracellular transport
In order to determine whether BBS proteins are involved in general intracellular transport, as opposed to solely an IFT function, we evaluated bbs loss-of-function zebrafish embryos for intracellular transport defects. To this end, we monitored melanosome transport in melanophores, which can be induced by epinephrine. Melanosomes associate with microtubules and are capable of being transported bidirectionally by either kinesin II (anterograde) or dynein (retrograde) motors (41). Knockdown of expression of each zebrafish bbs gene resulted in a significant delay in retrograde melanosome transport, whereas anterograde transport was not altered. Inhibition of retrograde transport indicates that BBS gene products have a role in intracellular transport, specifically retrograde vesicle and organelle transport. We mimicked the retrograde melanosome transport defect by pharmacological treatment with brefeldin A, a fungal metabolite that prevents retrograde intracellular transport. Although the exact role of BBS proteins in the transport process is not known, these proteins could play a role in the maintenance of the microtubule architecture, directly contribute to the function or efficiency of the molecular motors themselves, serve to load cargo onto the molecular motors or serve to detach cargo from actin filaments prior to transport. It is also possible that some BBS genes may play a regulatory role in one or more of these processes. For example, BBS3 belongs to a class of proteins that regulate a wide variety of cellular processes including intracellular transport (14).
A predominant role for BBS genes in retrograde transport when compared with anterograde transport is supported by known defects in melanosome intracellular dispersion in humans and mice. Known defects in anterograde transport result in failure to retain the peripheral distribution of melanosomes, leading to partial albinism and hair color abnormalities (44,52,53), findings not associated with BBS. Intact anterograde transport but defective retrograde transport, as demonstrated here, would not be expected to lead to hair color or albinism phenotypes. Melanosome transport is a general model for the study of intracellular transport of organelles and vesicles. The findings of this study lead to the conclusion that at least some components of the BBS phenotype result from a defect in unidirectional vesicle and organelle transport, specifically dynein-dependent transport towards the minus-end of microtubules. Our results do not exclude the possibility of a subtle effect on anterograde transport (22).
If bbs knock downs target a common pathway, we would predict that the KV defect should be tightly linked to the melanosome transport defect. In fact, the no KV class of bbs morphant (scored prior to 15 h.p.f.) has the greatest delay in transport (observed at 120 h.p.f.). In addition, it is these two specific defects that are rescued by the MO-resistant bbs RNA.
RP is a cardinal feature of BBS and results in blindness in human patients. Given the relatively normal cilia formation and then degeneration in KV, we would predict that the bbs MO-injected zebrafish would have normal retina development but later demonstrate photoreceptor degeneration. Our observation that photoreceptor outer segment morphogenesis is not severely impaired in bbs knockdown is consistent with Bbs2, Bbs4 and Bbs6 knockout mice data demonstrating normal retinal development but later apoptotic death of photoreceptors (21,24).
In summary, the data presented here demonstrate that all six BBS genes studied are necessary for the formation of the ciliated structure known as KV. Our data suggest that the BBS function does not have a general role in differentiation or development of ciliated cells or for the formation of cilia themselves, as other ciliated tissues such as the gut and eye are not impacted. But rather, our data are supportive of a functional role of BBS protein in the maintenance of cilia or in the survival of some ciliated cells, or both. Comprehensive knockdown of these proteins in zebrafish results in phenotypes consistent with a role in cilia function, and this work is the first to specifically identify a requirement for BBS function in KV cilia function and subsequent organ laterality defects. We further demonstrate that all BBS genes studied play a role in intracellular transport, with the most profound effect on retrograde transport. Our data demonstrating a significant delay in retrograde intracellular transport of melanosomes with knockdown of expression of each of the six BBS proteins suggest that BBS genes are involved in organelle and membrane-bound vesicle intracellular transport. We propose that all BBS proteins play a general role in retrograde intracellular trafficking of membrane-bound vesicles and organelles and that a defect in intracellular trafficking leads to cilia dysfunction and loss and in some cases, eventually results in death of specific cell types. The detailed mechanisms by which intracellular transport defects lead to specific components of the BBS phenotype will require additional study.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
The authors thank C. Searby for technical assistance, T. Westfall and H. Griesbach for zebrafish care and WMISH and M. Olvera for technical assistance with electron microscopy. This work was supported by National Institutes of Health Grants P50-HL-55006 (to V.C.S.) and RO1-EY-11298 (to V.C.S. and E.M.S.), RO3 HD045488-01 and ACS no. RSG DDC-106889 (to D.C.S.) and the Carver Endowment for Molecular Ophthalmology (E.M.S. and V.C.S.). V.C.S. and E.M.S. are investigators of the Howard Hughes Medical Institute.
Conflict of Interest statement. None declared.
These authors contributed equally.
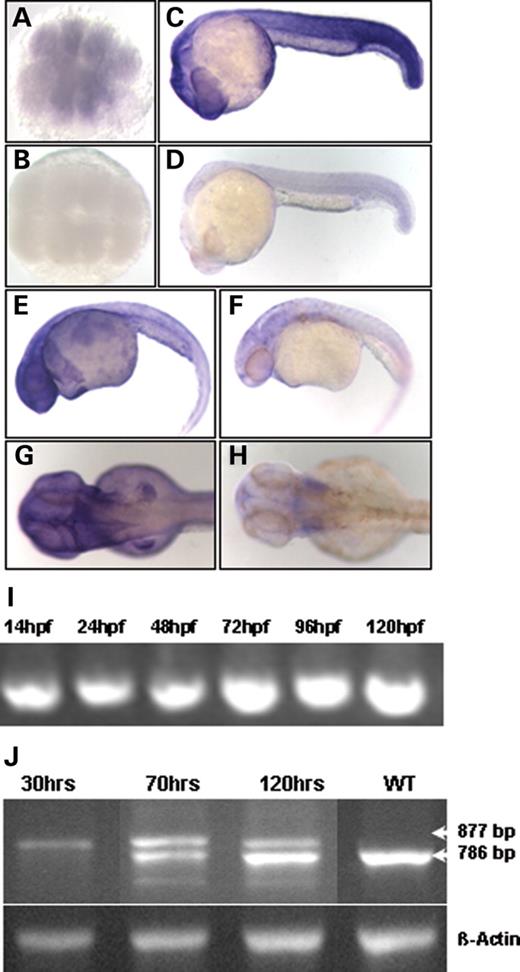
Figure 1. Zebrafish bbs expression is ubiquitous, and splice-block MO results in an altered transcript. Temporal and spatial expression of bbs7 analyzed by WMISH with antisense in (A) 16-cell, (C) 24 h.p.f., (E) 36 h.p.f. and (G) 48 h.p.f. stages and sense controls in (B) 16-cell, (D) 24 h.p.f., (F) 36 d.p.f. and (H) 48 h.p.f. stages. Animal pole view in (A) and (B), lateral view, anterior to the right in (C–H). (I) bbs7 developmental profile by RT–PCR from 14 to 120 h.p.f. (J) RT–PCR products from bbs7 splice-block MO-injected embryos. cDNA libraries were made from a pool of 10 embryos at 30, 70 and 120 h.p.f. The endogenous wild-type band migrates at 786 bp and the altered transcript at 877 bp. Bottom lane shows the β-actin control amplification.
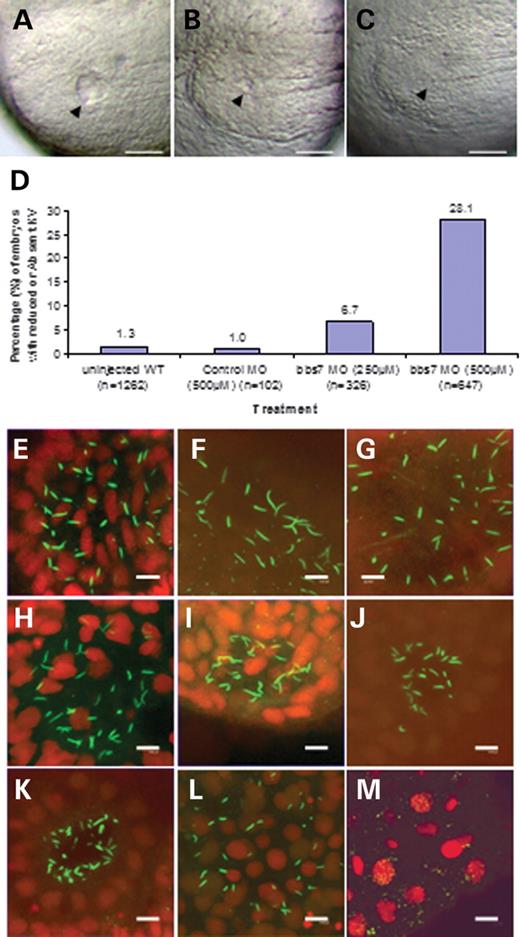
Figure 2.bbs knockdown induces KV cilia defects. Appearance of KV in live-mounted bbs7 morphants with (A) wild-type like, (B) reduced and (C) morphologically absent vesicle (scale bar of 100 µm). The arrowhead indicates vesicle location in the tail-bud posterior to the notochord, anterior to the right. (D) Graphical representation of the percentage of altered KV for each treatment; sample size is noted on the x-axis and the altered percentage is noted at the top of each bar. (E–M) Confocal microscopic images of anti-acetylated tubulin staining of KV cilia (green) counterstained for nuclei (red) and scale bar of 8.0 µm. Control morphant at (E) five to seven somite, (F) 8–10 somite and (G) 11–13 somite stages. bbs7 morphants with reduced KV at (H) five to seven somite, (I) 8–10 somite and (J) 11–13 somite stages. bbs7 morphant with no KV at (K) five to seven somite, (L) 8–10 somite and (M) 11–13 somite stages.
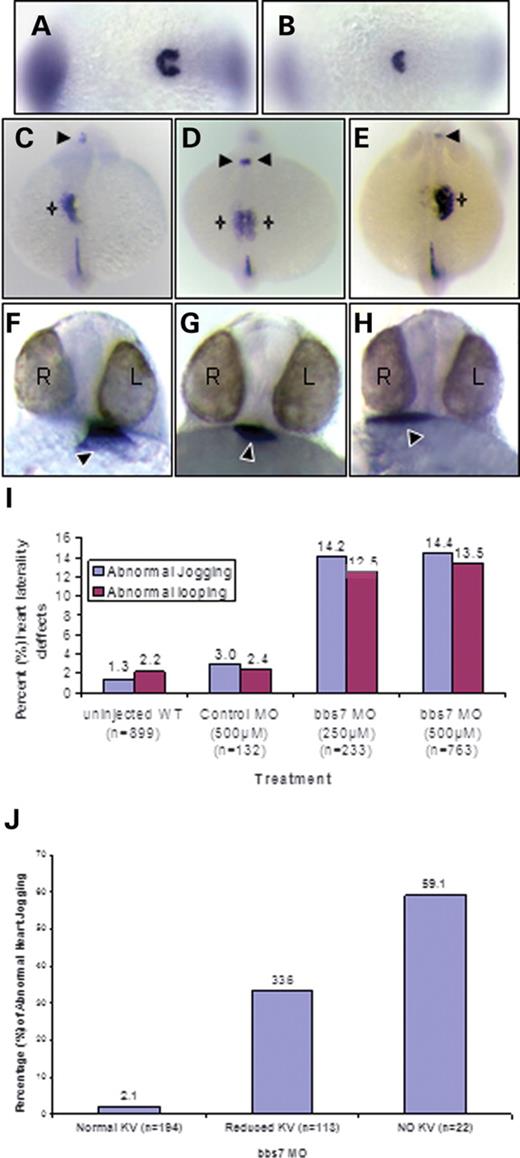
Figure 3.bbs knockdown predisposes embryos to laterality defects. WMISH of charon in 10–13 somite embryos: control (A) and bbs7 morphant (B) with no morphologically visible KV. Posterior view, anterior to the left. Dorsal view of lft-1 and lft-2 expression at the 21–23 somite stages in bbs7; no KV morphants showing (C) left-sided expression in the lateral plate (star) and brain (arrow), (D) bilateral expression and (E) right-sided expression in the lateral plate (star) and brain (arrow). nkx2.5 expression in bbs7 morphants displaying (F) left heart tube migration, (G) no migration and (H) reversed migration to the right. Black arrowhead notes the location of the heart tube relative to the eyes (L, left; R, right) in a ventral view. (I) Percentage of altered heart tube migration (jogging) in blue and looping in red for the total population of bbs7 morphants. (J) Jogging defects in the no KV class. Treatment and sample size noted on the x-axis and altered percentage noted at the top of each bar.
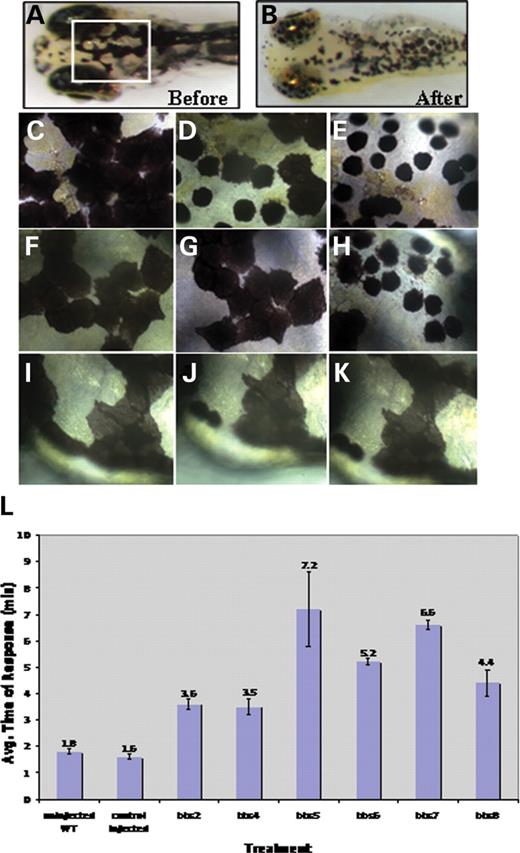
Figure 4.bbs knockdown results in epinephrine-induced melanosome retraction delay. Wild-type 5 d.p.f. larvae prior to treatment (A) and after epinephrine exposure (B). Box denotes the area at higher magnification (40×) (C–K). Wild-type 5 d.p.f. larvae prior to treatment (C), 1.0 min after epinephrine addition (D) and at the endpoint of 1.3 min (E). bbs7 morphant with reduced KV prior to treatment (F), at 4.9 min after epinephrine addition (G) and at 5.5 min (H). Endpoint was 7.15 min for this specific larva, not shown. Dorsal view of bbs7 morphant with no KV prior to treatment (I) at 11.25 min after epinephrine addition (J) and at 27 min (K). (L) Graphical representation with error bars demonstrating a significant delay of epinephrine-induced melanosome retrograde trafficking when compared with wild-type and controls. Treatment (control or bbs-morphant) is noted on the x-axis and average response time in minutes is noted on the y-axis.
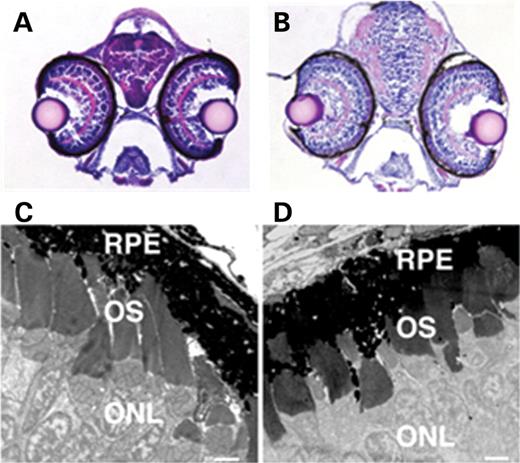
Figure 5. Eye histology and ultrastructure in bbs7 MO-injected embryos. Brightfield images of H&E stained transverse sections collected through the eyes of 5 d.p.f. (A) wild-type and (B) bbs7 morphant, photographed at 20×. Note the appearance of the lens and retina, with prominent inner plexiform layer (apparent as a pink semicircle in both embryos). Normal retinal differentiation was observed in these larvae. TEM analysis of 5–6 d.p.f. (C) wild-type and (D) bbs7 morphant ultrathin sections collected and labeled for retinal pigment epithelium (RPE), outer segments (OS) and outer nuclear layer (ONL). Normal outer segment morphogenesis was observed. Scale bar in (C and D) is 2 µm.
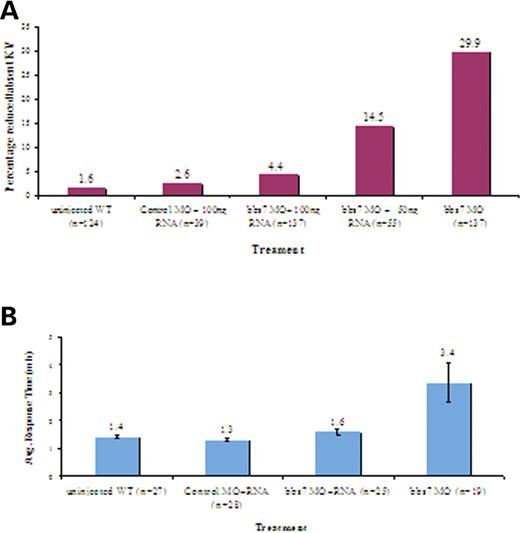
Figure 6. Rescue of knockdown phenotypes with bbs7 RNA. (A) Graphical representation of the percentage of embryos with reduced or no KV for uninjected WT (wild-type), control MO and bbs7 MO-injected embryos. Control MO-injected co-mixed with bbs7 RNA displayed KV defects relative to WT 1.6–2.6%. bbs7 MO injection results in ∼30% KV defects. Co-injection of bbs7 RNA with bbs7 MO results in partial rescue (14.5%) with 50 ng/µl of bbs7 RNA and 94% rescue with 100 ng/µl of bbs7 RNA. (B) Graphical representation demonstrating average response time of epinephrine-induced melanosome retrograde movement. Average response time with error bars noted at the top of each treatment. Note the reduced rate of response with co-injection of bbs7 RNA. Sample size noted in (). For all injections, bbs7 MO doses are 500 µm.
Zebrafish BBS clones
| Gene | Percentage of identity (similarity) to human | Predicted protein domain |
|---|---|---|
| BBS2 | 71 (80) | Novel |
| BBS4 | 76 (88) | TPRs |
| BBS5 | 90 (95) | Novel |
| BBS6 | 41 (63) | Chaperonin |
| BBS7 | 72 (81) | Novel |
| BBS8 | 72 (83) | TPRs |
| Gene | Percentage of identity (similarity) to human | Predicted protein domain |
|---|---|---|
| BBS2 | 71 (80) | Novel |
| BBS4 | 76 (88) | TPRs |
| BBS5 | 90 (95) | Novel |
| BBS6 | 41 (63) | Chaperonin |
| BBS7 | 72 (81) | Novel |
| BBS8 | 72 (83) | TPRs |
Zebrafish BBS clones
| Gene | Percentage of identity (similarity) to human | Predicted protein domain |
|---|---|---|
| BBS2 | 71 (80) | Novel |
| BBS4 | 76 (88) | TPRs |
| BBS5 | 90 (95) | Novel |
| BBS6 | 41 (63) | Chaperonin |
| BBS7 | 72 (81) | Novel |
| BBS8 | 72 (83) | TPRs |
| Gene | Percentage of identity (similarity) to human | Predicted protein domain |
|---|---|---|
| BBS2 | 71 (80) | Novel |
| BBS4 | 76 (88) | TPRs |
| BBS5 | 90 (95) | Novel |
| BBS6 | 41 (63) | Chaperonin |
| BBS7 | 72 (81) | Novel |
| BBS8 | 72 (83) | TPRs |
KV phenotype
| Treatment | Total | Percentage of reduced or absent KV |
|---|---|---|
| Wild-type (pooled from all experiments) | 1671 | 1 |
| Control-MO (500 µM) (for all bbs controls) | 458 | 3 |
| bbs2 (250 µM) | 144 | 38 |
| bbs4 (200 µM) | 160 | 35 |
| bbs5 (500 µM) | 40 | 16 |
| bbs6 (250 µM) | 489 | 29 |
| bbs7 (250 µM) | 326 | 7 |
| bbs7 (500 µM) | 647 | 28 |
| bbs8 (500 µM) | 114 | 18 |
| Treatment | Total | Percentage of reduced or absent KV |
|---|---|---|
| Wild-type (pooled from all experiments) | 1671 | 1 |
| Control-MO (500 µM) (for all bbs controls) | 458 | 3 |
| bbs2 (250 µM) | 144 | 38 |
| bbs4 (200 µM) | 160 | 35 |
| bbs5 (500 µM) | 40 | 16 |
| bbs6 (250 µM) | 489 | 29 |
| bbs7 (250 µM) | 326 | 7 |
| bbs7 (500 µM) | 647 | 28 |
| bbs8 (500 µM) | 114 | 18 |
KV phenotype
| Treatment | Total | Percentage of reduced or absent KV |
|---|---|---|
| Wild-type (pooled from all experiments) | 1671 | 1 |
| Control-MO (500 µM) (for all bbs controls) | 458 | 3 |
| bbs2 (250 µM) | 144 | 38 |
| bbs4 (200 µM) | 160 | 35 |
| bbs5 (500 µM) | 40 | 16 |
| bbs6 (250 µM) | 489 | 29 |
| bbs7 (250 µM) | 326 | 7 |
| bbs7 (500 µM) | 647 | 28 |
| bbs8 (500 µM) | 114 | 18 |
| Treatment | Total | Percentage of reduced or absent KV |
|---|---|---|
| Wild-type (pooled from all experiments) | 1671 | 1 |
| Control-MO (500 µM) (for all bbs controls) | 458 | 3 |
| bbs2 (250 µM) | 144 | 38 |
| bbs4 (200 µM) | 160 | 35 |
| bbs5 (500 µM) | 40 | 16 |
| bbs6 (250 µM) | 489 | 29 |
| bbs7 (250 µM) | 326 | 7 |
| bbs7 (500 µM) | 647 | 28 |
| bbs8 (500 µM) | 114 | 18 |
Heart jogging and looping
| Treatment | Total | Percentage of abnormal jogging (looping) |
|---|---|---|
| Wild-type | 1182 | 1.5 (1.4) |
| Control-MO (500 µM) | 269 | 1.4 (1.5) |
| bbs4 (200 µM) | 114 | 7.0 (8.8) |
| bbs5 (500 µM) | 40 | 18.4 (15.0) |
| bbs6 (250 µM) | 343 | 8.4 (5.2) |
| bbs7 (250 µM) | 233 | 14.2 (12.5) |
| bbs7 (500 µM) | 763 | 14.4 (13.5) |
| bbs8 (500 µM) | 114 | 20.2 (29.0) |
| Treatment | Total | Percentage of abnormal jogging (looping) |
|---|---|---|
| Wild-type | 1182 | 1.5 (1.4) |
| Control-MO (500 µM) | 269 | 1.4 (1.5) |
| bbs4 (200 µM) | 114 | 7.0 (8.8) |
| bbs5 (500 µM) | 40 | 18.4 (15.0) |
| bbs6 (250 µM) | 343 | 8.4 (5.2) |
| bbs7 (250 µM) | 233 | 14.2 (12.5) |
| bbs7 (500 µM) | 763 | 14.4 (13.5) |
| bbs8 (500 µM) | 114 | 20.2 (29.0) |
Heart jogging and looping
| Treatment | Total | Percentage of abnormal jogging (looping) |
|---|---|---|
| Wild-type | 1182 | 1.5 (1.4) |
| Control-MO (500 µM) | 269 | 1.4 (1.5) |
| bbs4 (200 µM) | 114 | 7.0 (8.8) |
| bbs5 (500 µM) | 40 | 18.4 (15.0) |
| bbs6 (250 µM) | 343 | 8.4 (5.2) |
| bbs7 (250 µM) | 233 | 14.2 (12.5) |
| bbs7 (500 µM) | 763 | 14.4 (13.5) |
| bbs8 (500 µM) | 114 | 20.2 (29.0) |
| Treatment | Total | Percentage of abnormal jogging (looping) |
|---|---|---|
| Wild-type | 1182 | 1.5 (1.4) |
| Control-MO (500 µM) | 269 | 1.4 (1.5) |
| bbs4 (200 µM) | 114 | 7.0 (8.8) |
| bbs5 (500 µM) | 40 | 18.4 (15.0) |
| bbs6 (250 µM) | 343 | 8.4 (5.2) |
| bbs7 (250 µM) | 233 | 14.2 (12.5) |
| bbs7 (500 µM) | 763 | 14.4 (13.5) |
| bbs8 (500 µM) | 114 | 20.2 (29.0) |
Melanosome retrograde and anterograde movement
| Treatment | Total | Average response time (min) | P-value | Average caffeine-induced dispersion in minutes (SE) |
|---|---|---|---|---|
| Wild-type | 77 | 1.7 | — | 4.1 (0.1) |
| Control-MO (500 µM) | 60 | 1.6 | 0.2 | — |
| bbs2 (250 µM) | 40 | 3.6 | 2.5×10−10 | 4.5 (0.4) |
| bbs4 (200 µM) | 39 | 3.5 | 7.6×10−9 | 4.3 (0.3) |
| bbs5 (500 µM) | 22 | 7.2 | 1.8×10−4 | — |
| bbs6 (250 µM) | 35 | 3.3 | 3.8×10−25 | 4.5 (0.3) |
| bbs7 (500 µM) | 87 | 6.6 | 4×10−7 | 4.0 (0.3) |
| bbs8 (500 µM) | 76 | 4.3 | 9.7×10−8 | — |
| Treatment | Total | Average response time (min) | P-value | Average caffeine-induced dispersion in minutes (SE) |
|---|---|---|---|---|
| Wild-type | 77 | 1.7 | — | 4.1 (0.1) |
| Control-MO (500 µM) | 60 | 1.6 | 0.2 | — |
| bbs2 (250 µM) | 40 | 3.6 | 2.5×10−10 | 4.5 (0.4) |
| bbs4 (200 µM) | 39 | 3.5 | 7.6×10−9 | 4.3 (0.3) |
| bbs5 (500 µM) | 22 | 7.2 | 1.8×10−4 | — |
| bbs6 (250 µM) | 35 | 3.3 | 3.8×10−25 | 4.5 (0.3) |
| bbs7 (500 µM) | 87 | 6.6 | 4×10−7 | 4.0 (0.3) |
| bbs8 (500 µM) | 76 | 4.3 | 9.7×10−8 | — |
Melanosome retrograde and anterograde movement
| Treatment | Total | Average response time (min) | P-value | Average caffeine-induced dispersion in minutes (SE) |
|---|---|---|---|---|
| Wild-type | 77 | 1.7 | — | 4.1 (0.1) |
| Control-MO (500 µM) | 60 | 1.6 | 0.2 | — |
| bbs2 (250 µM) | 40 | 3.6 | 2.5×10−10 | 4.5 (0.4) |
| bbs4 (200 µM) | 39 | 3.5 | 7.6×10−9 | 4.3 (0.3) |
| bbs5 (500 µM) | 22 | 7.2 | 1.8×10−4 | — |
| bbs6 (250 µM) | 35 | 3.3 | 3.8×10−25 | 4.5 (0.3) |
| bbs7 (500 µM) | 87 | 6.6 | 4×10−7 | 4.0 (0.3) |
| bbs8 (500 µM) | 76 | 4.3 | 9.7×10−8 | — |
| Treatment | Total | Average response time (min) | P-value | Average caffeine-induced dispersion in minutes (SE) |
|---|---|---|---|---|
| Wild-type | 77 | 1.7 | — | 4.1 (0.1) |
| Control-MO (500 µM) | 60 | 1.6 | 0.2 | — |
| bbs2 (250 µM) | 40 | 3.6 | 2.5×10−10 | 4.5 (0.4) |
| bbs4 (200 µM) | 39 | 3.5 | 7.6×10−9 | 4.3 (0.3) |
| bbs5 (500 µM) | 22 | 7.2 | 1.8×10−4 | — |
| bbs6 (250 µM) | 35 | 3.3 | 3.8×10−25 | 4.5 (0.3) |
| bbs7 (500 µM) | 87 | 6.6 | 4×10−7 | 4.0 (0.3) |
| bbs8 (500 µM) | 76 | 4.3 | 9.7×10−8 | — |
References
Green, J.S., Parfrey, P.S., Harnett, J.D., Farid, N.R., Cramer, B.C., Johnson, G., Heath, O., McManamon, P.J., O'Leary, E. and Pryse-Phillips, W. (
Bardet, G. (
Biedl, A. (
Elbedour, K., Zucker, N., Zalzstein, E., Barki, Y. and Carmi, R. (
Harnett, J.D., Green, J.S., Cramer, B.C., Johnson, G., Chafe, L., McManamon, P., Farid, N.R., Pryse-Phillips, W. and Parfrey, P.S. (
Mykytyn, K., Braun, T., Carmi, R., Haider, N.B., Searby, C.C., Shastri, M., Beck, G., Wright, A.F., Iannaccone, A., Elbedour, K. et al. (
Nishimura, D.Y., Searby, C.C., Carmi, R., Elbedour, K., Van Maldergem, L., Fulton, A.B., Lam, B.L., Powell, B.R., Swiderski, R.E., Bugge, K.E. et al. (
Katsanis, N., Ansley, S.J., Badano, J.L., Eichers, E.R., Lewis, R.A., Hoskins, B.E., Scambler, P.J., Davidson, W.S., Beales, P.L. and Lupski, J.R. (
Katsanis, N., Beales, P.L., Woods, M.O., Lewis, R.A., Green, J.S., Parfrey, P.S., Ansley, S.J., Davidson, W.S. and Lupski, J.R. (
Slavotinek, A.M., Stone, E.M., Mykytyn, K., Heckenlively, J.R., Green, J.S., Heon, E., Musarella, M.A., Parfrey, P.S., Sheffield, V.C. and Biesecker, L.G. (
Mykytyn, K., Nishimura, D.Y., Searby, C.C., Shastri, M., Yen, H.J., Beck, J.S., Braun, T., Streb, L.M., Cornier, A.S., Cox, G.F. et al. (
Badano, J.L., Ansley, S.J., Leitch, C.C., Lewis, R.A., Lupski, J.R. and Katsanis, N. (
Ansley, S.J., Badano, J.L., Blacque, O.E., Hill, J., Hoskins, B.E., Leitch, C.C., Kim, J.C., Ross, A.J., Eichers, E.R., Teslovich, T.M. et al. (
Chiang, A.P., Nishimura, D., Searby, C., Elbedour, K., Carmi, R., Ferguson, A.L., Secrist, J., Braun, T., Casavant, T., Stone, E.M. et al. (
Li, J.B., Gerdes, J.M., Haycraft, C.J., Fan, Y., Teslovich, T.M., May-Simera, H., Li, H., Blacque, O.E., Li, L., Leitch, C.C. et al. (
Fan, Y., Esmail, M.A., Ansley, S.J., Blacque, O.E., Boroevich, K., Ross, A.J., Moore, S.J., Badano, J.L., May-Simera, H., Compton, D.S. et al. (
Nishimura, D.Y., Swiderski, R.E., Searby, C.C., Berg, E.M., Ferguson, A.L., Hennekam, R., Merin, S., Weleber, R.G., Biesecker, L.G., Stone, E.M. et al. (
Stone, D.L., Slavotinek, A., Bouffard, G.G., Banerjee-Basu, S., Baxevanis, A.D., Barr, M. and Biesecker, L.G. (
Frydman, J., Nimmesgern, E., Erdjument-Bromage, H., Wall, J.S., Tempst, P. and Hartl, F.U. (
Avidor-Reiss, T., Maer, A.M., Koundakjian, E., Polyanovsky, A., Keil, T., Subramaniam, S. and Zuker, C.S. (
Mykytyn, K., Mullins, R.F., Andrews, M., Chiang, A.P., Swiderski, R.E., Yang, B., Braun, T., Casavant, T., Stone, E.M. and Sheffield, V.C. (
Blacque, O.E., Reardon, M.J., Li, C., McCarthy, J., Mahjoub, M.R., Ansley, S.J., Badano, J.L., Mah, A.K., Beales, P.L., Davidson, W.S. et al. (
Kim, J.C., Badano, J.L., Sibold, S., Esmail, M.A., Hill, J., Hoskins, B.E., Leitch, C.C., Venner, K., Ansley, S.J., Ross, A.J. et al. (
Nishimura, D.Y., Fath, M., Mullins, R.F., Searby, C., Andrews, M., Davis, R., Andorf, J.L., Mykytyn, K., Swiderski, R.E., Yang, B. et al. (
Fath, M.A., Mullins, R.F., Searby, C., Nishimura, D.Y., Wei, J., Rahmouni, K., Davis, R.E., Tayeh, M.K., Andrews, M., Yang, B. et al. (
Long, S. and Rebagliati, M. (
Turner, D.L. and Weintraub, H. (
Westerfield, M. (
Russell, S.R., Mullins, R.F., Schneider, B.L. and Hageman, G.S. (
Supp, D.M., Brueckner, M., Kuehn, M.R., Witte, D.P., Lowe, L.A., McGrath, J., Corrales, J. and Potter, S.S. (
Supp, D.M., Witte, D.P., Potter, S.S. and Brueckner, M. (
Essner, J.J., Amack, J.D., Nyholm, M.K., Harris, E.B. and Yost, H.J. (
Essner, J.J., Vogan, K.J., Wagner, M.K., Tabin, C.J., Yost, H.J. and Brueckner, M. (
Melby, A.E., Warga, R.M. and Kimmel, C.B. (
Cooper, M.S. and D'Amico, L.A. (
D'Amico, L.A. and Cooper, M.S. (
Amack, J.D. and Yost, H.J. (
Hashimoto, H., Rebagliati, M., Ahmad, N., Muraoka, O., Kurokawa, T., Hibi, M. and Suzuki, T. (
Chen, J.N. and Fishman, M.C. (
Skold, H.N., Aspengren, S. and Wallin, M. (
Barral, D.C. and Seabra, M.C. (
Marks, M.S. and Seabra, M.C. (
Blott, E.J. and Griffiths, G.M. (
Nascimento, A.A., Roland, J.T. and Gelfand, V.I. (
Gamse, J.T., Shen, Y.C., Thisse, C., Thisse, B., Raymond, P.A., Halpern, M.E. and Liang, J.O. (
Shen, Y.C. and Raymond, P.A. (
Fujinaga, Y., Wolf, A.A., Rodighiero, C., Wheeler, H., Tsai, B., Allen, L., Jobling, M.G., Rapoport, T., Holmes, R.K. and Lencer, W.I. (
Valderrama, F., Duran, J.M., Babia, T., Barth, H., Renau-Piqueras, J. and Egea, G. (
Brockerhoff, S.E., Rieke, F., Matthews, H.R., Taylor, M.R., Kennedy, B., Ankoudinova, I., Niemi, G.A., Tucker, C.L., Xiao, M., Cilluffo, M.C. et al. (
Kramer-Zucker, A.G., Olale, F., Haycraft, C.J., Yoder, B.K., Schier, A.F. and Drummond, I.A. (
Sun, Z., Amsterdam, A., Pazour, G.J., Cole, D.G., Miller, M.S. and Hopkins, N. (
Wilson, S.M., Yip, R., Swing, D.A., O'Sullivan, T.N., Zhang, Y., Novak, E.K., Swank, R.T., Russell, L.B., Copeland, N.G. and Jenkins, N.A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}