



Abstract
Hypomorphic mutations in the DNA repair enzyme RNase H2 cause the neuroinflammatory autoimmune disorder Aicardi-Goutières syndrome (AGS). Endogenous nucleic acids are believed to accumulate in patient cells and instigate pathogenic type I interferon expression. However, the underlying nucleic acid species amassing in the absence of RNase H2 has not been established yet. Here, we report that murine RNase H2 knockout cells accumulated cytosolic DNA aggregates virtually indistinguishable from micronuclei. RNase H2-dependent micronuclei were surrounded by nuclear lamina and most of them contained damaged DNA. Importantly, they induced expression of interferon-stimulated genes (ISGs) and co-localized with the nucleic acid sensor cGAS. Moreover, micronuclei associated with RNase H2 deficiency were cleared by autophagy. Consequently, induction of autophagy by pharmacological mTOR inhibition resulted in a significant reduction of cytosolic DNA and the accompanied interferon signature. Autophagy induction might therefore represent a viable therapeutic option for RNase H2-dependent disease. Endogenous retroelements have previously been proposed as a source of self-nucleic acids triggering inappropriate activation of the immune system in AGS. We used human RNase H2-knockout cells generated by CRISPR/Cas9 to investigate the impact of RNase H2 on retroelement propagation. Surprisingly, replication of LINE-1 and Alu elements was blunted in cells lacking RNase H2, establishing RNase H2 as essential host factor for the mobilisation of endogenous retrotransposons.
Introduction
Aicardi-Goutières syndrome (AGS) is a rare genetic autoimmune disease presenting in early childhood, which is characterized by chronic neuroinflammation resulting in brain calcification, leukoencephalopathy and cerebral atrophy. AGS shows a strong phenotypical overlap with systemic lupus erythematodus (SLE) with activation of the type I interferon system being a defining hallmark of both diseases (1). AGS also strongly resembles the sequelae of congenital viral infection. Mutations in the genes of TREX1, SAMHD1, ADAR1, IFIH1 and all three subunits of RNASEH2 have been identified in AGS patients (1,2). As all AGS genes encode proteins involved in nucleic acid metabolism, it is generally believed that an accumulation of unprocessed, endogenous nucleic acids constitutes the main pathogenic trigger for AGS (3). For the 3’-5’ exonuclease TREX1 and the dNTPase SAMHD1 it has been suggested that endogenous retrotransposons might represent the nucleic acid species accruing in AGS patients, as mobilisation of LINE-1 and Alu elements is strongly increased in cells lacking TREX1 or SAMHD1 (4,5). However, the exact molecular mechanism of how RNase H2-deficiency, which accounts for the bulk (> 50%) of all AGS patient mutations, leads to inappropriate activation of the immune system and thus disease development is not known (6).
RNase H2 is a trimeric enzyme consisting of the three subunits A, B and C, with RNase H2A bearing the catalytic site of the nuclease. RNase H2 is capable of removing the RNA moiety of RNA/DNA hybrids as well as initiating the excision of single ribonucleotides from a DNA duplex, a process that has been termed ribonucleotide excision repair (RER) (6,7). RNase H2 knockout mice, which show early embryonic lethality, have revealed the physiologic role of RNase H2 as an essential DNA repair enzyme executing RER in dividing cells (8,9). The genomic DNA of RNase H2 knockout embryos displays an increased burden of single ribonucleotides resulting in extensive DNA damage. In fact, it has turned out that DNA lesions arising from misincorporated ribonucleotides constitute the by far most frequent type of naturally occurring DNA damage (8,10). Previous studies have revealed DNA damage as a bona fide inducer of the type I interferon response (11,12). Mechanistically, it has recently been shown that in cells undergoing genotoxic stress nuclear DNA is released into the cytoplasm, where it activates components of the canonical DNA sensing machinery resulting in robust type I interferon expression (12–15). Engagement of the DNA sensor cyclic GMP-AMP synthase (cGAS) and the adapter protein stimulator of interferon genes (STING) hereby converts the presence of cytosolic DNA into a cellular signal culminating in the expression of type I interferons as well as numerous interferon-stimulated genes (ISGs) (16). Given the substantial DNA damage occuring in RNase H2 knockout cells, it is conceivable that damaged DNA represents the prime molecular trigger for the increased type I interferon production in RNase H2-dependent disease. It is, however, also possible that accumulating unprocessed RNase H2 substrates, such as RNA/DNA retrotransposition intermediates, stimulate the type I interferon axis, as has been speculated previously (17,18). As cytosolic DNA sensing via cGAS and STING governs the interferon signature seen in hypomorphic RNase H2 mice (19,20), it is highly likely that the nucleic acids accumulating in the absence of RNase H2 at some point enter the cytosol. Autophagy is a physiological, cellular process by which long-lived macromolecules and organelles are cleared from the cytoplasm. During autophagy cytosolic components are enclosed by a double membrane resulting in the formation of an autophagosome. Degradation of autophagosomal content subsequently takes place in the acidic environment of autophagolysosomes, which are generated upon lysosomal fusion (21). Mounting evidence suggests that autophagy can also serve as a mechanism to remove ectopic cytosolic DNA, which has first been shown for Mycobacterium tuberculosis-derived DNA and later also for damaged DNA released from the nucleus and subsequently routed to lysosomes for unspecific degradation via DNase II (15,22,23).
We show in this report that RNase H2-deficient cells accumulate cytosolic DNA aggregates, which strongly resemble micronuclei and activate the cGAS/STING pathway. Consistent with micronuclei, these cytoplasmic DNA structures contain damaged nuclear DNA and are surrounded by the nuclear envelope. Importantly, micronuclei have previously only been associated with genome instability, but not with eliciting an immune response. Here, we demonstrate that RNase H2-dependent micronuclei stimulate the type I interferon axis and propose a causative role for these distinct DNA aggregates in autoimmunity development. Furthermore, RNase H2-dependent micronuclei are cleared by autophagy in order to prevent excess activation of the innate immune system. Additional induction of autophagy by treatment with mTOR inhibitors such as rapamycin stimulated micronuclei removal and mitigated ISG upregulation in RNase H2-deficient cells. Finally, we demonstrate that retrotransposition of LINE-1 and Alu elements is largely abolished in human RNase H2 knockout cells. This is in contrast to TREX1- and SAMHD1-deficient cells, which show a strongly elevated mobilisation of endogenous retroelements, and therefore argues against retroelement-derived nucleic acids being the prime trigger of RNase H2-dependent disease.
Results
Time-dependent ablation of RNase H2 results in DNA damage and enhanced ISG expression
Genetic deletion of RNase H2 in mice is embryonic lethal and embryo-derived fibroblasts do not survive in cell culture (8,9). The drastic phenotype associated with RNase H2-deficiency has been attributed to severe DNA damage resulting in a p53-mediated growth arrest. Consequently, upon concomitant deletion of p53 embryonic death in RNase H2 null embryos was delayed and RNase H2−/− murine embryonic fibroblasts (MEFs) retained growth and viability (8). In order to investigate in more detail the molecular events triggered by RNase H2-deficiency we generated a tamoxifen-inducible RNase H2B knockout mouse line by intercrossing RNase H2Bflox mice with Cre-ERT mice. The resulting mouse line (RNase H2Bflox/flox, +Cre-ERT) allowed us to precisely abolish RNase H2 expression in a time-dependent manner, as administration of a low dose of 4-Hydroxytamoxifen (4-OHT) for 3 days led to the complete absence of both RNase H2 mRNA and protein in primary, non-immortalized MEFs (Fig. 1A, Supplementary Material, Fig. S1A). Importantly, it has been shown previously that genetic ablation of the B subunit is sufficient to disintegrate the whole enzyme complex. In keeping with this, immunofluorescence analysis using a polyclonal antiserum raised against all three murine subunits revealed the absence of the RNase H2 holoenzyme following 4-OHT treatment (Fig. 1B). For the sake of brevity, we termed these cells RNase H2ΔTam MEFs hereafter. Similar to RNase H2 knockout mice, RNase H2ΔTam cells displayed extensive DNA damage, as evidenced by increased phosphorylation of histone H2AX (γH2AX) as well as 53BP1 foci accumulation, both widely recognized marker proteins of DNA double strand breaks (Fig. 1C and D, Supplementary Material, Fig. S1B). Most importantly, MEFs devoid of RNase H2 showed increased expression of a panel of interferon-stimulated genes (ISGs) (Fig. 1E). As this “interferon signature” represents the most reliable biomarker of AGS to date (1,24), we concluded that RNase H2ΔTam MEFs can serve as a suitable cellular model for studying the consequences of RNase H2 deficiency, in particular with regard to disease development.
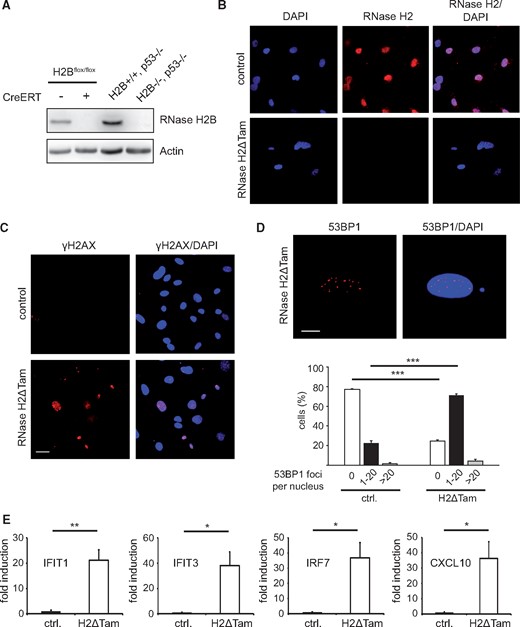
Genetic ablation of murine RNase H2 leads to extensive DNA damage and ISG upregulation. (A) Administration of a low dose of 4-OHT (100 nM) for 3 days resulted in complete abrogation of RNase H2B protein expression in primary MEFs derived from conditional RNase H2Bflox (RNase H2BloxP/loxP) embryos bearing a ubiquitous Cre recombinase (CreERT). RNase H2Bflox MEFs w/o Cre were used as control cells. For conciseness 4-OHT-treated RNase H2Bflox MEFs with CreERT were termed RNase H2ΔTam MEFs hereafter. RNase H2B/p53-double-KO MEFs generated in (8) served as negative control. RNase H2B protein was detected by Western Blotting using a specific rabbit antiserum raised against the murine holoenzyme (8), with actin serving as loading control. (B) Immunofluorescence analysis demonstrated loss of nuclear RNase H2 expression in RNase H2ΔTam MEFs. Nuclear DNA was counterstained with DAPI. Scale bar, 30 µM. (C) Widespread accumulation of DNA damage was observed in RNase H2ΔTam MEFs using immunofluorescence of phosphorylated histone H2AX (γH2AX). DAPI as nuclear counterstain. Scale bar, 30 µM. (D) RNase H2ΔTam MEFs exhibited elevated numbers of nuclear 53BP1 foci indicating increased levels of DNA damage. Top: Representative immunofluorescence picture showing numerous 53BP1 foci in a single RNase H2ΔTam nucleus. DAPI as counterstain. Scale bar, 10 µM. Bottom: Nuclear 53BP1 foci were counted and staged into three groups based on the number of foci per nucleus. Error bars are SEM. ***P<0.001, Tukey's multiple comparison test (n = 3, 100 nuclei/experiment). (E) Quantitative RT-PCR (qPCR) revealed strong mRNA upregulation of selected interferon-stimulated genes (ISGs) in RNase H2ΔTam MEFs compared to control MEFs. Error bars represent SEM. **P < 0.01, *P < 0.05, t-test (n = 3 independent experiments).
Accumulation of micronuclei-like DNA in the cytoplasm of cells lacking RNase H2
The molecular trigger of RNase H2-dependent disease is unknown. As most AGS genes encode proteins processing nucleic acids, it has been hypothesized that an accumulation of nucleic acids likely underlies the pathogenic type I interferon response characteristic for AGS (3,17,25). However, a long known link between DNA damage and activation of the type I interferon system exists that has only recently been attributed to an increased presence of cytosolic DNA (12,15). As cells devoid of RNase H2 exhibit massive DNA damage, we sought to investigate if RNase H2-deficient cells also accumulate cytosolic DNA. To that end, we screened the cytoplasm of RNase H2ΔTam MEFs for ectopic DNA using an antibody recognizing double-stranded (ds) DNA. Confocal microscopy revealed the presence of distinct dsDNA structures in the cytoplasm of both RNase H2ΔTam and control cells, which we – based on their subcellular localization – tentatively classified as either nuclear buds or perinuclear bodies (PNBs) (Fig. 2A). The fluorescence signal of the dsDNA-antibody almost invariably colocalized with the DNA dye DAPI (Supplementary Material, Fig. S1C). However, whereas in control MEFs extranuclear DNA aggregates were observed only scarcely, the number of nuclear buds as well as perinuclear bodies strongly increased in the absence of RNase H2 (Fig. 2A). To characterize the cytosolic DNA structures in more detail, we assessed whether cytoplasmic DNA displayed signs of DNA damage. Strikingly, >80% of cytosolic dsDNA aggregates in RNase H2-deficient cells contained damaged DNA, as evidenced by H2AX phosphorylation (Fig. 2B). We noticed a strong resemblance of these distinct DNA structures with micronuclei, cytosolic DNA aggregates typically arising during anaphase of mitosis and consisting of missegregated chromosome fragments. To investigate the subcellular origin of the cytosolic DNA accumulating in the absence of RNase H2 we stained cells for lamin B1, which is an integral nuclear envelope protein and thus a reliable marker for micronuclei. We observed that the majority of cytosolic DNA aggregates in RNase H2ΔTam MEFs were surrounded by Lamin B1 (95% of perinuclear bodies vs. 66% of nuclear buds), indicating that these DNA structures indeed represent micronuclei (Fig. 2C). The lower proportion of Lamin B1-positive nuclear buds in this context may indicate that micronuclei are released from the nucleus at regions of nuclear envelope instability or breakdown (26). Taken together, we show that upon RNase H2 deletion cells amassed cytosolic DNA, whereby the majority of DNA aggregates contained damaged DNA and likely represent micronuclei.
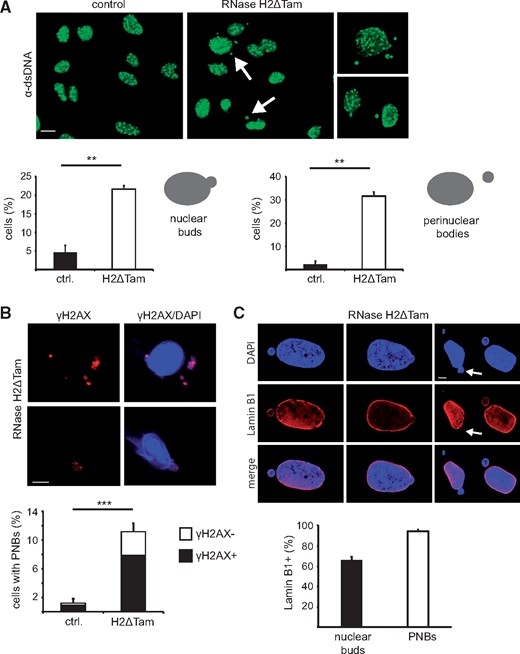
Accumulation of micronuclei-like cytosolic DNA in the absence of RNase H2. (A) Top: RNase H2ΔTam MEFs exhibited increased numbers of cytosolic dsDNA aggregates (highlighted by white arrows), as evidenced by confocal immunofluorescence using an antibody recognizing dsDNA (scale bar, 15 µM). Bottom: Quantification of cells showing cytosolic dsDNA, which based on subcellular localization was classified as either nuclear buds or perinuclear bodies. Cells with ≥1 cytosolic dsDNA aggregates were regarded as positive. Error bars are SEM. **P < 0.01, t-test (n = 3, 100 cells/experiment). (B) Excess cytosolic DNA in RNase H2ΔTam MEFs contained mostly damaged DNA. Top: Colocalization of extranuclear DAPI signals with cytosolic γH2AX in RNase H2ΔTam MEFs (scale bar, 10 µM). Bottom: Quantification of γH2AX-positive perinuclear bodies (PNBs) in RNase H2ΔTam and control MEFs. Error bars represent SEM. ***P < 0.001, t-test (n = 3, 100 PNBs/experiment). (C) Cytosolic DNA aggregates in RNase H2ΔTam cells are positive for the nuclear envelope component Lamin B1. Top: Confocal microscopy revealed broad colocalization of Lamin B1 with DAPI-positive cytosolic DNA. White arrows highlight nuclear buds lacking Lamin B1, possibly indicating regions of nuclear membrane disruption (scale bar, 5 µM). Bottom: Quantification of Lamin B1-covered cytosolic DNA structures in RNase H2ΔTam MEFs. Note the relatively lower proportion of Lamin B1-positive nuclear buds. Error bars are SEM (n = 3, 100 cells/experiment).
RNase H2-dependent micronuclei are linked to cellular proliferation and accompanied by a cGAS/STING-dependent innate immune response
RNase H2 functions as a DNA repair enzyme that mainly acts on replicating DNA and consequently its expression is particularly high in actively proliferating tissues (8). We sought to establish if the observed accumulation of damaged cytosolic DNA depends on the proliferative status of the cell and therefore forced RNase H2ΔTam MEFs out of the cell cycle by serum starvation. After having confirmed that most of the cells ceased proliferation and did not succumb to apoptosis (Supplementary Material, Fig. S1D) we gauged accumulation of micronuclei in G0-arrested RNase H2ΔTam cells. Compared to their proliferating counterparts the build-up of cytosolic DNA aggregates was significantly reduced in quiescent RNase H2ΔTam MEFs, which could be restored by serum re-addition (Fig. 3A). In keeping with this, we established that a considerable fraction of RNase H2-dependent micronuclei contained previously replicated DNA (Fig. 3B). We also occasionally found concentrations of both replicated and damaged DNA in micronuclei of RNase H2ΔTam MEFs (Fig. 3C).
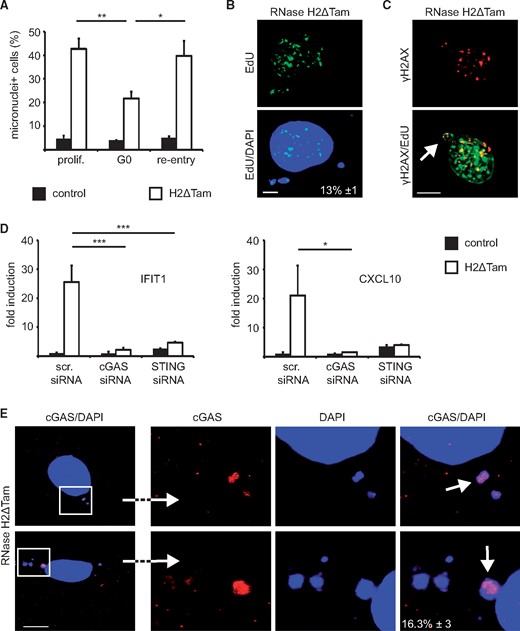
Generation of micronuclei in the absence of RNase H2 depends on cellular proliferation and is associated with cGAS/STING activation. (A) Accumulation of cytosolic DNA relies on cellular proliferation. RNase H2ΔTam and control MEFs were forced into GO by serum-starvation (0.5% FCS for 3 d) and micronuclei were counted as in Figure 2A. Re-addition of serum to serum-deprived MEFs restored micronuclei number (re-entry). Error bars are SEM, **P < 0.01, *P < 0.05, 1-way-ANOVA and Tukey's multiple comparison test (n = 3, 100 cells/experiment). (B) Micronuclei-like DNA aggregates include replicated DNA. RNase H2ΔTam MEFs were pulsed with the nucleoside analog EdU for 24 h and EdU labeling of cytosolic DNA was visualized by confocal microscopy. Proportion of EdU-positive micronuclei is indicated as percentage (white inset). SEM (n = 3, 100 micronuclei/experiment). (C) Freshly released micronuclei in RNase H2ΔTam MEFs occasionally contained both damaged (γH2AX+) and replicated (EdU+) DNA, as evidenced by confocal immunofluorescence analysis (scale bar, 10 µM). (D) Silencing of cGAS and STING blunted elevated ISG expression in the absence of murine RNase H2. IFIT1 and CXCL10 transcript levels in RNase H2ΔTam MEFs were measured by qPCR. Cells transfected with scrambled siRNAs (scr. siRNA) served as control. Error bars are SEM. ***P < 0.001, *P < 0.05, 1-way-ANOVA (n = 3). (E) cGAS localizes to micronuclei-like DNA aggregates in RNase H2-deficient MEFs. Confocal imaging showed colocalization of transfected cGAS-HA with DAPI-positive cytosolic DNA (scale bar, 10 µM). Number of cGAS-positive micronuclei is indicated as percentage (white inset). The three images on the right represent magnifications of the left figure. SEM (n = 3, 100 micronuclei/experiment).
Cytoplasmic DNA is recognized by the innate immune system via the intracellular nucleic acid sensor cGAS, which in turn activates the ER-resident adapter protein STING (16). It has previously been shown in this context that cytosolic DNA accumulating after genotoxic stress is sensed by the cGAS/STING pathway (12). Moreover, the type I interferon response ensuing TREX1 deficiency is mediated by cGAS and STING both in vitro and in vivo and two recent studies showed a similar cGAS/STING reliance in independent hypomorphic RNase H2 mouse models (19,20,27–29). We sought to confirm the involvement of the cGAS/STING axis also in our experimental setup and transfected RNase H2ΔTam MEFs with siRNA pools targeting either STING or cGAS. qPCR analysis revealed that the expression of STING and cGAS was largely abolished by RNAi (Supplementary Material, Fig. S2A). Importantly, gene silencing of either STING or cGAS blunted ISG expression in RNase H2ΔTam cells, confirming that ISG upregulation mainly depends on activation of the cGAS/STING pathway in our system (Fig. 3D). To gain further mechanistic insight we expressed an HA-tagged version of murine cGAS in RNase H2ΔTam MEFs and tracked cGAS localization by confocal fluorescence microscopy. We observed that a sizeable proportion of cytoplasmic DNA aggregates also stained positive for cGAS, indicating that DNA sensing in the absence of RNase H2 is mediated at least in part by cytosolic interaction of cGAS and micronuclei-like DNA (Fig. 3E).
Taken together, we provide evidence that 1) accumulation of RNase H2-dependent micronuclei is linked to cellular proliferation and 2) that ISG upregulation in the absence of RNase H2 is dependent on cGAS/STING signaling, which coincides with partial mobilization of cGAS to micronuclei.
RNase H2-dependent micronuclei are cleared by autophagy
Cells use autophagy to dispose of cytoplasmic waste and the removal of ectopic cytosolic DNA by autophagy has recently been reported in cells lacking the lysosomal nuclease DNase II (15). We sought to investigate whether accumulating micronuclei in RNase H2ΔTam MEFs are likewise targeted by autophagy for lysosomal degradation. To that end, we employed a mRFP-eGFP-LC3 tandem reporter construct, which due to quenching of the pH-sensitive GFP signal following lysosomal fusion allows specific detection of autophagosomes (RFP + GFP fluorescence) and acidic autophagolysosomes (only RFP) (30). Using confocal microscopy, we established that >80% of RNase H2-dependent micronuclei colocalized with autophagosomes, indicating that in the absence of RNase H2 cytosolic DNA is routed to lysosomes via autophagy (Fig. 4A). In keeping with this, blocking the fusion of autophagosomes with lysosomes using bafilomycin A1 led to even stronger micronuclei accumulation in RNase H2-deficient cells (Fig. 4B). Specifically, we only observed an increase in micronuclei, while the number of nuclear buds remained constant upon bafilomycin A1 treatment (Fig. 4B). However, this is fully consistent with the autophagic delivery of RNase H2-dependent micronuclei to lysosomes, as only this process and not the nuclear release of damaged DNA should be affected by bafilomycin A1. We sought to complement our results obtained with bafilomycin A1 by blocking early stage autophagy using Atg5-specific siRNAs, which resulted in almost complete silencing of Atg5 expression (Supplementary Material, Fig. S2B). Compared to control siRNAs, concomitant inhibition of autophagy in RNase H2ΔTam MEFs by Atg5 knockdown led to >10-fold increase in ISG expression (Fig. 4C). These results suggest a role for autophagy as a cell-intrinsic defense system restraining the innate immune response against ectopic self DNA, which otherwise - as in the case of RNase H2ΔTam MEFs with artificially reduced Atg5 expression – would result in excess activation of the type I interferon axis. We next wanted to know if cells lacking RNase H2 exhibit increased levels of autophagy or show an enlarged lysosomal compartment, which has recently been associated with TREX1-deficiency (31). However, although multiple experimental approaches were used, we were not able to detect increased lysosome numbers or activity in RNase H2ΔTam MEFs (Supplementary Material, Fig. S2C–E). Instead, Western Blot analysis assessing the lipidated form of LC3 (LC3II), which is associated with autophagosomes, revealed elevated steady-state levels of autophagy in the absence of RNase H2 (Fig. 4D). As our results suggested that excess cytosolic DNA is cleared by autophagy, we asked whether additional induction of autophagy would result in a reduction of RNase H2-dependent micronuclei. We therefore treated RNase H2ΔTam MEFs with the pharmacological mTOR inhibitors Rapamycin and Torin 1, which both have been reported as potent autophagy activators via their suppressive action on mTOR (32). Strikingly, administration of both drugs led to a significant reduction in micronuclei number as well as ISG expression in RNase H2-deficient cells already at low concentrations (Fig. 4E and F, Supplementary Material, Fig. S3A). As we used relatively low doses of both inhibitors in our experimental setup (in particular Rapamycin), we additionally verified their inhibitory effect on mTOR by assessing phosphorylation of the major mTOR substrate S6 kinase (S6K) (Supplementary Material, Fig. S3B). In conclusion, we established that micronuclei arising in the absence of RNase H2 are targeted by autophagy for lysosomal degradation. Moreover, additional induction of autophagy by pharmacological mTOR inhibition attenuated both micronuclei accumulation and ISG upregulation in cells lacking RNase H2.
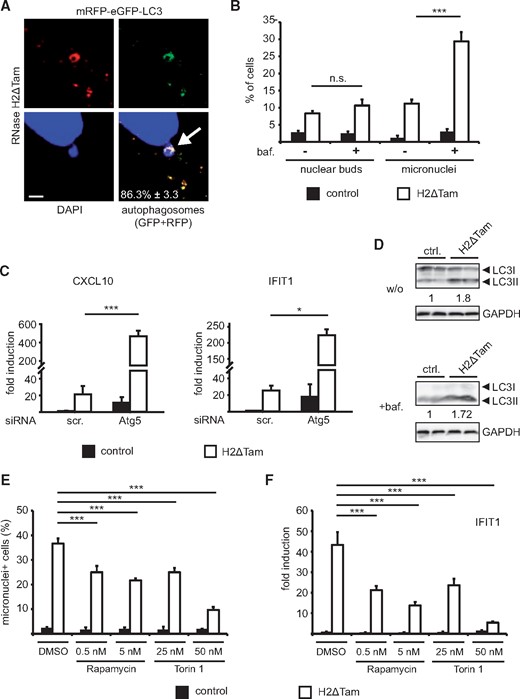
RNase H2-dependent micronuclei are targeted and removed by autophagy. (A) Cytosolic DNA aggregates accumulating in the absence of RNase H2 are targeted by autophagy. Confocal analysis revealed widespread colocalization of autophagosomal marker protein LC3 with DAPI-positive micronuclei. Expression of the pH-sensitive mRFP-eGFP-LC3 autophagy reporter in RNase H2ΔTam MEFs allowed discrimination between autophagosomes (RFP+GFP) and autolysosomes (RFP). In order to facilitate LC3 detection cells were starved in HBSS for 4 h before visualization. Quantification of LC3-positive micronuclei is expressed as percentage (white inset). SEM, n = 3, 100 micronuclei/experiment. Scale bar, 2.5 µM. (B) Treatment with bafilomycin A1 (baf., 10 nM for 24 h) resulted in micronuclei accumulation in RNase H2ΔTam MEFs, whereas number of nuclear buds remained unaffected. Bafilomycin A1 inhibits the fusion of autophagosomes with lysosomes. Error bars are SEM, ***P < 0.001, n.s. = not significant, 2-way-ANOVA and Bonferroni posttest (n = 3). (C) Abrogating autophagy by Atg5 silencing amplified ISG expression in RNase H2-deficient MEFs. CXCL10 and IFIT1 transcript levels were measured by qPCR 3 days after siRNA transfection and simultaneous 4-OHT treatment. Scrambled siRNAs served as negative controls. Error bars represent SEM, ***P < 0.001, *P < 0.05, 1-way-ANOVA (n = 3). (D) RNase H2ΔTam MEFs exhibited elevated levels of autophagy. Western Blot analysis revealed increased protein levels of the lipidated form of LC3 (LC3II) which is associated with autophagosomes. Cells were additionally treated with bafilomycin A1 (100 nM for 4 h) to inhibit lysosomal fusion and assess autophagic flux. Protein levels of LC3II were normalized to GAPDH expression and shown as x-fold increase over control. One representative experiment is shown (n = 2). (E,F) Induction of autophagy by pharmacological mTOR inhibition resulted in a marked decrease in micronuclei number and reduced ISG upregulation. E) RNase H2ΔTam and control MEFs were treated with indicated doses of Rapamycin and Torin 1 for 3 days (in parallel with 4-OHT) and micronuclei accumulation was analyzed as in Figure 2A. Solvent-treated cells served as controls (DMSO). Error bars are SEM, ***P < 0.001, 1-way-ANOVA and Tukey's multiple comparison test (n = 3, 100 cells/per experiment). F) RNase H2ΔTam and control MEFs were treated as in 4E and IFIT1 transcript levels were assessed by qPCR. Error bars represent SEM, ***P < 0.001, 1-way-ANOVA and Tukey's multiple comparison test (n = 3).
RNase H2 facilitates LINE-1 and Alu retrotransposition
As TREX1 and SAMHD1 have previously been identified as negative regulators of LINE-1 and Alu elements, endogenous retroelements have been suggested as the main source of aberrant nucleic acids accumulating in AGS. Since RNA/DNA hybrids are essential reverse transcription intermediates and at the same time bona fide RNase H2 substrates, we sought to establish whether RNase H2 likewise displays antiretroviral activity. As MEFs inherently display low retroelement activity and constitute a notoriously hard-to-transfect cell type, we generated HEK293T- and HeLa cells lacking RNase H2 via CRISPR/Cas9 genome editing. HEK293T- and HeLa cells exhibit high endogenous LINE-1 and Alu activity, respectively, and are therefore widely used cell lines in retroelement research. We targeted the first exon of the catalytic A subunit and verified correct targeting by Illumina Next Generation Sequencing. Absence of RNase H2A on protein level in independent HEK293T- and HeLa cell clones was confirmed by Western Blot analysis (Fig. 5A). We initially measured transcript levels of major retroelement classes but did not detect significant changes between RNase H2-knockout and parental cells (Supplementary Material, Fig. S3C.). To assess actual LINE-1 retrotransposition events we employed a plasmid-based reporter assay, where a luciferase gene is rendered functional only after a completed cycle of transcription, reverse transcription and integration (Supplementary Material, Fig. S4A). To our surprise and at odds with the hypothesis that increased retrotransposition causes AGS, we observed a dramatic reduction in LINE-1 mobilization in RNase H2-KO HEK cells compared to parental cells (Fig. 5B). We additionally confirmed the results with a second eGFP-based LINE-1 reporter assay using flow cytometry (Supplementary Material, Fig. S4B). As Alu elements rely on proteins encoded by the LINE-1 retrotransposition machinery, we investigated if RNase H2 likewise modulates replication of this non-autonomous retroelement class. Using a neomycin-based reporter system which similarly detects only completed Alu retrotransposition events, we demonstrated that Alu mobility is almost absent in RNase H2-deficient HeLa cells (Fig. 5C). In conclusion, we found that rather than restricting their activities, RNase H2 appears to be essential for the retrotransposition of LINE-1 and Alu elements.
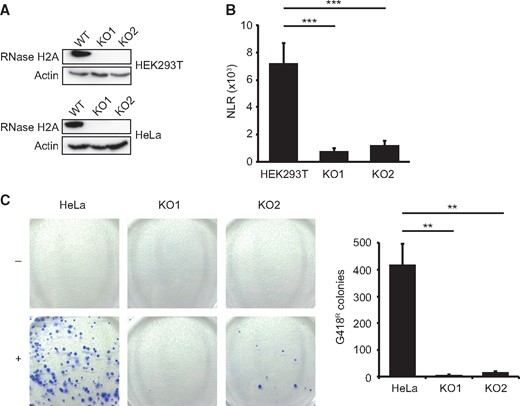
LINE-1 and Alu retrotransposition is blunted in RNase H2A-deficient HEK293T and HeLa cells. (A) The catalytic A subunit of RNase H2 was targeted in HEK293T and HeLa-HA cells using CRISPR/Cas9. Absence of RNase H2A protein in individual cell clones was confirmed by Western Blotting and Next Generation Sequencing (not shown). (B) De-novo LINE-1 retrotransposition is strongly reduced in HEK293T cell clones lacking RNase H2A. Cells were transfected with a firefly luciferase-tagged LINE-1 reporter plasmid. Luminescence was quantified 4 days following transfection and LINE-1 retrotransposition activity is expressed as normalized luminescence ratio (NLR). Luminescence signals are generated in transfected cells only after one completed round of LINE-1 retrotransposition (i.e. transcription, splicing, reverse transcription and integration into the genome, for details see Fig. S4A). Error bars represent SEM, ***P < 0.001, 1-way-ANOVA with Tukey's multiple comparison test (n = 6). (C) De novo Alu retrotransposition elements are almost absent in RNase H2A-deficient HeLa cell clones. Cells were transfected with a neomycin-tagged Alu reporter along with a LINE-1 ORF2-containing plasmid. G418-resistant colonies resulting from successful retrotransposition events are counted after 2 weeks of antibiotic selection. Cells transfected with empty vector (-) instead of Alu reporter plasmid (+) served as negative control. Left: Representative images of G418-resistant colonies stained with Trypan blue. Right: Quantification of G418-resistant colonies resulting from completed de-novo Alu retrotransposition events. Error bars are SEM, **P < 0.01, 1-way-ANOVA and Tukey's multiple comparison test (n = 3).
Discussion
AGS is a severe genetic, autoinflammatory disorder and mutations in RNase H2 account for more than 50% of disease cases (1). AGS displays high phenotypic overlap with SLE and rare RNase H2 gene variants have indeed recently been associated with SLE (33). While SLE represents a complex autoimmune disease of polygenic origin, AGS is caused by defined mutations in a limited number of genes and thus may serve as a simplified disease model for SLE. The molecular trigger of AGS is unknown. As most AGS genes code for proteins that modify nucleic acids, it is generally believed that in AGS unprocessed nucleic acids of endogenous origin accumulate and inappropriately stimulate receptors of the innate immune system (17,25,34). However, the identity of the nucleic acid species accumulating in RNase H2-compromised cells has not been established yet. Identifying the exact nature of these nucleic acids is of paramount importance, as they provide potential targets for therapeutic intervention. Using an inducible knockout system we demonstrated accumulation of distinct dsDNA species in the cytoplasm of primary murine embryonic fibroblasts lacking RNase H2 (RNase H2ΔTam MEFs) which was accompanied by elevated ISG expression. The majority of cytosolic DNA aggregates contained damaged DNA and were surrounded by nuclear envelope, indicating that these structures represent micronuclei. Cytosolic DNA is typically sensed by the cGAS/STING pathway (16). As TREX1-associated autoimmunity is mediated by this pathway and hypomorphic RNase H2 mice likewise exhibit a cGAS/STING-dependent interferon signature, we investigated whether ISG upregulation in our conditional RNase H2-knockout system was also reliant on cGAS/STING signaling. Indeed, we observed an almost complete reduction of ISG expression after cGAS or STING silencing. By localizing cGAS to RNase H2-dependent micronuclei we were additionally able to shed light on the underlying mechanism of DNA sensing. Similarly, mobilization of cGAS to cytosolic DNA has recently been reported for Mycobacterium tuberculosis-derived DNA upon autophagosomal escape and subsequent leakage into the cytoplasm (23). However, as micronuclei-associated chromatin in RNase H2ΔTam MEFs was encapsulated in the nuclear envelope, it remains to be established how exactly cGAS gains access to immunogenic DNA. It has in this context previously been shown that >60% of micronuclei suffer from nuclear envelope collapse during interphase, which would enable cytosolic proteins such as cGAS to engage the previously shielded DNA (26). Indeed, TREX1 has recently been pinpointed to disrupted but not intact micronuclei, although the relevance of this selective localization was not immediately obvious (35). Interestingly, as a surrogate for genomic instability micronuclei have already been described in the context of RNase H2-deficiency (8,36). However, in neither of the studies increased generation of micronuclei has been linked to immune signaling. In fact, a causal connection between micronuclei and activation of the innate immune system has to our knowledge not been established at all yet and may hence have significant ramifications beyond the context of RNase H2-dependent disease. Previous work suggests that both DNA damage and interferon signature in hypomorphic RNase H2 mice are due to defective removal of single ribonucleotides from genomic DNA (RER), as opposed to the degradation of RNA/DNA hybrids (19). We can therefore envision a scenario where the lack of RNase H2 in replicating cells initially results in the misincorporation of genomic ribonucleotides, which in turn leads to the development of widespread DNA damage, probably due to compensatory ribonucleotide excision by alternative, less precise DNA repair mechanisms (37). The increased appearance of nuclear buds in RNase H2-deficient MEFs indicates that the damaged DNA is – perhaps as a cellular disposal mechanism - expelled from the nucleus and subsequently accumulates in the cytoplasm, as recently proposed in (38). On a cautionary note, all known RNase H2 patient mutations are hypomorphic, which supports the notion that a complete reduction of RNase H2 activity is not compatible with human life (1). This also in line with the early embryonic lethality of RNase H2 knockout mice (8,9). It therefore remains to be seen whether our results gained from RNase H2 null cells can be readily translated into situations where the RNase H2 activity is only partially reduced such as AGS. However, as partial reduction of RNase H2 expression via RNAi resulted in increased micronuclei generation in HeLa cells (albeit without appreciation of their immunogenic potential) and both tissues and cells derived from hypomorphic RNase H2 mice exhibited an interferon signature, we regard it as likely that accumulating micronuclei also stimulate the innate immune system of hypomorphic RNase H2 cells (19,20,36).
Autophagy is a physiological process that clears the cytoplasm by routing old or defective material to lysosomes for unspecific degradation (21). A recent study has revealed that damaged cytosolic DNA is delivered to lysosomes via autophagy, where it is metabolized by the lysosome-resident nuclease DNase II (15). Accumulation of ectopic DNA in the cytoplasm of DNase II-deficient cells resulted in increased ISG expression, which was further amplified by additional blockade of autophagy (15). We therefore sought to investigate whether autophagy also plays a role in delivering RNase H2-dependent cytosolic DNA to lysosomes. Although we failed to detect an overall increase in lysosome number or activity (as has been reported for TREX1-null cells), we did observe elevated homeostatic levels of autophagy in RNase H2-deficient cells. Importantly, however, we could demonstrate that RNase H2-dependent micronuclei were specifically targeted by autophagy, as we observed a high degree of colocalization with LC3-positive autophagosomes. Consequently, blocking the fusion of autophagosomes with lysosomes resulted in an even higher amassment of micronuclei. Similarly, inhibition of autophagy in RNase H2-knockout cells by Atg5 silencing led to a dramatic increase in ISG expression, indicating that autophagy in this setting prevented excessive activation of the innate immune system. On the other hand, additional induction of autophagy by pharmacological mTOR inhibition resulted in the clearance of RNase H2-dependent micronuclei, which was accompanied by a significant reduction in ISG expression. Hence, our findings could open new avenues for therapeutic intervention, as pharmacological autophagy induction might represent a viable approach to facilitate cellular removal of immunostimulatory and possibly pathogenic DNA. Overall, our results point to DNA damage as the principal molecular trigger of the observed RNase H2-dependent type I interferon response. Extensive DNA damage has also been associated with TREX1- and SAMHD1-deficiency and it is thus conceivable that cytosolic accumulation of damaged DNA is not only restricted to cells lacking RNase H2 (39–41). In fact, in TREX1-knockout cells cytosolic DNA structures have been described strongly resembling the micronuclei-like extranuclear DNA aggregates in RNase H2ΔTam cells (14). However, both cellular origin and relation to autophagy are unclear for TREX1-dependent cytosolic DNA and whether DNA damage in a SAMHD1-deficient setting leads to cytosolic DNA accumulation is not known. Moreover, as many hereditary diseases caused by mutations in DNA repair or DNA damage response genes tend to manifest predominantly in the CNS (42,43), it is tempting to speculate whether the widespread DNA damage associated with cellular AGS models might underlie the strong neurological involvement of the disease.
It has been proposed that in RNase H2-compromised cells reverse transcription intermediates consisting of RNA/DNA hybrids may accumulate and inappropriately activate the innate immune system, resulting in the pathogenic type I interferon response characteristic for AGS (18). Consistent with this, it has been reported that RNA/DNA hybrids are able to elicit robust type I interferon expression via both TLR9- and cGAS-signaling (44,45). Our results demonstrating abolished LINE-1 and Alu element retrotransposition in human RNase H2-KO cells were therefore unexpected and suggest that RNase H2 rather operates to facilitate retrotransposition. Given that RNase H2 is endowed with two distinct enzymatic activities, it will be crucial to assess whether RNase H2 promotes retrotransposition either by cleaving RNA/DNA reverse transcription intermediates or removing single ribonucleotides from the retroelement cDNA. Nevertheless, our findings make it unlikely that nucleic acids derived from endogenous retroelements accumulate and trigger activation of the innate immune system in RNase H2-deficient cells.
Materials and Methods
Mice, MEF isolation and culture, knockout induction
RNase H2Bflox mice were kindly provided by Axel Roers (Institute of Immunology, Medical Faculty Carl Gustav Carus, University of Technology Dresden, Dresden, Germany). Generation of conditional RNase H2B knockout mice was described in (9). RNase H2Bflox mice were crossed to Cre-ERT mice for global gene inactivation upon tamoxifen induction (46). RNase H2Bflox/flox littermates w/o CreERT served as control animals. RNase H2ΔTam and control MEF lines were generated from individual E13.5 embryos. After removing the head and internal organs the embryos were mechanically dissociated in growth medium. Resulting suspensions were grown at 37 °C, 5% CO2 and 3% O2, and non-adherent cells removed after 24 h. Primary, non-immortalized MEFs were maintained in Dulbecco's modified Eagle's medium (DMEM, Sigma Aldrich) supplemented with 10% (v/v) FCS (PAN Biotech), 100 units/ml of penicillin (Sigma Aldrich) and 100 μg/ml of streptomycin (Sigma Aldrich). To induce Cre recombination in RNase H2ΔTam MEFs growth medium was supplemented with 100 nM 4-hydroxytamoxifen (4-OHT, Sigma Aldrich) solved in DMSO for 3 days. Control MEFs were treated with the same dose of 4-OHT. RNase H2B−/−;p53−/− and RNase H2B+/+;p53−/− MEF lines were a kind gift of Andrew P. Jackson (MRC Human Genetics Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, UK). The PCR program and primers used for genotyping are deposited in Supplementary Material, Table S1.
Reagents, plasmids, transfection, siRNAs
The following reagents were used as indicated in the figure legends: bafilomycin A1, puromycin, 4-hydroxytamoxifen (4-OHT), doxorubicin (all Sigma Aldrich), blasticidin (InvivoGen) and HBSS (10x, Invitrogen). The following plasmids were used: pUNO1-cGAS-HA3X (InvivoGen, #14A23-JC) and pMOWS-mRFP-eGFP-LC3 (cloned from ptfLC3 (Addgene, #21074) into retroviral vector pMOWS via EcoNI and EcoRI). MEF cells were transiently transfected with Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer's instructions and selected by antibiotics for 2 days. Prior to LC3 visualization cells were starved in HBSS for 4 h. For knockdown experiments MEFs were transfected with siRNAs targeting cGAS (Dharmacon, M-055608-01-0005), STING (Dharmacon, M-05528-01), Atg5 (Dharmacon, M-064838-02-0005) or non-targeting control pool (Dharmacon, D-001206-13-05) using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. siRNAs were transfected along with 4-OHT treatment and cells were harvested for RNA isolation after three days.
Immunocytochemistry, Western Blot and β-hexosaminidase assay
Cells were fixed with ice-cold acetone/methanol (1:2) for 10 min and blocked with 1% (w/v) BSA in PBS overnight at 4 °C. Cells were incubated with primary antibodies for 4 h and secondary antibodies for 2 h in a humidified chamber and subsequently embedded in Immu-Mount (Thermo Fisher Scientific) complemented with 5 µg/ml 4’,6-diamino-2-phenylindole (DAPI, Sigma Aldrich). Primary antibodies were: anti-dsDNA (HYB331-01, Santa Cruz, sc-58749), anti-pH2A.X (Ser 139, Cell Signaling, #2577), anti-HA-Tag (C29F4, Cell Signaling, #3724), anti-Lamin-B1 (Santa Cruz, sc-30264), anti-53BP1 (Cell Signaling, #4937) and anti-LAMP1 (DSHB, 1D4Bc). Rabbit anti-mouse RNase H2 antibody was kindly provided by Andrew P. Jackson (8). Secondary antibodies were: anti-mouse Alexa Flour 488 (Invitrogen) and anti-rabbit Cy3 (GE Healthcare). Staining with secondary Ab alone served as negative control. An Olympus FV1000 confocal laser scanning microscope was used for image acquisition. Western Blotting was performed as described in (47). The following primary antibodies were used: anti-γH2AX (Cell Signaling Technology, #2577), rabbit anti-mouse RNase H2 (8), anti-LC3 (MBL, PM036), anti-p62 (Biomol, PW9860), anti-LAMP2 (DSHB, Abl93c) and anti-RNase H2A (Origene, TA306706). To ensure equal loading amounts membranes were stripped and re-probed with anti β-actin (Cell Signaling Technology, #4967). Assessment of lysosomal β-hexosaminidase activity was performed as described in (48).
Quantitative PCR (qPCR) and flow cytometry
Total RNA was isolated using Gene Jet RNA Purification Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNA was synthesized using Revert Aid Reverse Transcriptase (Thermo Fisher Scientific) with oligo(dT)18 and random hexamer primers (1:2, Thermo Fisher Scientific). For the qPCR reaction SYBR Green qPCR Master Mix (Thermo Fisher Scientific), 0.5 µM target specific primer pairs (Supplementary Material, Table S2) and 10 ng cDNA template were used. Incorporation of SYBR Green was analyzed on a Lightcycler 480 II (Roche Applied Science) with the following program: initial denaturation at 95 °C for 10 min, 45 cycles of denaturation (10 s at 95 °C), annealing (20 s at 60 °C), and elongation (20 s at 72 °C). Relative mRNA expression was calculated using the ΔΔCp method with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) serving as house keeping control. For cell cycle analysis Click-iT EdU Flow Cytometry Assay Kit (Invitrogen) and SYTOX® AADvancedTM dead cell stain solution (Invitrogen) were used according to manufacturer’s instructions, with the EdU pulse lasting 4 h. Resulting fluorescence was detected using a FACSCanto flow cytometry system (BD biosciences).
Human RNase H2 knockout via CRISPR/Cas9 genome editing
The first exon of human RNASEH2A (ENST00000221486.4) was submitted to an online CRISPR Design Tool (http://crispr.mit.edu/) and guide RNA#4 was chosen due to its high cleavage efficiency towards the target sequence. A pair of oligonucleotides comprising gRNA#4 (forward: accGGCTCCTTGCGGCACACCGC, reverse: aacGCGGTGTGCCGCAAGGAGCC, gRNA#4 DNA sequence in capital letters) was annealed, phosphorylated and subsequently cloned into the SapI-digested guideRNA expressing plasmid LeGO-Cas9-iC-puro + cgRNA_SapI (generously provided by Boris Fehse, Research Dept. Cell and Gene Therapy, Department of Stem Cell Transplantation, University Medical Centre Hamburg-Eppendorf, Hamburg, Germany) as described in (49). The CRISPR/Cas9 plasmid was transfected into HEK293T and HeLa-HA cells by lipofection and cells were grown in the presence of 1 µg/ml puromycin for 72 h to enrich successful transfectants. For the generation of monoclonal knockout lines cells were seeded at low concentrations (100 cells per 10 cm dish), cultivated for 12 days and resulting colonies were expanded. Successful gene targeting was confirmed by deep sequencing of a PCR product comprising the predicted Cas9 cleavage site.
LINE-1 and Alu retrotransposition assays
To assess de-novo LINE-1 genomic integrations two independent LINE-1 plasmid-based reporter systems were employed. The dual-Luciferase LINE-1 assay was performed as described in (50). Briefly, the pYX017 plasmid, which contains the LINE-1RP element isolated from a patient with retinitis pigmentosa, was introduced into parental and RNase H2-knockout HEK293T cells by lipofection. Cells were grown for 4 d, lysed and luminescence of firefly luciferase-tagged LINE-1RP was quantified using the Dual-Luciferase Reporter Assay System (Promega). pYX017 also harbours a Renilla luciferase gene which allowed for normalization for transfection efficiency. Cells were transfected in parallel with the retrotransposition incompetent LINE-1 plasmid pYX015, which was used to calculate the normalized luminescence ratio (NLR), as described in (50). Plasmids used in the dual-Luciferase LINE-1 assay were kindly provided by Wenfeng An (Department of Pharmaceutical Sciences, South Dakota State University, Brookings, SD, USA). We additionally used an eGFP-tagged LINE-1 reporter according to (51). Briefly, parental and RNase H2-knockout HEK293T cells were transiently transfected with the plasmid 99PUR-RPS-EGFP (containing LINE-1RP fused to eGFP). 3 days after transfection eGFP-positive cells were quantified using flow cytometry and normalized to cells transfected with the retrotransposition incompetent plasmid 99PUR-JM111-EGFP. For detection of de-novo Alu retrotransposition events a neomycin-based Alu reporter system was employed according to (52). Briefly, HeLa cells lacking RNase H2A and parental HeLa cells were transfected with the Alu reporter plasmid (Alu neoTet) along with a plasmid encoding LINE-1 ORF2 (pcORF2). Co-transfection with pcORF2 is necessary, as Alu elements are strictly dependent on LINE-1 proteins for retrotransposition. Following transfection, cells were cultured for 7 days w/o antibiotics, split into a 10 cm dish and then cultured for further 14 days in the presence of G418 (700 µg/ml). The selection medium was renewed every 3 days. Cells were finally fixated with methanol/acetone and G418-resistant colonies were stained with 0.04% Trypan blue (Sigma Aldrich). Plasmids for the eGFP-based LINE-1 and Alu reporter assays as well as HeLa cells (substrain HA) were a kind gift of John Goodier (Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA).
Statistical analysis
All data are shown as mean ± SEM. Statistical analysis was performed with GraphPad Prism 5 using two-tailed Student’s t test, analysis of variance (ANOVA) test followed by Bonferroni’s post hoc test or Tukey's multiple comparison test. For all tests, p values less than 0.05 were considered statistically significant.
Supplementary Material
Supplementary Material is available at HMG online.
Acknowledgements
We thank Annett Lickert and Stefanie Schnell for excellent technical assistance. We are deeply grateful to Andrew P. Jackson, Boris Fehse, Axel Roers, John Goodier and Wenfeng An for providing essential material and reagents.
Conflict of Interest statement. None declared.
Funding
Deutsche Forschungsgemeinschaft, Bonn, Germany (Project grant RA 2404/1-1) and the Cluster of Excellence “Inflammation at Interfaces”.
References
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}