




Abstract
Boundaries between cellular compartments often serve as signaling interfaces during embryogenesis. The coronal suture is a major growth center of the skull vault and develops at a boundary between cells derived from neural crest and mesodermal origin, forming the frontal and parietal bones, respectively. Premature fusion of these bones, termed coronal synostosis, is a common human developmental anomaly. Known causes of coronal synostosis include haploinsufficiency of TWIST1 and a gain of function mutation in MSX2. In Twist1+/− mice with coronal synostosis, we found that the frontal–parietal boundary is defective. Specifically, neural crest cells invade the undifferentiated mesoderm of the Twist1+/− mutant coronal suture. This boundary defect is accompanied by an expansion in Msx2 expression and reduction in ephrin-A4 distribution. Reduced dosage of Msx2 in the Twist1 mutant background restores the expression of ephrin-A4, rescues the suture boundary and inhibits craniosynostosis. Underlining the importance of ephrin-A4, we identified heterozygous mutations in the human orthologue, EFNA4, in three of 81 patients with non-syndromic coronal synostosis. This provides genetic evidence that Twist1, Msx2 and Efna4 function together in boundary formation and the pathogenesis of coronal synostosis.
INTRODUCTION
Like most organs in developing metazoan embryos, the mammalian skull vault is a composite of cells with distinct embryological origins. In the mouse, the paired frontal bones are derived from neural crest, and the adjacent parietal bones from paraxial mesoderm (1,2). The coronal suture is established as the leading edges of the frontal and parietal mesenchymal cell populations come into apposition and overlap. Sandwiched between these overlapping sheets of osteogenic mesenchyme is a population of undifferentiated sutural mesenchyme of mesoderm origin (1). The coronal suture thus forms at the interface between the neural crest-derived osteogenic mesenchyme of the frontal bone and the mesoderm-derived osteogenic mesenchyme of the parietal bone. From its first appearance at E9.5 through at least the newborn stage, this interface remains distinct (1,3), suggesting that the coronal suture serves as a functional boundary. Supporting this possibility is the general finding that boundaries between different cell populations often serve as tissue organizers (4,5), together with studies showing that the coronal suture is a major growth center in the developing skull vault (6).
Craniosynostosis, a pathological condition in which bones of the skull vault fuse prematurely, often affects the coronal suture. Among the human genetic disorders in which coronal synostosis is a feature are Saethre-Chotzen syndrome, caused by heterozygous loss of function of the basic helix–loop–helix gene, TWIST1 (7,8); Boston-type craniosynostosis, caused by a gain of function mutation in the msh-class homeobox gene, MSX2 (9,10); and craniofrontonasal syndrome, caused by mutations in EFNB1 encoding ephrin-B1 (11,12). Analyses of mouse models, together with ex vivo approaches, have suggested that craniosynostosis may involve changes in the survival, proliferation or rate of differentiation of osteogenic cells within the suture (13–16). A number of investigations have also identified the underlying dura as an important source of signals controlling suture development (17,18). Despite these advances, neither the processes that control normal suture development nor the mechanisms underlying craniosynostosis are well understood.
Here, we investigate the influence of Twist1 on the neural crest-mesoderm boundary in the developing coronal suture. We show that the coronal suture is a stable boundary, which is defective in Twist1+/− mouse embryos. We demonstrate that Twist1 and Msx2 function together to maintain the integrity of this boundary, and that ephrin-Eph signaling has an important downstream role in this process. Consistent with the evidence that ephrin-Eph signaling is functionally important in coronal suture development, we identified heterozygous mutations in the ephrin-A4 human orthologue, EFNA4, in patients with non-syndromic coronal synostosis.
RESULTS
A boundary defect in Twist1+/− mutant embryos
We found, as previously reported (19), that Twist1+/− mutant mice exhibited synostosis of the coronal suture. Microcomputed tomography (μCT) analysis of Twist1+/− skulls at P21 showed unilateral or bilateral fusion of the frontal and parietal bones at the coronal suture in 13/15 mice (Fig. 1B and D). Of 15 wild-type littermates, none exhibited this phenotype (Fig. 1A and C). To trace the embryological origins of this defect, we examined the expression of the early osteoblast marker, alkaline phosphatase (ALP). No changes in the pattern of ALP expression were apparent at E12.5 or E13.5 (data not shown). However, at E14.5, whereas in wild-type embryos there was a discrete layer of non-ALP expressing cells between the ALP positive cells of the prospective frontal and parietal bones (Fig. 1E and G), in Twist1+/− mutants this layer was absent, and a disorganized ‘cloud’ of ALP-expressing cells filled the prospective suture (Fig. 1F and H).
We did not detect differences in apoptosis or proliferation between Twist1+/− and wild-type controls in the prospective frontal and parietal bones at E13.5, E14.5 and E16.5 (20, data not shown), prompting us to consider other mechanisms underlying the synostosis phenotype. The disorganized appearance of ALP-expressing cells within the coronal suture of Twist1+/− mice led us to hypothesize that the interface between neural crest and mesoderm-derived cells constitutes a functional boundary, and that a disruption of this boundary may be causally related to synostosis of the coronal suture. To test this hypothesis, we made use of the Wnt1-Cre/R26R neural crest marking system. Previously, we verified that the distribution of Wnt-1 transcripts was not altered in the neural tube of E9.5 Twist1 mutant embryos (21). As a part of the present study, we used an antibody against Cre recombinase to test for possible ectopic expression of the Wnt1-Cre transgene in the skull vault of E13.5 and E14.5 Twist1+/− embryos. No such expression was found (data not shown).
We examined Wnt1-Cre; R26R; Twist1+/− embryos at a series of developmental stages and found no changes in the distribution of lacZ positive cells of the frontonasal neural crest relative to adjacent non-neural crest cell populations at E10.5, E12.5 or E13.5 (data not shown). At E14.5, however, a substantial change was evident. In wild-type embryos, neural crest and non-neural crest cells were distinct, with the prospective parietal bone overlapping the prospective frontal bone (Fig. 2A). In Twist1+/− mutant embryos, by contrast, neural crest-derived cells were located ectopically in the presumptive parietal bone compartment, and the shape of the neural crest-mesoderm boundary was irregular (Fig. 2B, arrows; summarized in Supplementary Material, Fig. S1).
By E16.5, neural crest-derived cells occupied the endosteal layer of the parietal bone and were interspersed throughout the proximal and distal coronal suture of the Twist1 mutant (Fig. 2D and F). The parietal bone of E16.5 wild-type embryos was clear of neural crest cells (Fig. 2C and E). ALP staining of adjacent sections showed that wild-type embryos maintained a discrete layer of non-ALP expressing cells between the frontal and parietal bones (Fig. 2G), whereas in Twist1+/− mutants, this layer was nearly lost (Fig. 2H). At the newborn (P0) stage, the boundary between the neural crest-derived frontal bone and the mesodermally derived parietal bone remained distinct in wild-type mice (Fig. 2I). In P0, Twist1+/− mutants, the frontal and parietal bones were fused over a variable length of the coronal suture. Serial sections revealed that fusion in Twist1+/− mutants only occurred at sites of neural crest-mesoderm cell mixing (Fig. 2J; data not shown).
Altered Ephrin expression in the coronal suture boundary
Ephrin-Eph signaling is known to play a crucial role in the formation of boundaries in vertebrates, particularly in the processes of hindbrain and somite segmentation (22,23). A survey of the distribution of individual ephrin-A and ephrin-B ligands and their cognate receptors within the coronal suture led us to focus on ephrin-A2, ephrin-A4 and EphA4. Ephrin-A2 and ephrin-A4 proteins appeared in a single layer of cells on the ectocranial side of the prospective frontal bone immediately anterior to the coronal suture (Fig. 3A, C and G arrow). This layer corresponds to the surface where the parietal bone will later overlap the frontal bone. Comparison of this expression pattern with Wnt1-Cre/R26R labeled adjacent sections revealed that the ephrin-A2 and -A4 expressing layer was lacZ negative and thus composed of non-neural crest cells (Fig. 3G and H). Neither ephrin-A2 nor ephrin-A4 was detectable in the skull vault at E13.5 or E16.5 (data not shown). Therefore, these ephrin ligands were expressed transiently during the interval when the neural crest-mesoderm boundary defect became evident. EphA4, a receptor for ephrin-A2 and ephrin-A4, was detected on the ectocranial side of the prospective frontal bone in two layers immediately flanking the layer of ephrin-A4 expression (Fig. 3E). The upper layer of expression was lacZ negative in adjacent Wnt1-Cre/R26R labeled sections, whereas the lower layer was lacZ positive (data not shown). Thus, a discrete mesodermal layer expressing the ephrin-A2 and -A4 ligands establishes a boundary between the mesoderm and neural crest cells expressing EphA4. In Twist1 mutant mice, the layer of ephrin-A2 and -A4 expression retracted anteriorly (Fig. 3B and D), and the dual layers of EphA4 expression merged into a single layer (Fig. 3F).
Msx2 expression is expanded at the prospective coronal suture of Twist1+/− embryos
We showed previously that a gain of function mutation in MSX2 is associated with Boston type craniosynostosis (9,10). In addition, we found that Msx2 and Twist1 cooperate in frontal bone development (21). These findings led us to hypothesize that Twist1 and Msx2 may have an epistatic interaction during coronal suture development. In situ hybridization showed that Msx2 transcripts were concentrated in the calvarial mesenchyme of the frontal bone in wild-type embryos (Fig. 4A), but were distributed more broadly in and above the calvarial mesenchyme of E14.5 Twist1+/− embryos (Fig. 4B). Moreover, the expression of a 560bpMsx2-hsplacZ transgene, shown previously to recapitulate endogenous Msx2 expression accurately in wild-type embryos (24,25), was similarly expanded in Twist1+/− coronal sutures (Fig. 4C and D). Twist1 is expressed broadly in calvarial mesenchyme at E14.5 (20), and we have previously shown that Twist1 expression is not altered in calvarial tissues of Msx2−/− mice (21,26). These results suggest that directly or indirectly, Twist1 negatively regulates Msx2 expression in the coronal suture.
Reduced Msx2 gene dosage rescues the Twist1+/− coronal suture phenotype and ephrin-A2 and -A4 expression
We next sought to determine whether the expanded expression of Msx2 in the prospective coronal suture of Twist1+/− mice was functionally important in the development of the Twist1 boundary defect and craniosynostosis. If so, we would expect reduced Msx2 dosage in the context of the Twist1+/− mutant to mitigate these phenotypes. μCT analysis of P21 skulls from Msx2+/−; Twist1+/− mice showed that normal suture development was restored (Fig. 5A and B). This rescue occurred with high penetrance as the frequency of a fused coronal suture was substantially lower in double mutant mice (14%; 3/21), compared with Twist1+/− mice (86%; 13/15) (Fig. 5I). Wnt1-Cre/R26R analysis on Msx2+/− and Msx2+/−; Twist1+/− mutants at E14.5 and newborn stages showed that neural crest and mesodermal cells were distributed normally at both stages (Fig. 5C, D, G and H). Adjacent E14.5 sections stained for ALP further showed that reduced Msx2 gene dosage was capable of restoring a functional boundary between the osteogenic tissues of the prospective frontal and parietal bones (Fig. 5E and F). This rescue of the neural crest-mesoderm boundary in E14.5 Msx2+/−; Twist1+/− embryos was accompanied by a restoration of the wild-type expression patterns of ephrin-A2, ephrin-A4 (Fig. 6B and D), and EphA4 (Fig. 6F). Msx2+/− mutant embryos did not show changes in the distribution of these proteins (Fig. 6A, C and E).
Mutations of ephrin-A4 (EFNA4) in patients with non-syndromic craniosynostosis
The tight association between changes in ephrin-A2, ephrin-A4 and EphA4 expression, the frontal–parietal boundary defect, and the synostosis phenotype in the Twist1 mutant skull, strongly suggested that these proteins, either individually or in combination, have a functional role in coronal suture development. As a test of this hypothesis, we asked whether any of the human orthologues, EFNA2, EFNA4 or EPHA4, is mutated in individuals with coronal synostosis. We screened 81 DNA samples from patients with non-syndromic coronal craniosynostosis for changes in the coding sequences. No pathogenic mutations were detected in EFNA2 or EPHA4, but three distinct heterozygous nucleotide changes in EFNA4 (each predicted to disrupt function) were identified in different patients with unicoronal synostosis (Fig. 7A). Two of these (178C>T, 349C>A) encode the missense substitutions H60Y and P117T, respectively; the third change is a frameshifting indel 471_472delCCinsA, predicting 44 novel amino acids at the C-terminus of a previously reported alternatively spliced minor isoform (27). None of these variants was found in 370 control samples of Northern European origin (Fisher's exact test P=0.0056), nor in a further 151 unrelated craniosynostosis patients who either had syndromic features, or a different combination of affected cranial sutures.
To seek further evidence that these variants are pathogenic, we first tested the parents. Each variant was also present in a clinically unaffected parent, indicating that the phenotype is not fully penetrant (not shown). However, in the case of 349C>A (P117T), the mutation was absent in both parents of the carrier mother, showing that it had arisen de novo in her (Fig. 7B, arrow); correct biological relationships between the samples were confirmed. The location of the two missense substitutions suggested that they might affect receptor binding (see Discussion), so we transfected COS-7 cells with full-length wild-type or mutant EFNA4 cDNAs and measured binding using soluble EphA7 receptors (28). This showed complete (>98%) loss of binding for the P117T mutant and partial (65%) loss for the H60Y mutant (Fig. 7C). These binding defects were confirmed using in vitro ELISA assays (not shown). In the case of the frameshifting indel, we demonstrated using fibroblasts from the patient that the mutation is expressed in an alternatively spliced minor isoform of EFNA4 (Supplementary Material, Fig. S2).
DISCUSSION
The progressive subdivision of the embryo into distinct territories is a universal feature of metazoan development. Largely from work on Drosophila imaginal disks and the vertebrate hindbrain, this process is known to depend on mechanisms that produce stable boundaries by restricting the mixing of different cell populations (29–31). Here, we show that craniosynostosis in Twist1+/− mutant mice is associated with a defect in the boundary between neural crest and mesoderm within the developing coronal suture. Msx2 functions downstream of, and in opposition to, Twist1 in the control of this boundary. Together, Msx2 and Twist1 regulate the expression of the signaling ligands, ephrin-A2 and ephrin-A4. A critical role for Eph-ephrin signaling in boundary integrity and synostosis is supported by our identification of three distinct heterozygous mutations in EFNA4, encoding human ephrin-A4, in patients with non-syndromic coronal synostosis.
Cellular basis of synostosis in Twist1+/− mutant embryos
A deficiency in the neural crest-mesoderm boundary of the skull vault is consistent with previously documented anomalies in the distribution of neural crest cells in Twist1+/− embryos (32,33). Normally restricted to subectodermal mesenchyme, migrating neural crest cells invade the deeper paraxial mesoderm in Twist1−/− embryos, and late migrating subpopulations enter the cell-free zone between the first and second branchial arches (33). Intriguingly, over-expression of Twist1 can cause breast cancer cells to lose E-cadherin mediated cell contacts and undergo metastasis (34). That Twist1 may control cell-affinity is consistent with the intrusion of neural crest-derived cells into the sutural mesenchyme of the Twist1+/− embryo. Haploid loss of Twist1 function could modulate the activity of cell adhesion molecules such as cadherins, enabling sutural cells derived from neural crest to mix with those derived from mesoderm.
Several considerations lead us to infer that the boundary defect is causally related to synostosis. First, it is tightly correlated with ectopic differentiation and the synostosis phenotype. This is underscored by the finding that reduced Msx2 dosage in the Twist1 mutant rescues the boundary defect, as well as ectopic osteogenic differentiation and synostosis of the frontal and parietal bones. Second, our finding of changes in ephrin-A2, ephrin-A4 and EphA4 expression is consistent with a deficiency in the signaling interface between osteogenic cells of neural crest origin and sutural mesenchyme of mesodermal origin. This interface becomes the border between osteogenic mesenchyme and sutural cells that normally remain undifferentiated, and is likely to be of critical importance for the development of the suture. Finally, invading neural crest cells are potential sources of osteogenic signals: during the normal development of the coronal suture, neural crest cells form the dura and osteogenic mesenchyme of the frontal bone, both of which have osteoinductive properties (35–37). Consequently, invading neural crest cells could induce surrounding mesenchyme cells to change their fate and differentiate into osteoblasts.
Interaction between Msx2 and Twist1
That the expansion of Msx2 expression in Twist1 mutant sutures is functionally important is consistent with our previous finding that in humans, a gain of function mutation in the homeodomain of MSX2 causes Boston type craniosynostosis (9,10), as well as our demonstration that transgenic overexpression of Msx2 in mice causes overgrowth of calvarial sutures (38). Strong evidence that Msx2 functions in concert with Twist1 comes from our demonstration that reduced Msx2 dosage in the Twist1+/− embryo rescues the synostosis phenotype. This result also shows that Twist1 and Msx2, in contrast to their positive, cooperative interaction in the development of the frontal bone and frontal suture (21), have antagonistic effects in the coronal suture. They also have opposing effects on the expression of ephrin-A2, ephrin-A4 and EphA4, as reduced dosage of Twist1 causes the expression domains of these genes to contract, whereas reduced dosage of Msx2 in Twist1+/− embryos restore their expression to the wild-type pattern. We conclude that Msx2 is a key downstream effector of Twist1 in the control of the expression domains of ephrin-A2, ephrin-A4 and EphA4, as well as in the maintenance of the frontal–parietal boundary and the development of the coronal suture.
Function of ephrin signaling in the neural crest-mesoderm boundary and craniosynostosis
Major functions of ephrin-Eph signaling include inhibition of cell mixing between cellular compartments and the guidance of migrating cells or cellular processes to their targets (39). When cells or axons expressing an ephrin receptor (Eph) come in contact with a surface expressing the ligand, they are unable to cross and move into ligand-free territory, thus restricting the migration pathway (40). Such a mechanism limits the migration of trunk neural crest cells to the rostral half of individual somites (41,42). Interestingly, cranial neural crest cells in Efnb1 knockout mice invade territories normally devoid of neural crest (43), a defect strikingly similar to one described in Twist1−/− mutant embryos (33).
The expression domains of ephrin-A2, ephrin-A4 and EphA4 at the interface of the frontal and parietal bones are consistent with roles in either inhibition of cell mixing, or guidance of migratory cells to their targets. The layer of ephrin-expressing cells ectocranial to the frontal bone neural crest is in a layer of mesoderm that ultimately is interposed between the overlapped frontal and parietal bones. Expression of ephrin-A2, -A4 and EphA4 in adjacent cell layers could act to prevent underlying skeletogenic neural crest from crossing into the undifferentiated sutural mesenchyme. Ephrin-Eph signaling might also function in the specification of such sutural mesenchyme cells, or as part of a mechanism that directs skeletogenic neural crest to the growing surface of the frontal bone at the coronal suture.
EFNA4 mutations are associated with non-syndromic coronal craniosynostosis
Our identification of three heterozygous mutations of EFNA4 in patients with craniosynostosis provides genetic evidence that ephrin-A4 has a functional role in coronal suture development. One mutation, 349C>A (P117T), is located within the highly conserved dimerization interface, which is essential for ligand-receptor binding (44); an identical proline>threonine substitution at the equivalent residue of ephrin-B1 (proline 119) was previously reported in a patient with craniofrontonasal syndrome and unicoronal synostosis (11). We demonstrated that the P117T substitution in ephrin-A4 causes complete loss of binding to EphA7, a partner receptor; mutation of this highly conserved proline in the Caenorhabditis elegans homologue VAB-2 also resulted in loss of binding to the partner receptor, VAB-1 (45).
We also demonstrated that the 349C>A mutation has arisen de novo, which confirms the pathogenic nature of the P117T substitution. The 178C>T mutation predicts an H60Y substitution, located in a surface-exposed residue of the C-D loop that forms part of the low-affinity ligand/receptor tetramerization interface (44,46). We found that the H60Y substitution causes significantly reduced binding to EphA7, probably by interfering with the formation of this ligand/receptor heterotetramer, as observed for nearby ephrin-A5 mutations analyzed in vitro (47). The third mutation, a frameshifting indel 471_472delCCinsA, is predicted to add 44 novel amino acids at the C-terminus of a minor ephrin-A4 isoform that lacks the sequence necessary for glycosylphosphatidylinositol (GPI)-mediated anchoring to the cell membrane. This isoform was originally identified in activated B-lymphocytes and was speculated to have a chemoattractive or antagonistic role in ephrin-Eph interactions (27). Hence, this mutation may identify a novel mechanism for disrupting ephrin signaling.
Although each of the mutations exhibited incomplete penetrance (in each patient, the coronal suture on only one side was affected and an unaffected parent carried the same mutation), this is a recognized feature of other genetic forms craniosynostosis, such as those caused by specific mutations in FGFR2 (48) and FGFR3 (49). This phenotypic variability might be attributable to interaction with environmental factors, such as intrauterine fetal head constraint, as well as with the genetic background. Our finding that ephrin-A2 and -A4 are expressed almost identically in the area of the coronal suture suggests that these two genes might function redundantly. This is consistent with the general finding that knockouts of individual ephrin genes in mice often must be compounded with knockouts of coexpressing genes in order to generate a mutant phenotype (50,51). Functional redundancy is also likely to account for the mild male phenotype associated with hemizygous null mutations in the X-linked gene EFNB1 (11,12). Our findings suggest that other members of the ephrin family of ligands, such as ephrin-A4, step in as alternative mediators during cell signaling events in the developing cranial suture in males lacking functional ephrin-B1. Recent work showing that B-type ephrin ligands can signal through A-type receptors and vice versa makes this a clear possibility (28,46).
MATERIALS AND METHODS
Mouse mutants and genotyping
The Msx2 mutant was a kind gift from Dr Richard Maas, as was the Twist1 mutant from Dr Richard Behringer. Both single and double mutant lines were maintained in a C57Bl/6 background. The 560Msx2-hsplacZ transgenic mice were produced and genotyped as described previously (24). Genotyping of Msx2 and Twist1 mutants was done as previously described (21,32), as was the genotyping for Wnt1-Cre and R26R (1).
Microcomputed tomography (μCT)
μCT imaging was performed using a MicroCT 40 (Scanco Medical AG, Switzerland). P21 mouse skulls were scanned at high resolution [2048×2048 in-plane image matrix; 0.18 degree rotational step (DRS)] and a field of view (FOV) of 20 mm. Slices were collected at 10 µm increments and 3D renderings were then reconstructed from these 2D images using a threshold value of 160. All scans were conducted at an energy setting of 55 kVp, current intensity of 144 µA and an integration time of 200 ms/projection.
Histology, immunostaining, and in situ hybridization
Detection of ALP in tissue sections was carried out as described previously (38). Analysis of Wnt1-Cre/R26R and 560Msx2-hsplacZ reporter gene expression was carried out as described previously (21,24). Immunostaining of frozen sections was largely carried out as previously reported (21). Immunohistochemistry was performed using rabbit anti-ephrin-A2 (Zymed; 10 µg/ml), rabbit anti-ephrin-A4 (Zymed; 10 µg/ml) and goat anti-EphA4 (R&D Systems; 5 µg/ml) diluted in 1% BSA/PBS and incubated overnight at 4°C. Detection of goat primary antibody was performed by incubating rabbit anti-goat-HRP (Zymed; 1/250) for 1 h at room temperature, followed by visualization with 3,3-diamino-benzidinine (Zymed) substrate. Detection of rabbit primary antibody was performed similarly using goat anti-rabbit-HRP (Zymed). Section in situ was carried out as previously described (52).
Mutation screening of EFNA2, EFNA4 and EPHA4
Analysis of patient samples was approved by the Oxfordshire Research Ethics Committee and informed consent was given for all sampling procedures. DNA was obtained from whole blood, cultured fibroblasts or lymphoblastoid cell lines by phenol–chloroform extraction. Samples from 232 unrelated craniosynostosis patients, of whom 81 had coronal craniosynostosis (20 bicoronal, 61 unicoronal) were examined. For EFNA4, an additional 370 healthy controls of Northern European origin were analyzed. Previous genetic screening including FGFR2, FGFR3 and TWIST1 had not identified any causative mutations. The three patients heterozygous for EFNA4 variants each presented between 0.3 and 2.2 years with anterior plagiocephaly, suggestive of right unicoronal synostosis. This was confirmed by skull radiography or computed tomographic scanning and corrected by forehead advancement. No syndromic features were apparent in these patients and mental development was normal. The carrier parents did not exhibit any clinical features of craniosynostosis and the family history was negative. In the case of the 349C>A mutation, correct parental relationships between family samples was confirmed (>99% probability) by the concordant segregation of at least eight microsatellites of 78% average heterozygosity, each located on a different chromosome. Primer sequences used for the analysis of EFNA2, EFNA4 and EPHA4 are provided in Supplementary Material, Table S1 and methods for mutation screening and cDNA analysis in Supplementary Materials and Methods.
Binding studies
Mutations (178C>T, 349C>A) were first introduced into full-length human EFNA4 cDNA in pGEM-T (Promega) by PCR mutagenesis. These inserts were then subcloned into the BamHI/XbaI-digested plasmid pcDNA3 (+) (Invitrogen), and transfected into COS-7 cells using PolyFect (Qiagen) following the manufacturer's instructions. At 48 h post-transfection, cells were incubated with soluble EphA7/Fc receptor (R&D Systems; 5 µg/ml), stained with anti-human Fc (IgG1)-phycoerythrin labeled antibody (Beckman Coulter) and analyzed on a CyAn ADPTM flow cytometer apparatus (DakoCytomation) at an excitation wavelength of 488 nm and emission wavelength of 572 nm.
Accession codes
EFNA2: NM_001405 (cDNA), NT_011255 (genomic). EFNA4: NM_005227 and AJ006353 (alternative cDNA spliceforms), NT_004487 (genomic). EPHA4: NM_004438 (cDNA), AC079834 and AC010899 (genomic).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We thank Anthony Argentaro, Ann Atzberger, Kristina Mills, Yazmin Santiago, Indira Taylor and Steve Twigg for technical support and discussions, and Jo Byren, Ulrich Müller, John Mulliken and Stephanie Wisniewski for help with obtaining samples. This work was supported by NIH Grants DE12941 and DE12450 (R.E.M.) and a grant from the Wellcome Trust (A.O.M.W.). A.E.M. was supported by a fellowship from the American Heart Association (0010040Y). K.M.L. was supported by NIH Grant AR44528, and S.M.B. by an NIH National Research Service Award T32HL07895 from the National Heart, Lung and Blood Institute.
Conflict of Interest statement. None declared.
†The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
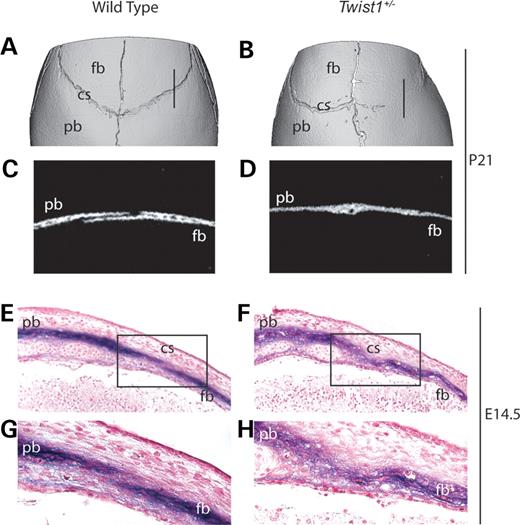
Figure 1. Synostosis and ectopic ALP expression in the coronal suture of Twist1+/− mutant mice. Three-dimensional μCT reconstructions of wild-type (A) and Twist1+/− (B) P21 skulls show unilateral fusion of the coronal suture in the Twist1 mutant. Two-dimensional μCT images of sagittal slices (10 µm) through the distal coronal suture show distinct frontal and parietal bones in the wild-type (C), whereas in the Twist1+/− (D), the bones are fused. Plane of section is indicated in (A) and (B). ALP staining of E14.5 paraffin sections mark the pre-osteogenic mesenchyme of the presumptive frontal and parietal bones in wild-type (E) and Twist1+/− mutant (F) embryos. High magnification views (G and H) of the boxed region in panels (E) and (F), respectively. Note the cloud-like appearance of ALP in the coronal suture of the Twist1+/− mutant (H). cs, coronal suture; fb, frontal bone; pb, parietal bone.
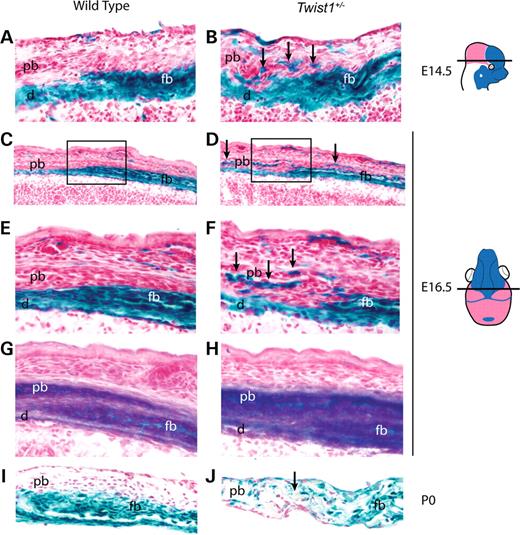
Figure 2. Defective frontal–parietal boundary formation in the Twist1+/− mutant coronal suture. Transverse sections (as depicted) of E14.5 Wnt1-Cre; R26R embryos were used to analyze the distribution of neural crest cells in wild-type (A) and Twist1+/− (B) coronal sutures. In the Twist1 mutant, neural crest cells invade the parietal bone and mid-suture mesenchyme (B, arrows). Coronal sections (as depicted) of E16.5 Wnt1-Cre; R26R embryos show the distribution of neural crest in wild-type (C) and Twist1+/− mutant (D) coronal sutures. Note the presence of neural crest in the mesoderm-derived parietal bone of the Twist1+/− mutant (D, arrows). High magnification views of (C, box) and (D, box), respectively, further show the precise boundary of neural crest-derived mesenchyme in the wild-type (E) coronal suture and the disruption of this boundary in the Twist1+/− (F, arrows) coronal suture. Adjacent sections (G and H) to (E) and (F), respectively, were stained for ALP to visualize the osteogenic mesenchyme. Coronal sections of P0 Wnt1-Cre; R26R mice (I) show that the neural crest-mesoderm boundary is maintained in the post-natal wild-type coronal suture. Cell mixing in the Twist1 mutant coronal suture (J) is associated with fusion at newborn (arrow). This mixing defect was not found in patent locations of the Twist1+/− mutant coronal suture (data not shown). d, dura; fb, frontal bone; pb, parietal bone.
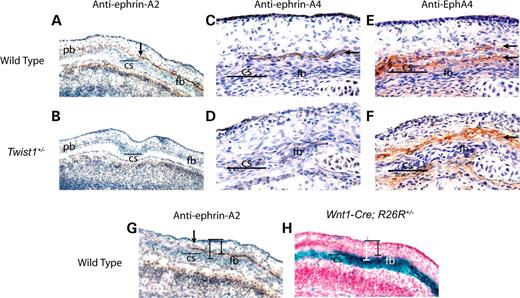
Figure 3. Expression of ephrin-A2, -A4 and EphA4 is altered in Twist1+/− mutant mice. An antibody against ephrin-A2 was used to stain frozen transverse sections of E14.5 wild-type and Twist1 mutants. In wild-type controls (A), ephrin-A2 is expressed in a layer above the prospective frontal bone. This expression extends to the coronal suture (A, arrow) and corresponds to the region of future overlap between the frontal and parietal bones. In the Twist1 mutant (B), there is a restriction of ephrin-A2 expression such that it does not extend into the region of future overlap. An antibody against ephrin-A4 shows that expression in wild-type embryos (C, arrow) is similar to that of ephrin-A2. In Twist1+/− (D), ephrin-A4 expression is also restricted to the anterior and does not extend into the coronal suture region. An adjacent section stained with antibodies against EphA4 (E, arrows) shows that ephrin-A4 expressing cells lie tightly between two layers of cells expressing EphA4. Accordingly, as seen in an adjacent section, EphA4 expression is reduced to one layer in the Twist1+/− coronal suture (F, arrow). Anti-ephrin-A2 staining (G) of an adjacent section to Wnt1-Cre; R26R staining (H) shows that ephrin expression is localized to a plane of cells located immediately above the frontal bone (bars). cs, coronal suture; fb, frontal bone; pb, parietal bone.
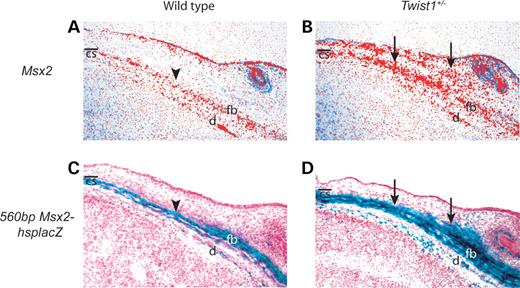
Figure 4. Altered expression of Msx2 and Msx2-lacZ transgenes in Twist1 mutant mice. In situ hybridization of transverse sections shows that Msx2 is expressed predominantly in the osteogenic mesenchyme of the frontal bone at E14.5 (A, arrowhead). This region of expression is expanded in and above the frontal bone osteogenic mesenchyme of Twist1+/− mice (B, arrows). Transverse frozen section of 560bpMsx2-hsplacZ transgenic embryo stained for lacZ at E14.5 shows expression normally restricted to the presumptive frontal bone (C, arrowhead). In the Twist1+/− background (D), 560bpMsx2-hsplacZ transgene expression was expanded in a similar pattern to that for endogenous Msx2 (B). cs, coronal suture; d, dura; fb, frontal bone.
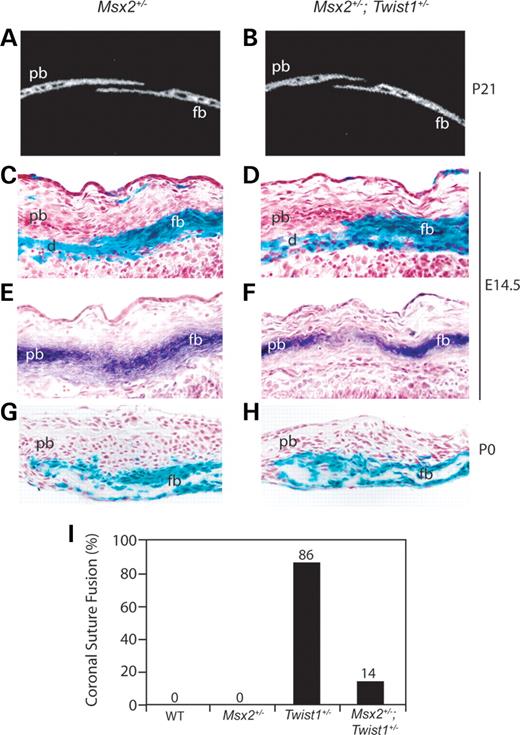
Figure 5. Reduced Msx2 dosage rescues the Twist1 coronal suture boundary defect. Two-dimensional μCT images of sagittal slices (10 µm) through the distal coronal suture of Msx2+/− (A) and Msx2+/−; Twist1+/− (B) mice at P21 show that the sutures are open. Wnt1-Cre; R26R analysis of E14.5 coronal sutures from Msx2+/− (C) and Msx2+/−; Twist1+/− (D) embryos show a normal distribution of neural crest cells. Adjacent sections stained for ALP in Msx2+/− (E) and Msx2+/−; Twist1+/− (F) embryos show that the sutures of double mutants have normal overlap and distinction of the presumptive frontal and parietal bones. Wnt1-Cre; R26R analysis of newborn (P0) coronal sutures show that the normal distribution of neural crest cells is maintained post-natally in Msx2+/− (G) and Msx2+/−; Twist1+/− (H) mice. The percentage of P21 animals with coronal suture fusion was calculated for each genotype (I): wild-type (0/15), Msx2+/− (0/9), Twist1+/− (86%; 13/15) and Msx2+/−; Twist1+/− (14%; 3/21). Therefore, rescue of coronal suture morphology is associated with a rescue of the neural crest-mesoderm boundary in Msx2+/−; Twist1+/− mice. d, dura; fb, frontal bone; pb, parietal bone.
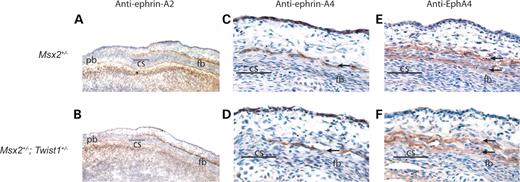
Figure 6. Rescue of ephrin-Eph boundary markers in the Msx2+/−; Twist1+/− coronal suture. Immunohistochemistry for ephrin-A2 on frozen transverse sections shows a layer of expression over the frontal bone, extending to the coronal suture in E14.5 Msx2+/− (A) and Msx2+/−; Twist1+/− (B) embryos. Similarly, ephrin-A4 expression remains unaltered above the frontal bone in Msx2+/− embryos (C, arrow) and is rescued in Msx2+/−; Twist1+/− embryos (D, arrow). Adjacent sections stained using an anti-EphA4 antibody show that expression is maintained in two layers above the frontal bone in Msx2+/− embryos (E, arrows), and this expression pattern is rescued in Msx2+/−; Twist1+/− embryos (F, arrows). cs, coronal suture; fb, frontal bone; pb, parietal bone.
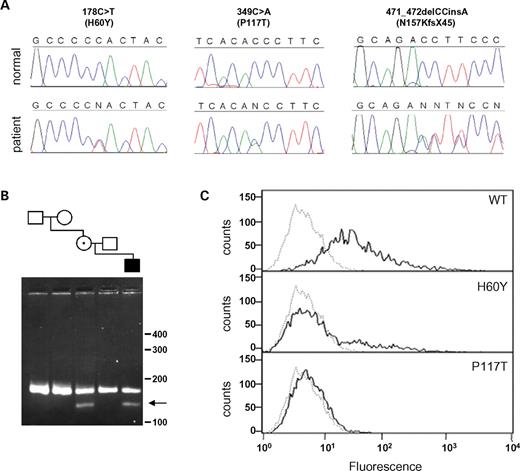
Figure 7. Heterozygous mutations of EFNA4 in patients with non-syndromic coronal synostosis. (A) DNA sequence chromatograms from three patients, are aligned with the corresponding normal sequence. (B) SexAI restriction digest of DNA from the family segregating the 349C>A mutation. Samples are ordered to correspond with the pedigree; the mutant fragment (arrow) is present in DNA from the patient and his mother, but not in the mother's parents. (C) Binding of cells transfected with wild-type and mutant ephrin-A4 proteins to EphA7/Fc receptor. The bound receptor was detected with anti-human Fc (IgG1)-phycoerythrin labeled antibody. Graphs show the distribution of fluorescence on 1×104 individual cells analyzed by flow cytometry of ephrin-A4-transfected (bold line) and non-transfected (dotted line) cells. The proteins were expressed at equal levels (not shown).
References
Jiang, X., Iseki, S., Maxson, R.E., Sucov, H.M. and Morriss-Kay, G.M. (
Trainor, P.A. and Tam, P.P. (
Matsuoka, T., Ahlberg, P.E., Kessaris, N., Iannarelli, P., Dennehy, U., Richardson, W.D., McMahon, A.P. and Koentges, G. (
Dahmann, C. and Basler, K. (
Cheng, Y.C., Amoyel, M., Qiu, X., Jiang, Y.J., Xu, Q. and Wilkinson, D.G. (
Opperman, L.A. (
el Ghouzzi, V., Le Merrer, M., Perrin-Schmitt, F., Lajeunie, E., Benit, P., Renier, D., Bourgeois, P., Bolcato-Bellemin, A.L., Munnich, A. and Bonaventure, J. (
Howard, T.D., Paznekas, W.A., Green, E.D., Chiang, L.C., Ma, N., Ortiz de Luna, R.I., Garcia Delgado, C., Gonzalez-Ramos, M., Kline, A.D. and Jabs, E.W. (
Jabs, E.W., Muller, U., Li, X., Ma, L., Luo, W., Haworth, I.S., Klisak, I., Sparkes, R., Warman, M.L., Mulliken, J.B. et al. (
Ma, L., Golden, S., Wu, L. and Maxson, R. (
Twigg, S.R., Kan, R., Babbs, C., Bochukova, E.G., Robertson, S.P., Wall, S.A., Morriss-Kay, G.M. and Wilkie, A.O. (
Wieland, I., Jakubiczka, S., Muschke, P., Cohen, M., Thiele, H., Gerlach, K.L., Adams, R.H. and Wieacker, P. (
Dry, G.M., Yasinskaya, Y.I., Williams, J.K., Ehrlich, G.D., Preston, R.A., Hu, F.Z., Gruss, J.S., Ellenbogen, R.G. and Cunningham, M.L. (
Yousfi, M., Lasmoles, F., Lomri, A., Delannoy, P. and Marie, P.J. (
De Pollack, C., Renier, D., Hott, M. and Marie, P.J. (
Bialek, P., Kern, B., Yang, X., Schrock, M., Sosic, D., Hong, N., Wu, H., Yu, K., Ornitz, D.M., Olson, E.N. et al. (
Opperman, L.A., Passarelli, R.W., Morgan, E.P., Reintjes, M. and Ogle, R.C. (
Opperman, L.A., Chhabra, A., Nolen, A.A., Bao, Y. and Ogle, R.C. (
Carver, E.A., Oram, K.F. and Gridley, T. (
Rice, D.P., Kim, H.J. and Thesleff, I. (
Ishii, M., Merrill, A.E., Chan, Y.S., Gitelman, I., Rice, D.P., Sucov, H.M. and Maxson, R.E., Jr. (
Cooke, J.E., Kemp, H.A. and Moens, C.B. (
Durbin, L., Brennan, C., Shiomi, K., Cooke, J., Barrios, A., Shanmugalingam, S., Guthrie, B., Lindberg, R. and Holder, N. (
Brugger, S.M., Merrill, A.E., Torres-Vazquez, J., Wu, N., Ting, M.C., Cho, J.Y., Dobias, S.L., Yi, S.E., Lyons, K., Bell, J.R. et al. (
Kwang, S.J., Brugger, S.M., Lazik, A., Merrill, A.E., Wu, L.Y., Liu, Y.H., Ishii, M., Sangiorgi, F.O., Rauchman, M., Sucov, H.M. et al. (
Antonopoulou, I., Mavrogiannis, L.A., Wilkie, A.O. and Morriss-Kay, G.M. (
Aasheim, H.C., Munthe, E., Funderud, S., Smeland, E.B., Beiske, K. and Logtenberg, T. (
Gale, N.W., Flenniken, A., Compton, D.C., Jenkins, N., Copeland, N.G., Gilbert, D.J., Davis, S., Wilkinson, D.G. and Yancopoulos, G.D. (
Micchelli, C.A. and Blair, S.S. (
Rauskolb, C., Correia, T. and Irvine, K.D. (
Xu, Q., Alldus, G., Holder, N. and Wilkinson, D.G. (
Chen, Z.F. and Behringer, R.R. (
Soo, K., O'Rourke, M.P., Khoo, P.L., Steiner, K.A., Wong, N., Behringer, R.R. and Tam, P.P. (
Yang, J., Mani, S.A., Donaher, J.L., Ramaswamy, S., Itzykson, R.A., Come, C., Savagner, P., Gitelman, I., Richardson, A. and Weinberg, R.A. (
Kirk, M.D. and Kahn, A.J. (
Mehrara, B.J., Most, D., Chang, J., Bresnick, S., Turk, A., Schendel, S.A., Gittes, G.K. and Longaker, M.T. (
Hobar, P.C., Schreiber, J.S., McCarthy, J.G. and Thomas, P.A. (
Liu, Y.H., Tang, Z., Kundu, R.K., Wu, L., Luo, W., Zhu, D., Sangiorgi, F., Snead, M.L. and Maxson, R.E. (
Poliakov, A., Cotrina, M. and Wilkinson, D.G. (
Robinson, V., Smith, A., Flenniken, A.M. and Wilkinson, D.G. (
Wang, H.U. and Anderson, D.J. (
Krull, C.E., Lansford, R., Gale, N.W., Collazo, A., Marcelle, C., Yancopoulos, G.D., Fraser, S.E. and Bronner-Fraser, M. (
Davy, A., Aubin, J. and Soriano, P. (
Himanen, J.P., Rajashankar, K.R., Lackmann, M., Cowan, C.A., Henkemeyer, M. and Nikolov, D.B. (
Chin-Sang, I.D., George, S.E., Ding, M., Moseley, S.L., Lynch, A.S. and Chisholm, A.D. (
Himanen, J.P., Chumley, M.J., Lackmann, M., Li, C., Barton, W.A., Jeffrey, P.D., Vearing, C., Geleick, D., Feldheim, D.A., Boyd, A.W. et al. (
Day, B., To, C., Himanen, J.P., Smith, F.M., Nikolov, D.B., Boyd, A.W. and Lackmann, M. (
Johnson, D., Wall, S.A., Mann, S. and Wilkie, A.O.M. (
Robin, N.H., Scott, J.A., Cohen, A.R. and Goldstein, J.A. Nonpenetrance in FGFR3-associated coronal synostosis syndrome.
Feldheim, D.A., Kim, Y.I., Bergemann, A.D., Frisen, J., Barbacid, M. and Flanagan, J.G. (
Lyckman, A.W., Jhaveri, S., Feldheim, D.A., Vanderhaeghen, P., Flanagan, J.G. and Sur, M. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}