



Abstract
Expansion of a polyglutamine tract in ataxin-3 (AT3) results in spinocerebellar ataxia type 3/Machado–Joseph disease, one of the nine polyglutamine neurodegenerative diseases. Understanding the normal functions of AT3 as well as its function in the context of expansion of the polyglutamine tract is critical for understanding the disease process. AT3 is a deubiquitylating enzyme with limited information on its cellular functions. We find that transfecting cells with AT3 increases cellular levels of endoplasmic reticulum-associated degradation (ERAD) substrates, CD3δ and TCRα, but does not alter levels of several non-ERAD substrates. AT3 increases the level of CD3δ by decreasing its degradation; pathogenic AT3 decreases degradation to a greater extent than wild-type AT3. Knock-down of endogenous AT3 decreases levels of CD3δ, suggesting that a normal function of AT3 is to regulate levels of ERAD substrates. AT3 binds VCP/p97, a key protein responsible for extracting ERAD substrates from the ER; binding is modulated by the size of the polyglutamine tract, and mutating a sequence adjacent to the polyglutamine tract inhibits the AT3–VCP interaction and AT3-dependent accumulation of CD3δ. AT3 and Ufd1 bind VCP in a mutually exclusive manner; AT3 decreases the interaction of VCP with Ufd1 as well as with ubiquitylated proteins. Using a reconstituted system, AT3 inhibits retrotranslocation of an ERAD substrate from the ER. These data suggest that a normal function of AT3 is to regulate flow through the ERAD pathway by modulating VCP-dependent extraction of proteins from the ER.
INTRODUCTION
Expansion of a polyglutamine tract in ataxin-3 (AT3) results in the fatal neurodegenerative disease, spinocerebellar ataxia type 3/Machado–Joseph disease (SCA3/MJD; 1). SCA3/MJD is a member of the polyglutamine neurodegenerative disease family that includes Huntington’s disease, spinal and bulbar muscular atrophy, dentatorubral pallidoluysian atrophy and the spinocerebellar ataxias 1,2,3,6,7 and 17 (2). Expansion of the polyglutamine tract results in mutant proteins acquiring a toxic gain of function; however, in most cases, functions of the wild-type polyglutamine proteins have not been identified. Recent studies suggest that identifying normal functions of polyglutamine neurodegenerative proteins is critical for understanding pathogenesis (3–6). Initial insights into AT3 function suggest that it has both nuclear and cytoplasmic functions (7,8). AT3 contains two to three functional ubiquitin-interacting motifs (UIMs;9–12) and is the first member of a new family of deubiquitylating (DUB) enzymes, the Josephin family (11,13). Recently, we showed that the DUB activity and UIMs of AT3 are required for aggresome formation of mutant cystic fibrosis transmembrane regulator (8). This suggests that AT3 may function in cellular pathways that deal with misfolded proteins.
Endoplasmic reticulum (ER)-associated degradation (ERAD) is a quality control system in the secretory pathway responsible for degrading misfolded proteins and unassembled polypeptides of protein complexes (14–16). ERAD involves a series of steps to extract proteins from the ER and deliver them to proteasomes. Proteins targeted for ERAD are deglycosylated, ubiquitylated, extracted from the ER and degraded by proteasomes (17). For each step, different proteins are recruited and assembled around the substrate targeted for degradation. The key protein essential for extracting substrates from the ER to the cytosol is the AAA ATPase Cdc48 in yeast and its counterpart, valosin-containing protein (VCP/p97), in mammals (18,19).
VCP is recruited from the cytosol to the ER by binding integral ER membrane proteins Derlin-1/2, SEL1L/Ubx2 and VCP-interacting membrane protein (VIMP) (20–23). Mammalian orthologs of yeast E3 ubiquitin ligases, Hrd1p/Hrd3p, are recruited to the complex by Derlin-1/2 and SEL1L/Ubx2 and are responsible for ubiquitylating the substrate (14,24–27). VCP recruits the heterodimer, Ufd1-Npl4, to form a subcomplex that is responsible for extracting the ubiquitylated substrate from the ER. Ufd1 binds directly to VCP, whereas Npl4 interacts indirectly by binding to Ufd1. Both VCP and Ufd1 bind ubiquitin chains and their interaction synergistically enhances binding to the ubiquitylated substrate (28–30). Ubiquitylation of the substrate serves as a recognition signal for VCP and Ufd1, and binding ubiquitin may also activate VCP, which results in it extracting the substrate from the ER (29).
Here we report that AT3 binds VCP and decreases its interaction with Ufd1 and polyubiquitin chains; this appears to decrease retrotranslocation of substrates from the ER, resulting in decreased degradation of ERAD substrates. We suggest that AT3 regulates flow through the ERAD pathway by adjusting the rate of extraction of ERAD substrates. Regulation of ERAD by AT3 is polyglutamine tract dependent; this raises the possibility that excessive binding between VCP and pathological AT3 shifts a physiological regulation to a pathophysiological ‘uncoupling’ of ERAD, which may contribute to disease pathogenesis.
RESULTS
AT3 increases the level of ERAD substrates by decreasing degradation
Hirabayashi et al. (31) identified VCP/p97 as an AT3-interacting protein; however, potential functions of this interaction were not investigated. We also identified VCP/p97, along with hsp90, hsc70, neurofilament light chain and tubulin, as AT3-interacting proteins (Supplementary Material, Fig. S1). Co-immunoprecipitations confirm that both endogenous and transfected AT3 interact with VCP (Supplementary Material, Fig. S1). To investigate potential cellular functions of this interaction, we focused on the well-characterized role of VCP in retrotranslocation and degradation of ERAD substrates (18,28,29,32).
Co-transfecting HEK293T cells with wild-type AT3 (AT3Q29) and ERAD substrates, TCRα or CD3δ (transmembrane subunits of the T cell receptor that do not assemble and are degraded in the absence of other subunits), resulted in increased protein levels for both ERAD substrates (Fig. 1B). AT3 is a DUB enzyme (11) and is reported to interact with the proteasome (10); therefore, AT3 may have pleiotropic effects and alter many proteasome substrates. Non-ERAD substrates were tested to determine whether expressing AT3 also increases their cellular levels. AT3 does not affect levels of the following non-ERAD substrates (Fig. 1C): (i) GFP-ODC, which contains the ornithine decarboxylase (ODC) degron that targets ODC for proteasome degradation independent of a polyubiquitin signal (33), (ii) Ubi-R-GFP, an N-end rule substrate (34) and (iii) Smac, a cytoplasmic protein ubiquitylated by IAP and targeted for proteasome degradation (35). Therefore, AT3 does not have a general effect on protein degradation but rather increases the levels of the ERAD substrates tested.
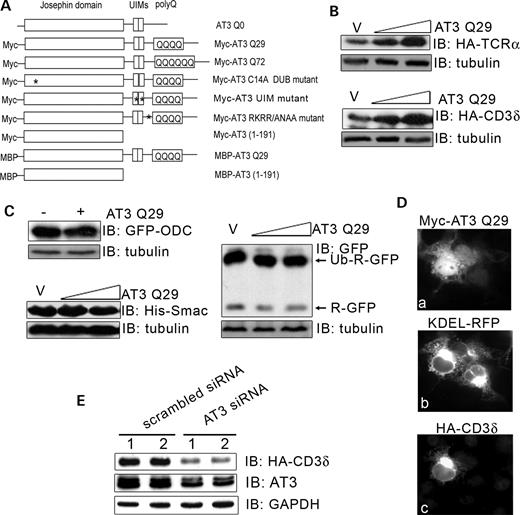
Ataxin-3 regulates cellular levels of ERAD substrates. (A) Constructs used in these studies; * represents inactivating mutations—either in the active site (C14A) or UIMs (L229/249A) or VCP-binding sequence (282RKRR/ANAA285). (B) HEK293T cells were co-transfected with an ERAD substrate, HA-TCRα or HA-CD3δ, and either vector (v) or wild-type AT3Q29 (0.4 or 0.8 µg); after 24 h, cells were processed and analyzed by immunoblots (IB) for HA-CD3δ, HA-TCRα or tubulin as a loading control. (C) Cells were transfected with AT3 and one of the following non-ERAD substrates: GFP-ODC (GFP containing the ornithine decarboxylase degron), Ubi-R-GFP (N-end rule substrate) or his-Smac (cytoplasmic protein) and processed for immunoblots. (D) The same field of Cos-7 cells transfected with myc-AT3, the ER marker, KDEL-RFP and HA-CD3δ; (a) anti-myc fluorescent staining; (b) RFP fluorescence; (c) anti-HA fluorescent staining. Note that CD3δ remains associated with the ER in the AT3-transfected cell. (E) Duplicate dishes of cells were co-transfected with HA-CD3δ and either scramble siRNA or siRNA against AT3; after 48 h, cells were processed and immunoblotted for endogenous AT3, HA-CD3δ or GAPDH as a loading control (in the AT3 blot, both proteins in the doublet represent AT3).
Localization studies were performed on cells to determine whether expressing AT3 increases the level of CD3δ by altering its distribution. CD3δ co-localizes with the ER marker, KDEL-RFP, in AT3-transfected cells (Fig. 1D). Therefore, AT3 does not increase the level of CD3δ by changing its normal localization and diverting it away from the ER and the ERAD pathway, but rather causes a build-up of CD3δ associated with the ER.
Although transfected AT3 increases the level of ERAD substrates, a key issue is whether a normal function of endogenous AT3 is to regulate levels of ERAD substrates. We tested the function of endogenous AT3 on CD3δ, using siRNA knock-down of cellular AT3. AT3 decreases ∼50% following treatment with siRNA for 48 h, which results in a striking decrease in the level of CD3δ (Fig. 1E). This suggests that AT3 normally functions to regulate the level of ERAD substrates.
AT3 could increase the level of CD3δ protein by increasing translation or by decreasing its degradation. Transfecting cells with increasing amounts of AT3 does not increase the amount of CD3δ or GFP metabolically labeled with [35S]methionine/cysteine during a 30 min period, indicating that AT3 is not increasing translation of CD3δ (Fig. 2A). The effect of AT3 on degradation of CD3δ was examined by inhibiting protein synthesis with cycloheximide in cells transfected with CD3δ or co-transfected with AT3. CD3δ degradation is attenuated in cells transfected with either wild-type or pathological AT3 compared with cells transfected with the vector control (Fig. 2B and C). These data show that AT3 increases the level of CD3δ by decreasing degradation and that pathological AT3 inhibits degradation more effectively than wild-type AT3.
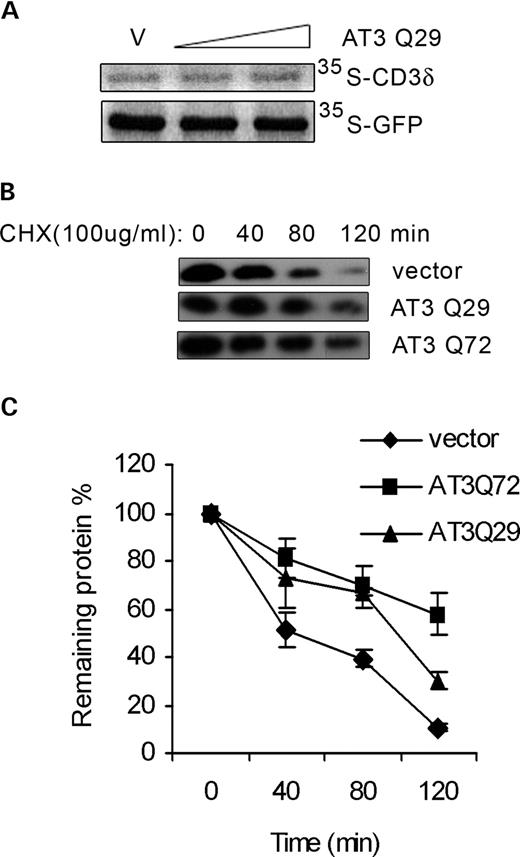
AT3 decreases degradation of CD3δ. (A) HEK293T cells were transfected with HA-CD3δ or GFP and vector or AT3 and then metabolically labeled with 35S-Met/Cys for 30 min and CD3δ or GFP immunoprecipitated. Samples were electrophoresed on an SDS–PAGE gel and exposed to a phosphoimager plate; AT3 did not affect the amount of either protein translated during the 30 min period. (B) Cells were co-transfected with CD3δ and either vector, AT3Q29 or AT3Q72 for 24 h and then treated with 100 µg/ml cycloheximide (CHX) to block protein synthesis. At the indicated times following CHX treatment, cells were harvested, processed and immunoblotted for HA-CD3δ. AT3 increases the level of CD3δ; therefore, to have similar levels of CD3δ for each condition, the percentage of cell lysates used for blots were the following: vector, 25%; AT3Q29, 16.67% and AT3Q72, 8.33%. (C) To quantitate data in (B) and two additional independent experiments, the ratio of CD3δ to GAPDH (as a loading control) was determined for each sample and time. Data represent the mean±SD of three experiments; P<0.05 for comparison of vector and AT3Q29 (80 and 120 min) or AT3Q72 (40, 80 and 120 min); P<0.05 for AT3Q29 compared with AT3Q72 at 120 min.
The effect of DUB activity, UIMs and polyglutamine tract of AT3 on the accumulation of CD3δ and interaction with VCP
Previously, we showed that AT3 is a DUB enzyme containing UIMs (11). Therefore, AT3 could alter degradation of ERAD substrates by removing polyubiquitin chains. To determine whether its DUB activity or UIMs are required to increase CD3δ levels, cells were transfected with the DUB-deficient mutant, AT3C14A, or UIM-deficient mutant, AT3L229/249A. Both mutants increased accumulation of CD3δ; however, at expression levels similar to wild-type AT3, both mutants are somewhat less effective (Fig. 3A), indicating that neither DUB activity nor UIMs are required to alter degradation of CD3δ, but each has a small modulatory effect. It is unlikely that the small modulatory effect of AT3 DUB activity is due to the removal of ubiquitin from CD3δ because AT3 does not decrease the level of ubiquitylated CD3δ (Fig. 3B). If anything, the level of ubiquitylated CD3δ increases in AT3 transfected cells (Fig. 3B).
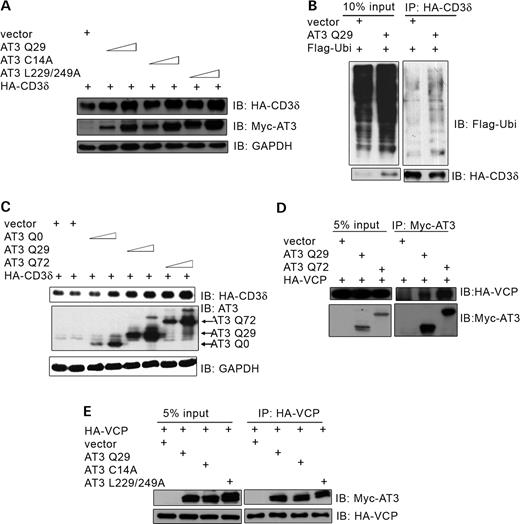
Effect of the active site, UIMs and polyglutamine tract of AT3 on accumulation of CD3δ and binding to VCP. (A) HEK293T cells were co-transfected with HA-CD3δ and either vector, AT3Q29, AT3C14A (active site mutant) or AT3L229/249A (UIM mutant that does not bind ubiquitin) and immunoblots processed for HA-CD3δ, myc-AT3 or GAPDH (control). (B) HA-CD3δ, Flag-ubiquitin and either AT3Q29 or vector control were transfected into cells, and HA-CD3δ immunoprecipitated and immunoblotted for Flag-ubiquitin. (C) Cells were transfected with HA-CD3δ and vector or AT3 containing a polyglutamine tract with 0 (AT3Q0), 29 (AT3Q29) or 72 (AT3Q72) glutamines. Cells were collected, processed and immunoblotted for HA-CD3δ, AT3 or GAPDH (control). (D) Cells were co-transfected with HA-VCP and either vector, myc-AT3Q29 or myc-AT3Q72 and immunoprecipitated with anti-myc and blotted for HA-VCP or myc-AT3; more VCP interacts with pathological AT3 than wild-type AT3. (E) Cells were co-transfected with HA-VCP and either vector, AT3Q29, AT3C14A or AT3L229/249A. Following immunoprecipitation of HA-VCP, samples were processed and blotted for myc-AT3 or HA-VCP; wild-type AT3, AT3C14A and AT3L229/249A interact similarly with VCP.
To examine the role of the polyglutamine tract in regulating CD3δ levels, cells were co-transfected with CD3δ and AT3 with polyglutamine tracts containing 0, 29 or 72 glutamines (below the wild-type range, wild-type range and pathological range, respectively). CD3δ accumulation increases as the number of glutamines in the tract increases (Fig. 3C). For instance, at similar expression levels, AT3Q72 (Fig. 3C, lane 7) increases the level of CD3δ more than AT3-Q0 (Fig. 3C, lane 4). These data are consistent with data in Fig. 2, showing that pathological AT3 decreases degradation of CD3δ more effectively than wild-type AT3. The ability of AT3 to bind VCP may provide a link to its decreasing degradation of CD3δ. To determine whether pathological AT3 shows enhanced binding to VCP, cells were transfected with AT3Q29 or AT3Q72. Compared with wild-type AT3, the pathological protein binds VCP to a greater extent (Fig. 3D); this is consistent with it increasing CD3δ to a higher level than wild-type AT3. The DUB activity and UIMs of AT3 have a very small modulatory effect on increasing the level of CD3δ; therefore, it is not surprising that mutating the active site or UIMs of AT3 have little or no effect on the interaction of AT3 and VCP (Fig. 3E).
Following completion of this study, a sequence in AT3 adjacent to the polyglutamine domain was identified, which binds VCP (36). We used this information to determine whether the AT3–VCP interaction was required for accumulation of CD3δ. The interaction of AT3 and VCP is greatly decreased (but not totally blocked) when mutations are made in this sequence (Fig. 4A). The same mutation in AT3 (282RKRR→ANAA285) greatly decreases but does not totally block the build-up of CD3δ (Fig. 4B). Together, these data suggest that AT3 increases cellular levels of CD3δ through interactions with VCP and that this is modulated by the polyglutamine domain and, to a lesser extent, by DUB activity and UIMs of AT3.
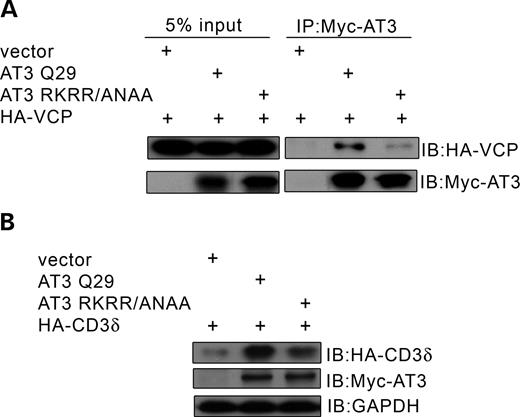
A mutation in AT3 that disrupts binding to VCP decreases accumulation of CD3δ. (A) HEK293T cells were co-transfected with HA-VCP and either vector, AT3Q29 or AT3RKRR/ANAA (mutation of the sequence that binds VCP); myc-AT3 was immunoprecipitated and blotted for HA-VCP or myc-AT3. Interaction of VCP with mutant AT3 is greatly decreased. (B) Cells were co-transfected with HA-CD3δ and either vector, AT3Q29 or AT3RKRR/ANAA and immunblots processed for HA-CD3δ, myc-AT3 or GAPDH (control). The mutation that decreases binding of AT3 to VCP also decreases accumulation of CD3δ.
AT3 decreases Ufd1 binding to VCP
The interaction of AT3 with VCP and the ability of AT3 to increase accumulation of ERAD substrates raise the possibility that AT3 may interfere with the VCP/Ufd1-Npl4 complex, which extracts ERAD substrates from the ER (29). Ufd1 interacts directly with VCP, whereas Npl4 interacts indirectly with VCP through its binding to Ufd1 (37). Transfecting HEK293T cells with AT3 decreases the amount of Ufd1 associated with VCP (Fig. 5A and B). Conversely, increasing the amount of transfected Ufd1 decreases the amount of AT3 co-immunoprecipitated with VCP (Fig. 5C), suggesting that AT3 and Ufd1 compete for binding to VCP. Consistent with this possibility are studies showing that transfecting AT3 decreases the amount of VCP co-immunoprecipitated with Ufd1 (Fig. 5D). The VCP/Ufd1-Npl4 complex associates with the ER membrane and retrotranslocates ERAD substrates; therefore, the ability of AT3 to disrupt the interaction of VCP and Ufd1 should decrease the amount of Ufd1 associated with microsomal membranes. Transfecting cells with AT3 decreases Ufd1 associated with microsomal membranes but does not affect the amount of VCP bound to microsomes (Fig. 5E). Importantly, overexpressing AT3 does not alter the amount of endogenous VCP bound to microsomal membranes or binding of VCP to the ER transmembrane ligase, hHrd1 (Fig. 5F and G).
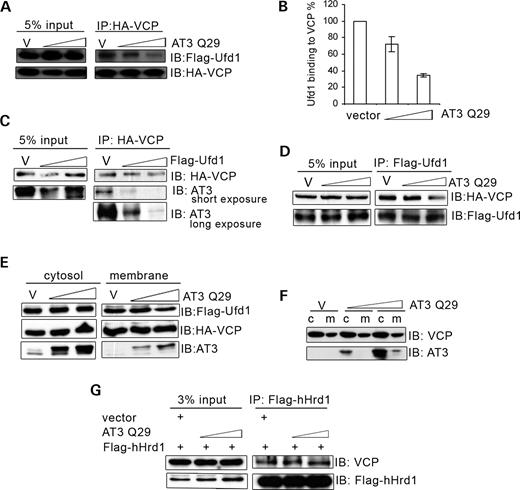
AT3 decreases interaction of Ufd1 and VCP. (A) HEK293T cells were transfected with HA-VCP, Flag-Ufd1 and either vector (v) or AT3Q29 (0.3 or 0.5 µg). HA-VCP was immunoprecipitated and blotted for Flag-Ufd1 and HA-VCP; AT3 decreases the amount of Ufd1 interacting with VCP. (B) Quantitation of blot in (A) along with two additional blots from independent experiments showing that AT3 decreases binding of Ufd1 to VCP. Bars represent mean±SD of three experiments, expressed as a percent of vector control; P<0.05, AT3 compared with vector. (C) HA-VCP, myc-AT3Q29 and either vector (v) or Flag-Ufd1 were transfected into cells, and HA-VCP immunoprecipitated. Lysates were processed for immunoblots of HA-VCP and myc-AT3; increasing Ufd1 decreases interactions of AT3 with VCP (more easily seen with long exposure for AT3). (D) Cells were transfected with Flag-Ufd1, HA-VCP and either vector (v) or AT3. Flag-Ufd1 was immunoprecipitated and processed for immunoblots of HA-VCP and Flag-Ufd1; the higher concentration of AT3 decreases VCP associated with Ufd1. (E) Cells transfected with Flag-Ufd1, HA-VCP and either vector (v) or myc-AT3 were harvested and separated into cytosolic and microsomal fractions and immunoblotted for Flag-Ufd1, HA-VCP or AT3; the higher level of AT3 decreases Ufd1 but not VCP associated with microsomes. Note that cytosolic levels of Ufd1 are much greater than microsome-associated levels; therefore, loss of microsome-associated Ufd1 is not reflected in a perceptible increase in cytosolic Ufd1. (F) Vector or AT3Q29 was transfected into cells and lysates fractionated into cytosol and microsomal membranes. Immunoblots for endogenous VCP or transfected AT3 show that AT3 does not alter membrane-associated endogenous VCP. (G) Cells were transfected with Flag-hHrd1 and either vector (v) or AT3Q29. Flag-hHrd1 was immunoprecipated and immunoblotted for endogenous VCP or transfected hHrd1; AT3 does not alter the amount of endogenous VCP associated with hHrd1.
AT3 impairs retrotranslocation of an ERAD substrate from the ER to the cytosol
The interaction of VCP and Ufd1 synergistically enhances their binding to ubiquitylated ERAD substrates (29). AT3 decreases the interaction of Ufd1 with VCP; therefore, AT3 may also decrease the amount of ubiquitylated protein associated with VCP. Transfecting cells with increasing amounts of AT3 decreases ubiquitylated proteins associated with VCP (Fig. 6A). Ubiquitin positive material is not present on blots from immunoprecipitations carried out in RIPA buffer, which dissociates proteins bound to VCP (data not shown); this indicates that the presence of ubiquitin in Figure 6A is due to ubiquitylated proteins bound to VCP and not due to VCP being ubiquitylated. AT3 with mutations that inactivate its DUB activity or UIMs still decrease binding of ubiquityltated proteins to VCP, indicating that decreased binding to VCP is not due to AT3 cleaving or binding to ubiquitin chains (Fig. 6A). These data along with those in Figure 5 suggest that AT3 causes accumulation of ERAD substrates in the ER by decreasing binding of Ufd1 to VCP, which results in decreased binding of ubiquitylated proteins to the VCP/Ufd1-Npl4 complex and impaired retrotranslocation.
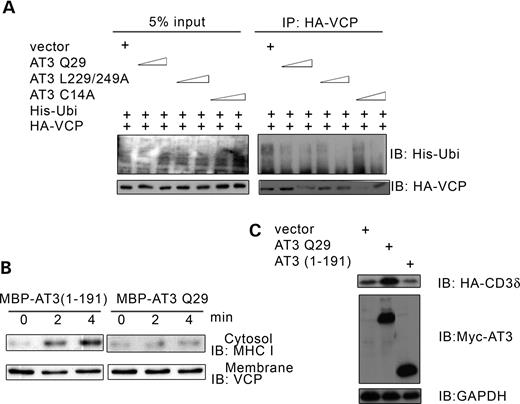
AT3 decreases binding of ubiquitylated proteins to VCP and impairs retrotranslocation of MHC I HC. (A) HEK293T cells were transfected with HA-VCP, His-ubiquitin and either vector, AT3Q29, AT3L229/249A or AT3C14A (0.3 or 0.5 µg). HA-VCP was immunoprecipitated and the amount of associated ubiquitylated proteins was determined by immunoblotting for his-ubiquitin. (B) Cytosol and microsome fractions from Daudi cells were prepared as described by Koopmann et al. (53), and retrotranslocation of MHC I HC was carried out according to the procedure of Albring et al. (39). Cytosol was incubated with MBP-AT3(1-191) or MBP-AT3Q29 in the presence of MG-132 for 2 h on ice, then 20 mm ATP was added and the cytosol warmed to 37°C, followed by adding pre-warmed microsomes from 5×106 cells. The mixture was incubated at 37°C for 0, 2 or 4 min and microsomes pelleted by centrifugation. The amount of MHC I HC retrotranslocated into the cytosol and endogenous VCP associated with microsomes was determined using immunoblots. (C) HEK293T cells were co-transfected with HA-CD3δ and either vector, AT3Q29 or AT3(1-191) and immunoblots processed for HA-CD3δ, myc-AT3 or GAPDH (control). AT3(1-191) does not increase accumulation of CD3δ.
Retrotranslocation of CD3δ cannot be followed in a reconstituted system because its extraction depends on proteasome activity, which rapidly degrades CD3δ (38). However, Albring et al. (39) defined a reconstituted in vitro system in which the ERAD substrate, MHC I HC, is retrotranslocated and accumulates in the cytosol even when proteasomes are inhibited. Addition of bacterially expressed MBP-AT3 to this reconstituted in vitro system inhibits translocation of MHC I HC into the cytosol compared with addition of the control protein MBP-AT3 (1-191), representing the Josephin domain of AT3 (Fig. 6B). Consistent with the observation that AT3 (1-191) does not inhibit translocation of MHC I HC, it does not cause accumulation of CD3δ (Fig. 6C) and does not bind VCP (data not shown). The ability of AT3 to inhibit extraction of a substrate in the reconstituted system is consistent with data in Figure 1D, showing that AT3 increases accumulation of CD3δ in the ER. Taken together, our data suggest that AT3 binds to ER-associated VCP and destabilizes the VCP–Ufd1 interaction, resulting in decreased binding to ubiquitylated ERAD substrates, which decreases their extraction from the ER.
DISCUSSION
Following synthesis in the ER, proteins in the secretory pathway undergo cycles of folding and/or assembly to ensure proper conformation prior to transport to the plasma membrane and other organelles (40). A significant fraction of these proteins do not adopt a stable structure. To maintain cellular homeostasis, proteins that do not fold or assemble properly are extracted from the ER by a VCP/p97-dependent process and degraded by proteasomes. AT3 binds VCP and decreases extraction of ERAD substrates. An important question is whether AT3 regulates degradation of all ERAD substrates or only a subset of ERAD substrates. In the present study, AT3 did not affect degradation of non-ERAD substrates tested; however, both ERAD substrates used in the study were transmembrane proteins. It will be interesting to determine whether AT3 also regulates degradation of soluble/luminal ERAD substrates.
VCP in complex with Ufd1-Npl4 recognizes ubiquitin chains on a substrate targeted for degradation; VCP then uses its ATPase activity to extract the protein from the ER. Our data show that AT3 binds VCP and decreases its interaction with Ufd1. This appears to decrease the interaction of VCP with ubiquitylated proteins, resulting in decreased retrotranslocation of targeted proteins from the ER to the cytosol and a build-up of proteins in the ER. AT3 does not alter the ATPase activity of VCP (B. Burnett, X. Zhong, and R. Pittman, unpublished data) or its interaction with ER membrane proteins; therefore, its primary effect on substrate extraction appears to be through decreasing interactions of VCP with Ufd1 and ubiquitylated proteins. AT3 and Ufd1 appear to compete for binding to VCP (Fig. 5A and C). Under conditions where AT3 and Ufd1 are expressed at similar levels, Ufd1 competes more effectively for binding to VCP (data not shown); however, knock-down of endogenous AT3 has a striking effect on the level of CD3δ, suggesting that under normal cellular conditions, AT3 competes effectively for binding to VCP.
Cellular functions of AT3 and regulation by the polyglutamine tract
We previously showed that AT3 DUB activity and UIMs are required for aggresome formation as well as for interaction of AT3 with the dynein motor complex (8), which is required for transporting misfolded proteins to the microtubule-organizing center for aggresomes formation. Currently, it is not clear whether there is a link between the present study showing that AT3 regulates flow through the ERAD pathway and its regulation of aggresome formation. However, it seems reasonable that a subset of cellular proteins might function to monitor and coordinate various degradation pathways. Although there are no strong data to suggest that AT3, or other cellular protein, functions as a ‘sentinel protein’ to monitor and coordinate degradation pathways, the ability of AT3 to regulate both the ERAD pathway and aggresome formation and to interact with key UPS regulators such as VCP, Rad23 and HDAC6 makes it a potential candidate for such a regulatory protein.
Mutating the active site or UIMs in AT3 does not alter binding to VCP and has only a very small effect on the accumulation of CD3δ. In contrast, AT3 with an expanded polyglutamine tract shows a much stronger interaction than wild-type AT3 with VCP; this is consistent with the in vitro study of Hirabayashi et al. (31), showing that pathological AT3 binds VCP more effectively.
Pathological AT3 also causes a larger accumulation of CD3δ. The enhanced effect of pathological AT3 on the level of CD3δ is likely due to its enhanced interaction with VCP, resulting in decreased substrate extraction from the ER. AT3 without a polyglutamine tract increases accumulation of CD3δ protein, but to a lesser extent than AT3 containing a polyglutamine tract either in the normal or pathological range. This suggests that the polyglutamine tract is not directly involved in binding VCP but may alter the conformation of the domain responsible for binding. Data from Bevivino and Loll (41) showing that the length of the polyglutamine tract in AT3 regulates the amount of helical structure in the protein is consistent with this possibility. The very recent study by Boeddrich et al. (36) identified a region contiguous with the polyglutamine tract as a binding site for VCP; mutations in this region greatly decrease binding of AT3 to VCP (36) (Fig. 4A and B). An alternative possibility for the enhanced effect of pathological AT3 on accumulation of CD3δ is that it forms aggregates and sequesters VCP or inhibits proteasomes. Inhibiting proteasomes seems unlikely because, similar to wild-type AT3, pathological AT3 does not increase levels of non-ERAD substrates (not shown). Aggregations and sequestering proteins such as VCP seem unlikely in that in vitro studies show that pathological AT3 binds VCP more effectively than wild-type AT3 under conditions in which aggregation does not occur (31), and immunofluorescent and biochemical data show that full length AT3Q72 used in these studies does not aggregate in HEK293T cells (42,43). Future studies using mutations in pathological AT3 that decrease binding to VCP will help resolve this issue. Increased binding of pathological AT3 to VCP and its greater effect on inhibiting ERAD raise the possibility that excessive binding between VCP and pathological AT3 over an extended time or during increased cellular stress shifts a physiological regulation to a pathophysiological uncoupling of ERAD, which may contribute to disease pathogenesis.
AT3 regulation of the ERAD pathway may extend beyond modulating extraction of substrates
It is interesting that the E3/E4 ligase, Ufd2/E4B, polyubiquitylates both wild-type and pathological AT3 and increases their degradation (44). Following extraction of substrates from the ER, Ufd2 associates with the VCP complex to extend the short ubiquitin chains on targeted proteins; this likely increases their recognition and degradation by the proteasome (45). Rather than just reflecting normal turnover of AT3, ubiquitylation of AT3 by Ufd2 and subsequent degradation described by Matsumoto et al. (44) may reflect an ongoing homeostatic regulatory mechanism to restore flow in the ERAD pathway back to normal when there is excess AT3 binding VCP, or possibly in the presence of pathological AT3 that binds VCP tightly and does not release appropriately. Alternatively, AT3 may not only regulate extraction of substrates by VCP but may also regulate additional processes in the escort pathway through its interaction with Ufd2 or VCP. For instance, AT3 may stabilize proteins by editing or binding and protecting ubiquitin chains on substrates or by controlling the length of ubiquitin chains to optimize degradation (46). This possibility is consistent with our data showing that DUB activity and UIMs of AT3 are not required for binding VCP or increasing levels of CD3δ; however, they both have a small but consistent effect on accumulation of CD3δ. Relevant to this possibility is the recent study by Rumpf and Jentsch (47) showing that the fate of ERAD substrates depends on recruitment of different partners by Cdc48/VCP. Recruitment of Ufd2 results in proteasomal degradation, whereas recruitment of Ufd3 protects substrates by blocking binding of Ufd2 to VCP; recruitment of the DUB enzyme, otubain-1, protects substrates by removing ubiquitin (47). Through its interaction with Ufd2 and/or VCP, AT3 may perform similar downstream functions as otubain-1 in the escort pathway, in addition to its modulating VCP-dependent extraction of substrates.
AT3–VCP interaction and accumulation of ERAD substrates: homeostatic modulation or stopping flow in the pathway
An important issue is whether the interaction of AT3 with VCP is part of a homeostatic mechanism that modulates traffic through the ERAD pathway or a process to stop flow in the pathway during excess cellular stress. Although AT3 may be part of a surveillance network that responds to cellular stresses, several observations are more consistent with it being involved in ERAD homeostasis. Knock-down of endogenous AT3 results in lower levels of CD3δ protein than in the presence of normal endogenous AT3. This likely reflects changes in an ongoing homeostatic process rather than the inability to decrease/stop flow in the ERAD pathway in response to stress. Several unpublished observations are more consistent with AT3 functioning to regulate flow in the pathway as part of homeostasis rather than stopping flow as part of a cellular response: (i) overexpressing wild-type AT3 causes little or no unfolded protein response (UPR) over 48 h; if AT3 stopped flow in the ERAD pathway over that period of time, it should induce UPR, (ii) AT3 slows but does not block degradation of CD3δ and extraction of MHC I HC and (iii) inhibiting proteasome activity with MG-132 for 10 h causes a much larger build-up of CD3δ than overexpressing high levels of AT3 for 24–48 h.
If AT3 decreases extraction of substrates from the ER as part of a homeostatic process to regulate flow in the ERAD pathway, a key question is ‘what function does this serve’? Potential functions for AT3 would be to monitor flow through this pathway and decrease extraction to: (i) allow additional cycles of protein folding, (ii) increase sorting of a subset of ER proteins into an alternate quality control pathway: movement out of this ‘sorted’ ER compartment would be slower than proteasome degradation and result in a modest build-up of protein in the ER, or (iii) decrease extraction when excess substrates are present at the proteasome (Fig. 7). AT3 may function as one of many sensors for proteasome load/function and relay this information to the VCP-dependent ERAD pathway. This would be consistent with the ability of AT3 to interact with Rad23 (10,48). In yeast, RAD23 binds ubiquitylated proteins and delivers them to proteasomes (49,50), and in mammals, a similar role is proposed (10,51). Therefore, AT3 bound to Rad23 at the proteasome could monitor proteasome traffic and delivery of ubiquitylated proteins. AT3 could release from Rad23 in the presence of a modest build-up of polyubiquitylated proteins and increase its interaction with VCP; this would decrease delivery of ERAD substrates to the proteasome. In addition, this should allow additional time for protein folding, for proteasomes to degrade proteins and to divert some misfolded ER proteins into other quality control pathways (Fig. 7).
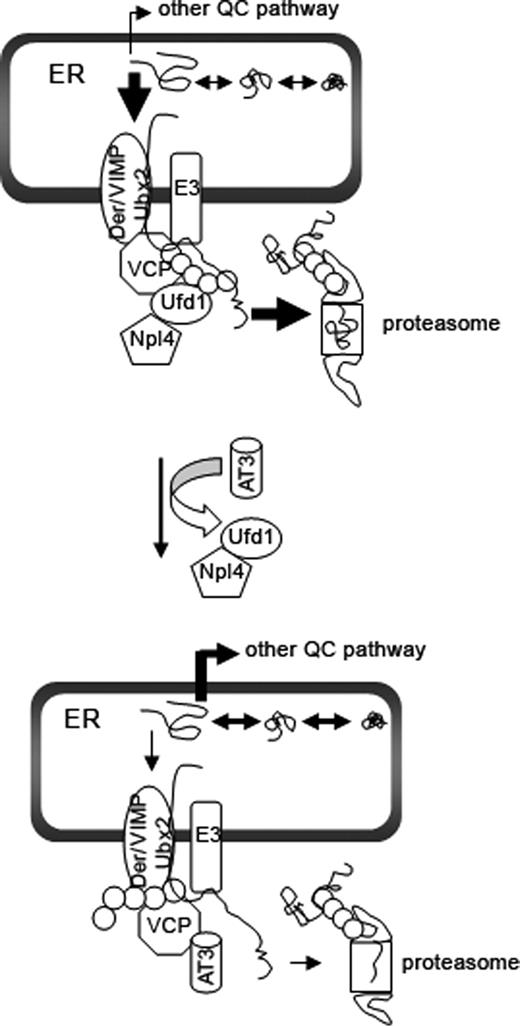
Model for AT3 regulation of the ERAD pathway. Through its interaction with VCP (and likely other proteins), AT3 may function as one of a number of proteins that monitor and regulate flow through the ERAD pathway as part of normal homeostasis. Upper panel. The VCP/Ufd1-Npl4 complex associates with ER proteins including Derlin-1/2, VIMP, Ubx proteins and E3 ligases (E3). An ER protein that does not fold properly is ubiquitylated while still associated with the ER: this allows the VCP/Ufd1-Npl4 complex to bind to it. VCP then extracts the ubiquitylated protein from the ER and through a series of steps delivers the ubiquitylated protein to the proteasome for degradation. In the absence of AT3, misfolded proteins are rapidly extracted from the ER and shuttled to the proteasome for degradation (indicated by bold arrows). Lower panel. Endogenous and transfected AT3 compete with Ufd1 for binding to VCP; displacing Ufd1 from VCP impairs its binding to ubiquitylated ERAD substrates and slows their extraction from the ER, which decreases delivery to proteasomes. In the presence of AT3, more proteins remain in the ER and may undergo additional cycles of folding or may be sorted into other quality control (QC) pathways.
MATERIALS AND METHODS
Plasmids and antibodies
The following constructs were previously described (8,11): MBP-AT3Q29, MBP-AT3(1-191), myc-AT3Q29, myc-AT3Q72, myc-AT3C14A, myc-AT3Q29L229/249A, myc-AT3 (1-191), AT3 siRNA and scrambled siRNA. The sequence in AT3 involved in binding VCP was mutated (282RKRR → ANNA285) using the QuickChange site-directed mutagenesis kit (Stratgene) according to the manufacturer’s directions using the following primers: forward: 5′-CTT ACT TCA GAA GAG CTT GCG AAT GCA GCA GAA GCC TAC TTT GAA AAA C-3′ and reverse: 5′-G TTT TTC AAA GTA GGC TTC TGC TGC ATT CGC AAG CTC TTC TGA AGT AAG-3′. Mouse Ufd1 in pCMV-Sport6 (Open Biosystems) was cloned into pCMV-Tag4A. KDEL-RFP (ER marker), pEGFPN1 and pd1EGFPN1 (contains the ornithine decarboxylase degron) were purchased from Clontech. AT3 cDNA with zero glutamines was from Henry Paulson (University of Iowa) and TCRα cDNA was provided by Ron Kopito (Stanford University). CD3δ and pCIneo-flag-hHrd1 cDNAs were provided by Shengyun Fang (University of Maryland Biotechnology Institute). His-ubiquitin cDNA was obtained from Cecile Pickart (Johns Hopkins), and HA-tagged VCP cDNA was provided by Akira Kakizuka and Gen Sobue (Kyoto University and Nagoya University, Japan). Ub-R-GFP cDNA was from Maria Masucci (Karolinska Institute, Sweden). HA antibody used for immunofluorescence was from Jeffrey Field (University of Pennsylvania). Hybridoma expressing monoclonal antibody, HC-10, against MHC I HC was a kind gift from Peter Cresswell (Yale University) and Joel Taurog (University of Texas Southwestern). The following antibodies were commercially available or made in our laboratory: Flag, HA, myc (Sigma), 1H9 (Chemicon), anti-MJD (made in our laboratory; 52), GAPDH (Immuno Chemical), GFP, VCP (BD Bioscience), tubulin and His (Santa Cruz) and myc polyclonal antibody for immunostaining (Cell Signaling).
Cell culture, transfections and immunoprecipitations
HEK293T and COS-7 cells were cultured in DMEM containing 10% fetal bovine serum (FBS), 100 unit/ml penicillin, 100 µg/ml streptomycin and 1 mm glutamine. Daudi cells (obtained from ATCC) were maintained in RMPI containing 10% FBS and Pen/Strep, as described previously. Cells were transfected with FuGENE 6 (Roche) according to the manufacturer's protocol. In some experiments, two different concentrations of cDNA were used (dose response). For these experiments, 0.4 or 0.8 µg cDNA/well of a six-well dish was used with co-transfections and 0.3 or 0.5 µg/well was used with triple transfections; total cDNA/well was kept constant at 1 µg. Immunoprecipitations were performed as previously described (7). Briefly, cleared cell lysates in lysis buffer [30 mm Tris, pH 8.0, 150 mm KAc, 5 mm Mg(Ac)2, 0.5% NP-40, 1 mm DTT and protease inhibitors] were incubated with the appropriate primary antibody and protein A or G beads (Invitrogen) overnight at 4°C. Beads were washed with lysis buffer, and bound proteins separated by SDS–PAGE and analyzed by immunoblotting. siRNA knock-down of endogenous AT3: vector-based scramble RNA or siRNA against AT3 in pRNAin-H1.2 (GeneScript) (8) was transfected into cells along with CD3δ using FuGENE6. After 48 h, cells were collected and tested for levels of endogenous AT3 and CD3δ by blotting with 1H9 or HA, respectively.
Translation and degradation assays
Metabolic labeling with [35S]methionine/cysteine
HEK293T cells were co-transfected with increasing concentrations of AT3Q29 cDNA (0.4 or 0.8 µg/well of a six-well dish) and either CD3δ or EGFP. After 24 h, cells were incubated with methionine/cysteine-free medium for 2 h to deplete intracellular pools, translation measured by labeling for 30 min with 0.1 mCi/ml [35S]methionine/cysteine, the reaction quenched by adding 10 mm methionine/cysteine and cells lysed in RIPA buffer (150 mm NaCl, 50 mm Tris, pH 8.0, 1% NP-40, 1% deoxycholic acid, 0.1% SDS and protease inhibitors). Labeled proteins were immunoprecipitated using anti-HA (for CD3δ) or anti-GFP and processed for SDS–PAGE. Gels were dried and exposed to a phosphorimager screen and scanned using a Typhoon 840 (Pharmacia).
Degradation assay using cycloheximide chase
Cells were co-transfected with CD3δ and pcDNA3 vector, myc-AT3Q29 or myc-AT3Q72 and treated with 100 µg/ml cycloheximide or vehicle for indicated times. Cells were harvested directly into SDS sample buffer and processed for SDS–PAGE and immunoblots for CD3δ.
Cell fractionation
HEK293T cells were harvested 24 h after transfection and resuspended in buffer (50 mm Tris, pH 8.0, 1 mm 2-mercaptoethanol, 1 mm EDTA, 0.32 m sucrose and protease inhibitors). After 10 min on ice, cells were triturated 20 times using a 26 gage needle. Following centrifugation at 1000g, the supernatant was centrifuged at 100 000g for 30 min at 4°C. The pellet from the 100 000g centrifugation represented the microsomal fraction, and the supernatant represented the cytosol fraction.
In vitro retrotranslocation assay
Cytosol and microsome fractions from Daudi cells were prepared as described (53) and stored at −80°C. Retrotranslocation of MHC I HC from microsomes was carried out according to the procedure of Albring et al. (39) in the following buffer: 130 mm KCl, 10 mm NaCl, 1 mm CaCl2, 2 mm EGTA, 2 mm MgCl2, 5 mm HEPES, pH 7.3. Briefly, 15 µl cytosol (5 µg/µl) was incubated with 10 µg MBP-AT3(1-191) or MBP-AT3Q29 in the presence of 10 µm MG-132 for 2 h on ice, and 20 mm ATP was added and the cytosol warmed to 37°C, followed by adding pre-warmed microsomes from 5×106 cells. The mixture was incubated at 37°C for various times up to 4 min. Microsomes were pelleted by centrifugation at 14 000g for 10 min at 4°C, and the supernatant collected to determine the amount of MHC I HC transported into the cytosol using western blots and HC-10 antibody.
Immunofluorescence
COS-7 cells were grown on glass bottom microwell dishes (MatTech). Twenty-four hours after transfection with myc-AT3, HA-CD3δ and KDEL-RFP, cells were fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.1% Triton X-100. Cells were blocked with 2% goat serum in PBS, incubated with anti-myc (polyclonal) or anti-HA (monoclonal) for 1 h at room temperature, washed with PBS and incubated with goat secondary antibodies conjugated with Alexa Fluor 350 or Alexa 488 (Molecular Probes). Fluorescent cells were analyzed using a Nikon TE-2000U microscope.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We thank Pang Yao, Paul Taylor, Yihong Ye and Mark Forman for critical reading of the manuscript and a special thanks to Barrington Burnett for input throughout the course of this study. We thank the following investigators for constructs or antibodies: Hank Paulson, Ron Kopito, Shengyun Fang, Cecile Pickart, Akira Kakizuka, Gen Sobue, Maria Masucci, Jeffrey Field, Peter Cresswell, Joel Taurog and Honglin Zhou. This study was funded by NIH grant NS42625 to R.N.P.
Conflict of Interest statement. There is no conflict of interest for either author.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}