





Abstract
The combined processes of gene duplication, nucleotide substitution, domain duplication, and intron/exon shuffling can generate a complex set of related genes that may differ substantially in their expression patterns and functions. The APETALA2-like (AP2-like) gene family exhibits patterns of both gene and domain duplication, coupled with changes in sequence, exon arrangement, and expression. In angiosperms, these genes perform an array of functions including the establishment of the floral meristem, the specification of floral organ identity, the regulation of floral homeotic gene expression, the regulation of ovule development, and the growth of floral organs. To determine patterns of gene diversification, we conducted a series of broad phylogenetic analyses of AP2-like sequences from green plants. These studies indicate that the AP2 domain was duplicated prior to the divergence of the two major lineages of AP2-like genes, euAP2 and AINTEGUMENTA (ANT). Structural features of the AP2-like genes as well as phylogenetic analyses of nucleotide and amino acid (aa) sequences of the AP2-like gene family support the presence of the two major lineages. The ANT lineage is supported by a 10-aa insertion in the AP2-R1 domain and a 1-aa insertion in the AP2-R2 domain, relative to all other members of the AP2-like family. MicroRNA172-binding sequences, the function of which has been studied in some of the AP2-like genes in Arabidopsis, are restricted to the euAP2 lineage. Within the ANT lineage, the euANT lineage is characterized by four conserved motifs: one in the 10-aa insertion in the AP2-R1 domain (euANT1) and three in the predomain region (euANT2, euANT3, and euANT4). Our expression studies show that the euAP2 homologue from Amborella trichopoda, the putative sister to all other angiosperms, is expressed in all floral organs as well as leaves.
Introduction
The APETALA2 (AP2) domain (sometimes also called the AP2/ethylene-responsive element–binding factor [ERF] domain or ERF/AP2 domain) defines a large gene family of DNA-binding proteins called AP2/ERF. AP2/ERF genes are divided into classes based on the number of AP2 domains that are present (fig. 1). One class encodes a protein containing two AP2 domains (AP2-like) and includes AP2 (Jofuku et al. 1994), AINTEGUMENTA (ANT) (Elliott et al. 1996; Klucher et al. 1996), and Glossy15 (GL15) (Moose and Sisco 1996) (fig. 1). A second class encodes a protein with only one AP2 domain (ERF-like) and includes ERFs (Ohmetakagi and Shinshi 1995), TINY (Wilson et al. 1996), AtEBP (Buttner and Singh 1997), and ABI4 (Finkelstein et al. 1998) (fig. 1). A third class of AP2/ERF genes, RAV1 and RAV2 (Kagaya, Ohmiya, and Hattori 1999), encodes proteins that have two different DNA-binding domains, AP2 and B3 (Giraudat et al. 1992) (fig. 1).
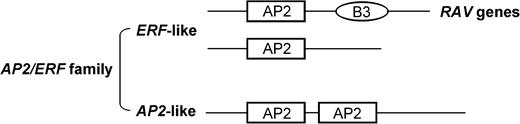
Structure of AP2/ERF genes. ERF-like genes contain one AP2 domain and AP2-like genes contain two AP2 domains. Because RAV genes contain another DNA-binding domain, B3, RAV genes are sometimes treated as a third group in the AP2/ERF family (Kagaya, Ohmiya, and Hattori 1999).
Arabidopsis AP2 is the most well-studied gene in AP2/ERF family. AP2 encodes a putative transcription factor (Jofuku et al. 1994; Riechmann and Meyerowitz 1998) and plays a central role in the establishment of the floral meristem (Irish and Sussex 1990; Huala and Sussex 1992; Bowman et al. 1993; Schultz and Haughn 1993; Shannon and Meekswagner 1993), the specification of floral organ identity (Komaki et al. 1988; Bowman, Smyth, and Meyerowitz 1989; Kunst et al. 1989), and the regulation of floral homeotic gene expression (Bowman, Drews, and Meyerowitz 1991; Drews, Bowman, and Meyerowitz 1991; Jack, Brockman, and Meyerowitz 1992; Mandel et al. 1992) in Arabidopsis. There are two AP2 domains in AP2, and each AP2 domain contains 68 amino acids (aa) with an 18-aa core region that forms an amphipathic α-helix (Jofuku et al. 1994; Allen et al. 1998). These domains are essential for AP2 function (Jofuku et al. 1994).
In addition to AP2, other genes encoding the AP2 domain have been well studied in Arabidopsis. These include ANT (AP2-like), a gene that regulates ovule development and floral organ growth (Elliott et al. 1996; Klucher et al. 1996), and CBF1 (ERF-like), a gene that binds to the C-repeat/Dehydration Response Element, a cis-acting DNA regulatory element that stimulates transcription in response to low temperature and water deficit (Stockinger et al. 1997). The AP2-like genes whose functions have been determined by mutant analyses (e.g., Arabidopsis AP2, ANT, and maize GL15) act as key regulators in developmental processes, whereas the ERF-like genes (e.g., tobacco ethylene-responsive element–binding protein [EREBP]-2) appear to be involved in responses to biotic and environmental stress, although their precise functions are largely unknown (Riechmann and Meyerowitz 1998).
The AP2 domain has been considered plant specific (Riechmann and Meyerowitz 1998). However, recent studies based on an extensive computer-assisted search showed that homologues are present in the cyanobacterium Trichodesmium erythraeum, the ciliate Tetrahymena thermophila, and the viruses Enterobacteria phage Rb49 and Bacteriophage Felix 01 (Magnani, Sjölander, and Hake 2004; Wuitschick et al. 2004; reviewed in Wessler 2005). These nonplant proteins bearing an AP2 domain are predicted to be HNH (or in some cases, HNN; histidine and asparagine) endonucleases. Magnani, Sjölander, and Hake (2004) hypothesized that a horizontal transfer of an HNH-AP2 endonuclease from bacteria into plants may have led to the origin of the AP2/ERF family.
Riechmann and Meyerowitz (1998) reviewed the molecular and biochemical characteristics of some AP2/ERF proteins and performed a phylogenetic analysis of the AP2/ERF genes using the partial Arabidopsis genome sequence available at the time (approximately 14% of the entire genome). Their study included 6 AP2-like genes and 34 ERF-like genes, but many additional AP2/ERF genes have since been identified.
Following the complete sequencing of the Arabidopsis genome, Reichmann et al. (2000) searched for AP2/ERF in the Arabidopsis genome and found 144 AP2/ERF genes. Sakuma et al. (2002) classified AP2/ERF genes in Arabidopsis as members of five classes based on similarities in their DNA-binding domains: AP2 subfamily (14 genes), RAV subfamily (6 genes), DREB subfamily (55 genes), ERF subfamily (65 genes), and others (the fifth group; 4 genes). However, some subfamilies did not form monophyletic groups in their phylogenetic tree, the relationships among subgroups were not clear, and no measures of internal support for clades (e.g., bootstrap values) were provided.
In addition to Arabidopsis, the fully sequenced genome of rice is now available for analysis of all genes of the AP2-like gene family (Goff et al. 2002; Yu et al. 2002). Furthermore, several AP2-like genes in various flowering plants and gymnosperms have been identified, and their patterns of expression have been studied (e.g., Moose and Sisco 1996; Maes, Van Montagu, and Gerats 1999; Maes et al. 2001; Vahala, Oxelman, and von Arnold 2001; Boutilier et al. 2002; Shigyo and Ito 2004). Phylogenetic analyses of all members of the AP2-like gene family detected in the Arabidopsis and rice genomes, as well as homologues reported from other diverse taxa, have not yet been performed.
Our goals in this study were to (i) clarify the phylogeny of the AP2-like gene family and (ii) assess the pattern of domain evolution by tracing structural changes (domains, motifs, and gaps) onto the sequence-based phylogenetic tree for this gene family. We initially treated RAV genes as members of ERF-like genes because both classes have a single AP2 domain and therefore may be more closely related to each other than either is to the AP2-like genes, which are the focus of this study. We test this hypothesis in this study. We also report the expression of AP2 homologues of Amborella trichopoda (Am.tr.AP2), the putative sister to all other extant angiosperms (e.g., Mathews and Donoghue 1999; P. S. Soltis, D. E. Soltis, and Chase 1999; Kim et al. 2004).
Although the name “AP2/ERF” has been frequently used for this gene family in recent studies (Fujimoto et al. 2000; Sakuma et al. 2002; Magnani, Sjölander, and Hake 2004; Xue and Loveridge 2004), the names AP2/EREBP (Ohmetakagi and Shinshi 1995; Riechmann and Meyerowitz 1998; Vahala, Oxelman, and von Arnold 2001; Li and Chye 2004; Shigyo and Ito 2004) and RAP2 (related to AP2) (Okamuro et al. 1997) have also been used. Because it appears to be the most widely used, the family name AP2/ERF is used in this study.
Materials and Methods
Data Collection
We searched for AP2-like genes in the (i) Arabidopsis thaliana and Oryza sativa genome databases at The Institute for Genomic Research (TIGR; http://www.tigr.org), (ii) expressed sequence tag (EST) collection of the Floral Genome Project (FGP; http://www.floralgenome.org) (D. E. Soltis et al. 2002; Albert et al. 2005), (iii) EST contigs of PlantGDB (http://www.plantgdb.org), (iv) EST set for Physcomitrella (http://www.moss.nibb.ac.uk/), and (v) GenBank. Predicted protein sequences from the fully sequenced genomes of Arabidopsis (version 3; ftp://ftp.tigr.org/pub/data/a_thaliana/) and rice (version 2; ftp://ftp.tigr.org/pub/data/Eukaryotic_Projects/o_sativa/) were downloaded from TIGR for a total of 27,117 sequences from Arabidopsis and 80,975 sequences from rice. TIGR's rice database contains the International Rice Genome Sequencing Project Bacterial Artificial Chromosome/P1-derived Artificial Chromosome clones which have inherent duplication due to overlap. Therefore, we eliminated identical versions of loci, reducing the number of sequences to 63,673. All 90,790 sequences were blasted against each other using BlastP 2.4 (Altschul et al. 1990) with an E value cutoff of 1 × 10−5. The TribeMCL package (Enright, Van Dongen, and Ouzounis 2002) was then used at high stringency (inflation = 5.0) to cluster the proteins into putative protein families.
For the AP2-like gene family, 18 Arabidopsis sequences and 35 rice sequences were detected. (Recently, version 3 of the rice [O. sativa] genome was released and many gene locations were changed compared to version 2. To avoid future confusion, we followed the names of version 3 in this paper although we extracted our rice data from version 2.) The number of Arabidopsis AP2-like genes that we detected differed from those reported by Reichmann et al. (2000; 14 sequences) and Sakuma et al. (2002; 14 sequences) but was the same as that found by Magnani, Sjölander, and Hake (2004). Using BlastP, 16 ESTs (eight unigenes) of Welwitschia, Amborella, Nuphar, Persea, Liriodendron, and Eschscholzia were detected in the FGP EST database. We obtained 31 aligned contigs (264 ESTs) from 12 taxa from PlantGDB. Two sequences were detected in the Physcomitrella EST library. Using Se-Al (http://evolve.zoo.ox.ac.uk/software/html?name=SeAl), we predicted the aa sequences from ESTs and EST contigs and determined the appropriate frame for translation by searching for the longest continuous aa sequences and then deleting segments before the initiation codon and after the termination codon at the 5′ and 3′ ends. All inferred aa sequences of Arabidopsis, rice, and ESTs were blasted using BlastP in GenBank with a cutoff of 5 × 10−3; we eliminated those sequences that were detected multiple times.
Some translational errors resulting from improper estimation of splicing sites were expected in the genes from the rice genome due to unfinished sequence assembly. All 17 genes that we found with extraordinarily long, unique insertions or deletions were from the rice genome. Because of potential errors in sequencing and assembly, we eliminated these 17 sequences from the 35 rice sequences detected in the initial search. After the preliminary alignment of 109 sequences, we eliminated 37 sequences that were shorter than 120 aa because short sequences do not provide sufficient informative characters for the reconstruction of the phylogeny.
We selected one or two representatives of each sublineage of Arabidopsis ERF-like genes (including RAV genes) as outgroups based on the results of a previous phylogenetic analysis that focused on the relationships among ERF genes (Sakuma et al. 2002) and added three ERF homologues of Physcomitrella to the outgroup. Our final data set (Supplementary material 1 online) included 72 genes and contained 18 Arabidopsis and 18 rice sequences, eight EST unigenes from the FGP database (Amborella, Eschscholzia, Liriodendron [two sequences], Nuphar [two sequences], Persea, and Welwitschia), 19 EST unigenes from the PlantGDB (Glycine [four sequences], Hordeum [three sequences], Lycopersicon [two sequences], Medicago, Solanum [three sequences], Sorghum, Triticum [three sequences], and Zea [two sequences]), two ESTs of Physcomitrella, 17 aa sequences from GenBank (Antirrhinum [two sequences], Brassica [two sequences], Hordeum, Hyacinthus, Malus, Nicotiana, and Petunia [two sequences], Picea [two sequences], Pinus [three sequences], Pisum, and Zea), and 17 outgroup sequences.
To investigate domain evolution of AP2-like genes, we analyzed a matrix containing only the domain regions of the AP2-like genes, with the domain of ERF-like genes (including RAV genes) as the outgroup. Some of the 18 Arabidopsis sequences that we obtained did not contain a complete AP2-R2 domain (i.e., At2g41710, At2g28550, At2g39250, At3g54990, and At5g60120) and were deleted from the domain analysis (see Discussion). We also deleted ESTs that did not contain sequence across the entire domain. The matrix for the domain analyses (Supplementary material 2 online) contained 84 domains from 42 AP2-like genes and 17 domains from ERF-like genes as outgroup sequences (Supplementary material 2 online).
Alignment and Phylogenetic Analyses
The aa alignment was first conducted using ClustalX (version 1.83) (Thompson et al. 1997) using all default options. During the initial aa alignment, we recognized that the 5′ and 3′ ends of the genes (predomain and postdomain sequences, respectively; fig. 1) were very difficult to align. To improve our alignment of these regions, we divided the sequences into three subgroups based on the initial alignment: (i) outgroup sequences (ERF genes including RAV genes, which have one AP2 domain), (ii) sequences having a 10-aa insertion in the AP2-R1 domain (see Results), and (iii) sequences lacking a 10-aa insertion in the AP2-R1 domain. We aligned sequences within each subgroup first and then combined them into a global alignment using the “profile alignment” method in ClustalX (file to file alignment). This alignment was adjusted manually. The final alignment of 99 aa sequences contained 888 characters. The alignment of aa sequences was converted into an alignment of DNA sequences using the program AA2DNA (http://www.bio.psu.edu/People/Faculty/Nei/Lab/software.htm) for DNA analyses. We phylogenetically analyzed several different alignments in which we adjusted the alignment of the predomain and postdomain regions to address the impact of alignment on the topology.
Maximum parsimony analyses of aa and DNA matrices were conducted on both the entire gene and the AP2 domain region separately using PAUP* 4.0b10 (Swofford 2001). The heuristic search strategy involved 100 random addition replicates with TREE bisection-reconnection branch swapping, saving all optimal trees. To assess support for each node, bootstrap analyses (Felsenstein 1985) were performed using 500 resamplings and 10 random addition replicates with TREE bisection-reconnection branch swapping, saving all optimal trees. However, in the analyses of the aa matrix of the domain region, we saved only 10 trees per replicate because the limit of computer memory was reached when we saved all optimal trees.
Prior to conducting a Bayesian analysis (Huelsenbeck and Ronquist 2001), we selected the best model of molecular evolution using Modeltest (version 3.06; Posada and Crandall 1998). The GTR + I + Γ model of DNA substitution, which assumes general time reversibility (GTR), a certain proportion of invariant sites (I), and a gamma distribution to accommodate rate variation among sites (Γ), was selected for both the full and domain-only DNA matrices. Bayesian analyses were conducted using MrBayes (version 3.04b; Huelsenbeck and Ronquist 2001). For each analysis, we ran four chains, sampling one tree every 1,000 generations for 1,000,000 generations starting with a random tree. Stationarity was reached at approximately generation 250,000 in the full DNA matrix and 150,000 in the domain-only matrix; thus, the first 250 trees in the full DNA matrix and 150 trees in the domain-only matrix were considered the “burn in” of the chain, and phylogenetic inferences were based on those trees sampled after generations 250,000 and 150,000, respectively.
Hypothesis Tests of Models of Domain Evolution
We used a likelihood ratio test coupled with parametric bootstrapping to test alternative models of domain evolution in AP2 genes (see fig. 8). Model A involves domain duplication prior to the duplication and divergence of two gene lineages. Model B posits gene duplication and divergence prior to independent domain duplication in each gene lineage. These models differ in the relationships among domain groups, and the more distal relationships are not relevant. Therefore, to reduce computation time in the hypothesis tests, we reduced the original matrix and tree from 84 terminals to 15 terminals by randomly selecting for inclusion three genes from each major clade (euAP2, ANT, and ERF genes). To reduce the possible effect of different sequence sampling, we made three different reduced data sets in this manner and performed the following analyses using each one.
The results of the phylogenetic analysis for the full data set support Model A (fig. 6; Supplementary material 3 online). To test whether the data statistically support Model A and reject Model B, we first built a constraint tree consistent with Model B using MacClade (W. Maddison and D. Maddison 1992). Using the GTR + I + Γ model of molecular evolution and the parameter values estimated using Modeltest, we conducted a maximum likelihood (ML) analysis enforcing the Model B constraint. From the ML tree, we reestimated base frequencies and parameter values and repeated the analysis, again using the GTR + I + Γ model and enforcing the Model B constraint. The ML tree from this analysis was used to reestimate base frequencies and parameter values for the GTR + I + Γ model to simulate 100 data sets using SEQ-GEN (Rambaut and Grassly 1997), with the size of the data sets identical to the original reduced data set (i.e., 15 terminals, 231 bp). Each of the 100 data sets was analyzed by ML using the GTR + I + Γ model with the parameter values and base frequencies that were used to simulate the data and a heuristic search strategy with 10 random addition replicates and TBR branch swapping, and the −ln likelihood tree scores were saved from each analysis. We then analyzed the 100 simulated data sets in the same manner, but enforcing the constraint of Model A, and saved the −ln likelihood tree scores. These analyses of the simulated data allowed us to calculate the null distribution of the likelihood ratio test statistic, δ = (ln L1 − ln L0), where ln L1 is the −ln likelihood tree score of the unconstrained analysis using data simulated under the assumption that Model B is true and ln L0 is the −ln likelihood tree score of the tree obtained from the analysis constrained with Model A, with the data simulated under Model B. We then tested whether δ calculated from analysis of the real (reduced) data set fell within this null distribution of δ. This entire analysis was repeated for each of the three reduced data sets.
Expression Study of A. trichopoda
Total RNAs were extracted from leaves and floral organs following Kim et al. (2004). Reverse transcription of RNAs was performed following the manufacturer's recommendation using SuperScript II RNnase H-reverse transcriptase (Invitrogen, Carlsbad, Calif.). Using the AP2 homologue of A. trichopoda detected from the EST library generated by the FGP (Albert et al. 2005), we designed specific primers for the expression study: AF1AP2 (5′-GCAAGTCTACCTAGGAGGGTTTGA-3′) and AF2AP2 (5′-TTCCCAACGCCCACATTT-3′). For relative quantitative reverse transcription–polymerase chain reaction (RT-PCR), we performed multiplex PCR using gene-specific primer pairs, 18S rDNA primers (as an internal control), and 18S rDNA primer competimers following the protocol in the QuantumRNA Classic 18S Kit (Ambion, Austin, Tex.). Because the copy number of 18S rDNA is much greater than the copy number of the target gene, we adjusted the ratio between 18S rDNA primers and their competimers to get comparable strength of bands of 18S products and target gene products. We used 25 ng of RNA for each PCR and performed 25 cycles of PCR. The completion of this number of cycles is late in the exponential phase of PCR and is therefore optimal for relative quantitative RT-PCR. PCR products were loaded in a 2% agarose gel and visualized with ethidium bromide.
Results
Gene Structure
The most highly conserved regions of the AP2/ERF family were two repeats of the putative DNA-binding AP2 domain (repeated units 1 and 2, abbreviated as R1 and R2) and the region linking these two repeats. In contrast, the predomain and postdomain sequences (fig. 2) were highly variable. We recognized two major groups in the AP2-like genes based on the presence/absence of a 10-aa insertion in the R1 domain (figs. 2 and 3): the ANT group having a 10-aa insertion and the euAP2 group that lacks this insertion. In the R2 domain, a 1-aa insertion was detected in genes of the ANT group (figs. 2 and 3).
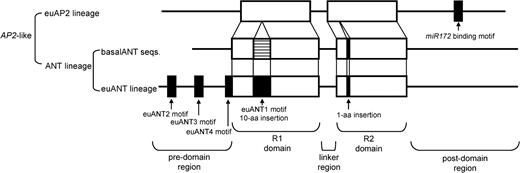
Detailed structure of AP2-like genes. Open boxes indicate AP2 domains, and black boxes indicate lineage-specific motifs or insertions. Hatched region indicates that portion of the euANT1 motif that is not conserved in the basalANT sequences.

A part of the alignment of R1 and R2 domains in AP2-like genes and the corresponding domain of ERF-like genes. Conserved sequences for the AP2/ERF gene family (>95%) are indicated as “*” at the bottom of the alignment. Conserved sequences for the AP2-like genes (>95%) are indicated as “#” at the top of the alignment.
The microRNA172 (miR172)-binding sequence in the postdomain region was restricted to the sequences of the euAP2 group of the AP2-like family (fig. 4). Predicted aa sequences of the miR172-binding sequence were highly conserved (miR172-binding motif: AAASSFG[S/P]).
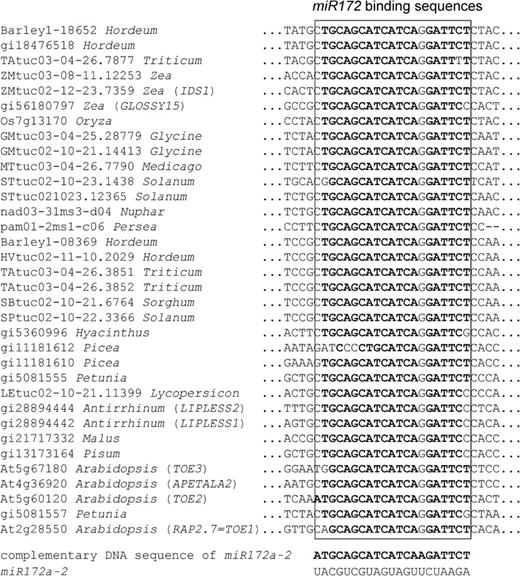
Putative binding site of miR172a-2 in the genes of the euAP2 lineage. The sequence that matches the complementary DNA sequence of miR172a-2 is highlighted in bold.
Within the ANT group, we identified the euANT subgroup based on conserved motifs, including a 10-aa insertion in the R1 domain (the euANT1 motif: NSC[K/R][K/R]EGQ[T/S]R) and three insertions in the predomain region (the euANT2, 3, and 4 motifs: WLGFSLS, PKLEDFLG, and TFGQR) (fig. 2 and Supplementary material 4 online). The sequences of the euANT group also possess a relatively long (127–307 aa) predomain region. The remaining sequences in the ANT lineage (i.e., the basalANT group) have a short predomain region (44–81 aa), and all three motifs are absent.
To address the question of AP2 domain evolution, we aligned the R1 and R2 domains of each AP2-like gene with the domain in the 17 ERF-like genes, which represent all sublineages of Arabidopsis ERF-like genes (fig. 3). R1 and R2 of the AP2-like genes and the domain of the ERF-like genes were easily aligned: 17 residues were strongly conserved (>95%) in the domains of both ERF-like and AP2-like genes. However, this number decreased when we included more ERF-like genes. Thirty-two residues were conserved (>95%) among all AP2-like genes. AP2-like genes have a longer domain than those of the ERF-like genes, which resulted in three gaps in ERF-like genes when aligned (fig. 3, boxes B, C, and D). An 8-aa gap in the R2 domain of the euAP2 and ANT groups was recognized in the same position as the 10-aa insertion in the R1 domain (fig. 3, box B). This 8-aa gap was shared with sequences of the ERF-like genes, with the exception of the RAV genes (fig. 3, box B). Instead, RAV genes shared a 10-aa gap with the R1 domain sequence of euAP2 genes (fig. 3, box A). A 1-aa gap found in the R2 domain of euAP2 genes was also found in ERF-like genes, including RAV genes.
Phylogenetic Analyses
Parsimony analysis of the aa sequence data set generated 39 shortest trees of 13,522 steps (consistency index [CI] = 0.56, retention index [RI] = 0.56) (fig. 5). Major clades recognized in the strict consensus tree of aa sequences exactly matched the groups recognized in structural analyses (fig. 5). We defined these clades as the ANT and euAP2 sublineages of the AP2-like gene family with 84% and 90% bootstrap support, respectively, in the aa analysis. However, the euANT clade, a subclade of the ANT clade that corresponds to the euANT group containing the euANT1, 2, 3, and 4 motifs, received <50% bootstrap support.
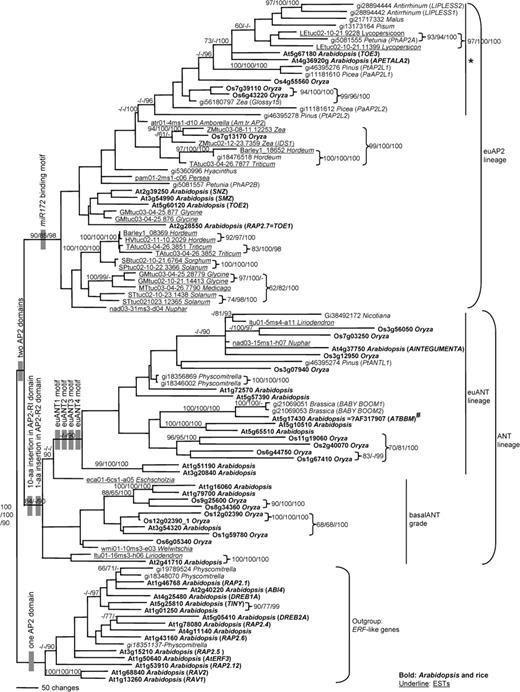
Strict consensus of 39 maximum parsimony trees using aa sequences, shown as a phylogram. Structural characters that support major lineages are indicated at the nodes. Numbers above the branches are bootstrap values from the aa analysis, bootstrap values from the DNA analysis, and the posterior probabilities from the Bayesian analysis (100×). Only values over 50% in the bootstrap analyses and over 90% in the Bayesian analysis are indicated. Asterisk indicates the genes having conserved regions (B and C) recognized by Vahala, Oxelman, and von Arnold (2001) (see Discussion). “#” indicates that only 4 aa changes and a 3-aa insertion out of 529 aligned aa were different between At5g17430 from the Arabidopsis genome database and published AtBBM (Boutilier et al. 2002). These may be alleles of the same gene or sequence errors in a single allele.
The basalANT group, a group of sequences with relatively short predomain sequences and lacking the euANT motifs, does not form a clade in the strict consensus tree. Instead, these sequences form a grade that subtends the euANT clade. The placement of these sequences (fig. 5) suggests that the short predomain region is the ancestral condition in the ANT lineage.
When the aligned aa data set was converted to a nucleotide data set, phylogenetic analysis recovered one shortest tree of 31,129 steps (CI = 0.22, RI = 0.46) with a topology similar to that of the strict consensus tree of the aa analysis: two major clades, euAP2 and ANT, were recovered in the AP2 lineage. Bootstrap support for each node in the nucleotide tree was generally lower than in the aa tree. The euAP2 clade had 85% bootstrap support based on analysis of aligned nucleotides. However, the ANT clade received <50%. Bayesian inference using nucleotide sequences produced a tree similar to that obtained with parsimony analysis of the aa matrix: euAP2 and ANT clades were recognized with posterior probability values of 0.98 and 0.90, respectively. The euANT clade was also recognized with a posterior probability of 0.90. Details of the phylogenetic trees were sensitive to the alignment of the pre- and postdomain regions. However, the same major clades were always recognized, regardless of alignment (data not shown).
Two domains (AP2-R1 and AP2-R2) in the AP2-like genes and the domain of the ERF-like genes were analyzed phylogenetically to address the pattern of domain evolution. Parsimony analyses of this matrix generated 140 shortest trees (length = 594) in the aa analysis and 31 shortest trees (length = 2,960) in the DNA analysis. Strict consensus trees from the aa analysis and the DNA analysis were almost identical to each other (summarized in fig. 6). Five major clades, corresponding to the R1 and R2 domains of the euAP2 and ANT genes plus the ERF-like genes, were recognized. The ANT-R1 clade grouped with the euAP2-R1 clade, and the ANT-R2 clade grouped with the euAP2-R2 clade in the DNA analysis (complete tree is shown in Supplementary material 3 online). The phylogenetic relationships among these domains were moderately to highly supported, with most clades receiving bootstrap support over 70% in the analyses of both nucleotides and aa sequences (fig. 6). The only exception was the ERF clade, which received 60% support in the analysis of aa sequences. Bayesian inference showed the same domain relationships: the R1 domains of both lineages grouped together as did the R2 domains of both lineages. Posterior probabilities of R1 and R2 clades were 0.85 and 1.00, respectively.
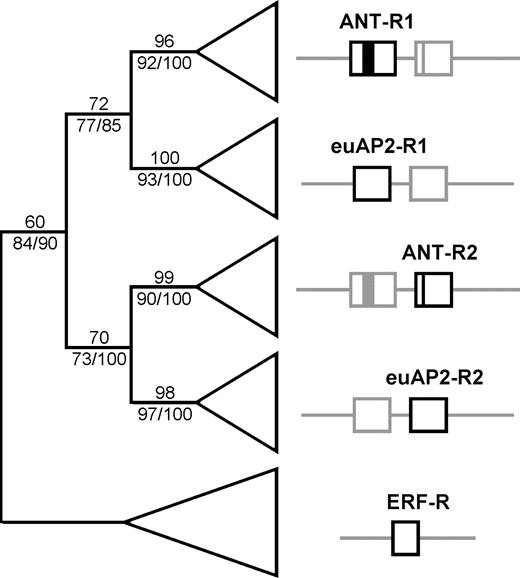
Summarized phylogenetic relationships among R1 and R2 domains of both lineages (euAP2 and ANT) using aa and DNA analyses. Numbers above the branches are bootstrap support values from the aa analysis. Numbers below the branches are bootstrap support values from the DNA analysis and the posterior probabilities from the Bayesian analysis (100×).
Expression of AP2 in the Basal Angiosperm Amborella trichopoda
Am.tr.AP2 was placed in the euAP2 clade in the phylogenetic tree (fig. 5). In mature flowers, nearly identical levels of mRNA of Am.tr.AP2 were detected in each floral organ (i.e., tepals, stamens, and carpels) from both male and female flowers (fig. 7). A similar level of expression was also detected in leaf tissue (fig. 7).
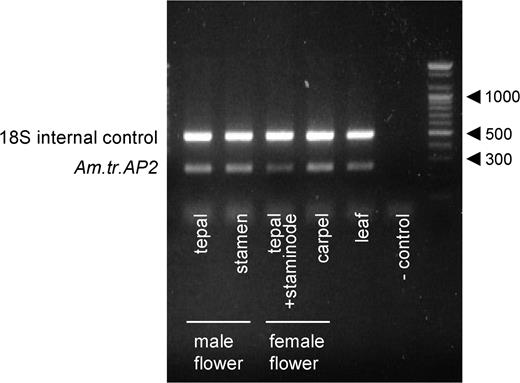
Expression of Am.tr.AP2 in each floral organ and leaf tissue, based on relative quantitative RT-PCR.
Discussion
Previous studies identified a length difference in the R1 domain between AP2 and ANT in Arabidopsis (Klucher et al. 1996), compared AP2 and ANT with related sequences of Arabidopsis, Brassica, and maize (Riechmann and Meyerowitz 1998; Boutilier et al. 2002), analyzed Arabidopsis AP2-like and ERF-like genes phylogenetically (Sakuma et al. 2002), and revealed two major lineages, ANT and euAP2, of AP2-like genes. Our phylogenetic analyses based on all available AP2-like genes confirmed these results.
The evolution of gene structure in the AP2-like gene family is summarized in figure 5. The euAP2 lineage has acquired the miR172-binding motif in the postdomain region. The ANT lineage has acquired a 10-aa insertion and a 1-aa insertion in the R1 and R2 domains, respectively. The euANT lineage has acquired one motif in the R1 domain (euANT1) and three motifs (euANT2–4) in the predomain region.
Divergence Time Between AP2-like and ERF-like Lineages
The AP2/ERF gene family has a limited taxonomic distribution. Homeobox genes and MADS-box genes, many members of which also control plant development (Riechmann and Meyerowitz 1998), are present in animals as well as yeast, whereas AP2/ERF genes have been reported only from eudicot and monocot angiosperms (e.g., Jofuku et al. 1994; Riechmann and Meyerowitz 1998; Maes et al. 2001; Boutilier et al. 2002) and gymnosperms (Ito and Meyerowitz 2000; Vahala, Oxelman, and von Arnold 2001; Shigyo and Ito 2004). In our study, we also detected AP2-like genes in basal angiosperms (Amborella, Nuphar, Liriodendron, and Persea) and a moss (Physcomitrella). Although the sequences of Physcomitrella were already in the EST database, the presence of AP2/ERF genes in Physcomitrella has not been previously published.
Riechmann and Meyerowitz (1998) hypothesized that the common ancestor of AP2-like genes predated the “dicot”/monocot divergence because these genes were found in both Arabidopsis (dicot) and maize (monocot). (Note that the traditional angiosperm group dicots is not monophyletic and has been dropped from angiosperm classification [e.g., APGII 2003] and the eudicot clade encompasses most traditional dicots and approximately 75% of all angiosperm species [Drinnan, Crane, and Hoot 1994].) In our database searches and phylogenetic analyses, ERF-like genes from Physcomitrella were also identified. The presence of Physcomitrella genes in both the AP2-like and ERF-like lineages in the AP2/ERF gene family suggests that the gene duplication that gave rise to these two major lineages preceded the divergence of mosses and tracheophytes. Cooksonia, a Silurian fossil plant, is generally accepted as the oldest known tracheophyte (Lang 1937; Edwards and Davies 1976; Edwards and Feehan 1980; Gifford and Foster 1988). The oldest samples of Cooksonia-type tracheids occur in the uppermost Ludlow Series of Silurian strata, about 418 Myr B.P. (Edwards and Davies 1976). Therefore, we hypothesize that the split between AP2-like and ERF-like lineages occurred at least 418 Myr B.P.
Binding Site of MicroRNA
MicroRNAs (miRNAs) are noncoding RNAs of ∼21 nt in length that have been identified in both animals and plants (Lagos-Quintana et al. 2001; Lau et al. 2001; Lee and Ambros 2001; Llave et al. 2002; Mourelatos et al. 2002; Park et al. 2002; Reinhart et al. 2002; Kasschau et al. 2003; Kidner and Martienssen 2004). A recent study showed that miR172 causes early flowering and disrupts the specification of floral organ identity when overexpressed in Arabidopsis (Park et al. 2002); it also acts in cell fate specification as a translational repressor of AP2 in Arabidopsis flower development (Chen 2004). MiRNAs appear to regulate target genes by binding to complementary sequences located in the transcripts produced by these genes (Aukerman and Sakai 2003). Aukerman and Sakai (2003) hypothesized that the regulatory target of miR172-like miRNAs is a “subfamily of AP2 genes” in Arabidopsis based on functional studies and the sequence conservation observed between maize (a monocot) and Arabidopsis (a eudicot), and Schmidt et al. (2003) identified that some Arabidopsis genes (AP2, RAP2, 7 [TOE1], TOE2, TOE3, SMZ, and SNZ) have potential target sites for miRNA172.
Vahala, Oxelman, and von Arnold (2001) noted the presence of a conserved region that we show here corresponds to the miR172-binding motif. These workers compared the sequences of nine AP2/ERF-like genes from three eudicot, one monocot, and one gymnosperm species. All these sequences shared three conserved motifs, “B, C, and D”; conserved motif “A” corresponds to the region of the AP2 domain and the linker. Conserved region D, located in the postdomain region, is the same as our “miR172-binding motif.” However, their motifs B and C, located in the predomain region, were only found in some sequences of the euAP2 lineage (fig. 5).
Our analyses of the AP2-like gene family showed that the binding site of miR172 is restricted to members of the euAP2 lineage and is not present in all AP2-like genes (figs. 4 and 5). This result suggests that the type of regulation by miR172 reported in Arabidopsis may be present in the genes throughout the euAP2 lineage. Furthermore, we hypothesize that this type of gene-regulation mechanism by miR172 originated before the divergence of extant angiosperms and gymnosperms because both angiosperm and gymnosperm sequences are present in the euAP2 lineage. Extant seed plants originated approximately 290–309.2 Myr B.P. (Mapes and Rothwell 1984, 1991), and most evidence indicates a very early split between the living gymnosperms and the line leading to angiosperms (reviewed in P. S. Soltis et al. 2002). Therefore, the gene-regulation mechanism by miR172 may predate this age.
Floyd and Bowman (2004) demonstrated that the target sequence of two miRNAs (miR166 and miR166b) known to regulate genes in the class-III homeodomain-leucine zipper (HD-Zip) gene family in Arabidopsis is conserved in homologous sequences from all lineages of land plants, including bryophytes, lycopods, ferns, and seed plants. In addition, the mRNAs from these genes are cleaved in the same miRNA-binding site in representatives of each land plant group as in Arabidopsis. These findings indicate that the regulation of genes in the HD-Zip gene family by these miRNAs dates back more than 400 Myr B.P.
Insertion of 10 aa in the ANT Genes
Previous studies of the three-dimensional protein model of AP2/ERF genes showed three β-sheets and one α-helix structure in the AP2 domain (Allen et al. 1998; Krizek 2003). When we compare aligned sequences with the three-dimensional protein model, most of the conserved residues in the entire AP2/ERF gene (fig. 2) are found in a part of the α-helix region and the first β-sheet region. We confirmed that the 10-aa insertion found in the first domain of ANT genes is placed between the second and third β-sheets in the three-dimensional protein model and this region is found on the surface of the protein in the space-filling model (data not shown). We may expect that residues of this motif in ANT sequences form a longer linker of the two β-sheets than those of euAP2 sequences, and these extruded residues may provide a specific function to the sequences of the ANT lineage.
Domain Evolution
Gene duplication is one of the major evolutionary factors leading to functional diversification and speciation (e.g., Stebbins 1966; Ohno 1970; Levin 1983; Lynch and Conery 2000). Likewise, portions of genes, such as specific exons or domains, may be duplicated within a gene, further complicating the history of gene evolution. Just as we compare gene trees with species trees to coinfer patterns of organismal and genic evolution, we may compare gene trees to “domain trees” to address the pattern of domain evolution.
Two possible models of AP2 domain evolution are shown in figure 8. Model A shows that the duplication of the domain (a and b) preceded the duplication and divergence of two gene lineages (A and B). A phylogenetic tree of the domains based on this model contains a clade of each domain, each with both gene lineages represented (Aa + Ba and Ab + Bb). If the duplication and divergence of the gene lineages preceded independent domain duplications in each lineage, we would expect clades of each gene lineage, each with two domain types (Aa + Ab and Ba + Bb) (Model B). From a parsimony perspective, there are fewer evolutionary steps in Model A than in Model B. Two steps (one domain duplication event and gene lineage splitting) are needed in Model A, whereas three steps (gene lineage splitting and independent duplication of the domain in each gene lineage) are needed in Model B. In addition to Models A and B, we may hypothesize a third possibility involving horizontal domain transfer, which is comparable to the horizontal gene transfer observed in mitochondrial genes in angiosperm evolution (e.g., Bergthorsson et al. 2003, 2004; Won and Renner 2003). This model is more complicated than Models A and B. In this case, one domain cannot form a clade in the phylogenetic tree. Our phylogenetic analyses strongly support Model A (compare figs. 5, 6, and 8) as the likely evolutionary pathway for duplication of the binding domain in the AP2/ERF family.
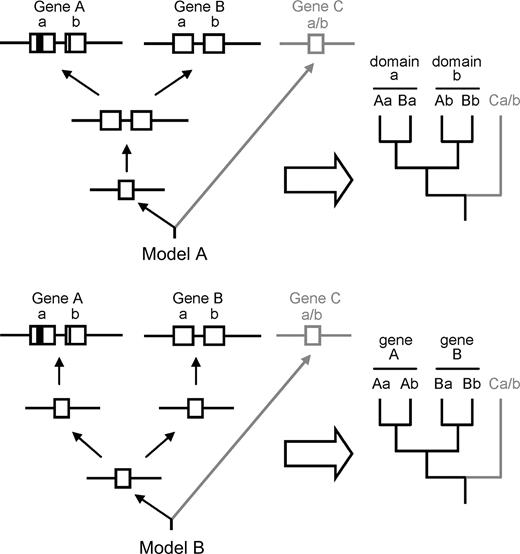
Possible models of domain evolution. Model A: domain duplication preceded the split of gene lineages. Model B: the split of gene lineages preceded independent domain duplications.
To test the possibility of the alternative hypothesis of domain evolution (Model B), we performed a likelihood ratio test using parametric bootstrapping. For each of the three reduced data sets we tested, we rejected the alternative hypothesis (Model B) that gene duplication preceded independent domain duplications (Supplementary material 5 online).
Some sequences included in AP2-like genes in our phylogenetic analyses contain only one AP2 domain. For example, much of the sequence for the AP2-R2 region reported in At2g41710 did not closely match the AP2-R2 domain of other genes. In addition, At2g28550, At2g39250, At3g54990, and At5g60120 contain a large gap in each AP2-R2 domain (Supplementary material 6 online). Because the AP2-R2 domain region of these genes still contains a part of the conserved sequence of the AP2-R2 domain (Supplementary material 6 online) and phylogenetic analyses placed these genes in the AP2 lineage instead of the ERF lineage, the data suggest that the loss of the domain in these genes occurred following the split of the euAP2 and ANT lineages.
Phylogenetic Position of RAV Genes in the AP2/ERF Gene Family
In all of our phylogenetic analyses (fig. 5; Supplementary material 3 online) and in the study of Sakuma et al. (2002), two RAV genes form a clade and are placed between the AP2-like genes and other ERF genes. Although these genes are included in the ERF lineage in figure 5, this is because of our outgroup choice. We set RAV genes and other ERF genes as the outgroup because our focus was on AP2-like genes in our phylogenetic analyses. To address the phylogenetic position of RAV genes in the AP2/ERF gene family, an appropriate outgroup (i.e., rooting point) is needed that is completely outside of these three gene groups. Three scenarios for the evolution of RAV genes (fig. 9C, D, and E) are possible depending on where the root of the tree is placed (fig. 9B). The first scenario (fig. 9C) suggests that the ERF-like classes of genes, when RAV genes are included, are paraphyletic. The second scenario (fig. 9D) proposes a monophyletic origin of ERF-like genes and RAV genes. Following this possibility, it is reasonable to include RAV genes in the ERF-like gene group. The third scenario suggests that RAV genes are sister to the rest of the AP2/ERF gene family (fig. 9E). Recent global phylogenetic analysis of AP2/ERF genes including the fifth group of Arabidopsis genes (Sakuma et al. 2002) and a few sequences from ciliates, bacteria, and viruses (Magnani, Sjölander, and Hake 2004) support the topology of our second scenario (fig. 9D).
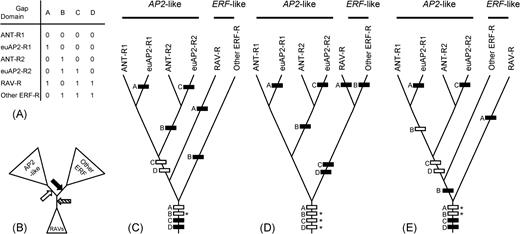
The phylogenetic position of RAV genes and evolution of gaps in domains. (A) Gap coding based on figure 3; “0,” absent; “1,” present. (B) Summarized relationship among AP2-like genes, RAV genes, and other ERF-like genes shown by the unrooted tree: RAV genes are placed between AP2-like genes and other ERF genes. However, rooting at the position of the solid arrow generates tree (C), rooting at the open arrow generates tree (D), and rooting at the hatched arrow generates tree (E). Ancestral character states were reconstructed for trees (C), (D), and (E) by MacClade version 3.04 (W. Maddison and D. Maddison 1992) with the accelerated transformation optimization. Black, “present”; white, “absent”; and white bars with asterisk designate characters with equivocal ancestral state reconstructions. For each gap character with an equivocal ancestral state, two patterns of evolution are possible; we show only the case in which the gap is absent in the ancestor.
Some gaps in the domain regions of AP2/ERF genes appear to have arisen more than once, based on our phylogenetic analyses. The occurrence of some gaps (e.g., gap C in the euAP2 and in ERF lineages) does not agree with the relationships suggested by phylogenetic analyses of domain sequences (figs. 3 and 6). However, when we coded these gaps as characters and analyzed them together with either aa or DNA sequence data, the tree topology was not changed (tree not shown) from those obtained without gap characters. We reconstructed gap evolution by plotting these gaps onto the three possible topologies for the AP2/ERF gene family (fig. 9C and D). Gaps A, B, and C each evolved (gain or loss) at least twice in all evolutionary scenarios, showing the complicated history of gap evolution in the AP2/ERF gene family.
Expression Data and Phylogenetic Relationship
Expression studies of several genes of the AP2/ERF family have shown various patterns in different plant organs. For example, (i) AP2 of Arabidopsis is expressed at the RNA level in all four types of floral organs (sepals, petals, stamens, and carpels) and in developing ovules (Jofuku et al. 1994), (ii) ANT of Arabidopsis is expressed in developing ovules and in the primordia of other floral and vegetative organs (Elliott et al. 1996; Klucher et al. 1996), (iii) four ERF-like genes of Arabidopsis (RAP2.1, RAP2.2, RAP2.3, and RAP2.4) are differentially expressed in flower, leaf, inflorescence, stem, and root tissues (Okamuro et al. 1997), (iv) expression of PhAp2A (Petunia) is similar to AP2 but PhAp2B and PhAp2C (Petunia) showed different patterns during flower development than PhAp2A (Maes et al. 2001), and (v) two euAP2 genes of Picea (PaAP2L1 and PaAP2L2) are differentially expressed in different organs: for example, PaAP2L2 is expressed in roots but PaAP2L1 is not (Vahala, Oxelman, and von Arnold 2001). Direct comparisons of patterns and levels of expression in these examples are difficult because each study used different methods and had a different focus.
An EST sequence of Amborella was embedded in the euAP2 lineage. Two Arabidopsis euAP2 genes, AP2 and At5g67180 (TOE3), appear to be coorthologues of Amborella AP2 in the phylogenetic tree (fig. 5). The Amborella gene and AP2 exhibit similar expression patterns, with expression in all floral organs. Many additional Arabidopsis genes are also included in the euAP2 lineage (e.g., At2g39250 [SNZ], At3g54990 [SMZ], At5g60120 [TOE2], At2g28550 [TOE1], and At5g67180 [TOE3]), but the roles of these genes are different from AP2. Two euAP2 genes of Picea (PaAP2L1 and PaAP2L2) appear in similar positions in the phylogenetic tree (fig. 5) but differ in expression pattern and possible function. Whereas both genes are expressed in leaves, stems, and female cone buds, PaAP2L2 is expressed in the root and male cone buds and PaAP2L1 is not. The correspondence between expression pattern and phylogenetic relatedness of AP2-like genes requires further study. The size of functionally similar groups seems smaller than the size of the major lineages (euAP2, ANT, and euANT) recognized in this study, suggesting functional diversification within lineages. Comprehensive studies of expression data and intensive phylogenetic information for AP2-like genes will give us a clearer indication of the roles these genes play in the developmental processes of different species as well as the function of AP2-like genes in the evolutionary history of angiosperms.
Charles Delwiche, Associate Editor
This work was supported by the FGP (NSF grant PRG-0115684) and a Biocomplexity grant (NSF grant DEB-0083659/0196412). We are grateful to David Lorence of the National Tropical Botanical Garden, Kauai, Hawaii, for material of Amborella. We thank Jack Sullivan, Edward Braun, Christy Edwards, and Matthew Gitzendanner for helpful discussion and implementations of the parametric bootstrap analysis and David Oppenheimer and Steven Manchester for helpful comments and discussion.
References
Albert, V. A., D. E. Soltis, J. E. Carlson et al. (24 co-authors).
Allen, M., K. Yamasaki, M. Ohme-Takagi, M. Tateno, and M. Suzuki.
Altschul, S., W. Gish, W. Miller, E. Myers, and D. Lipman.
Aukerman, M., and H. Sakai.
Bergthorsson, U., K. L. Adams, B. Thomason, and J. D. Palmer.
Bergthorsson, U., A. O. Richardson, G. J. Young, L. R. Goertzen, and J. D. Palmer.
Boutilier, K., R. Offringa, V. Sharma et al. (12 co-authors).
Bowman, J. L., J. Alvarez, D. Weigel, E. M. Meyerowitz, and D. R. Smyth.
Bowman, J. L., G. N. Drews, and E. M. Meyerowitz.
Bowman, J. L., D. R. Smyth, and E. M. Meyerowitz.
Buttner, M., and K. B. Singh.
Chen, X.
Drews, G. N., J. L. Bowman, and E. M. Meyerowitz.
Drinnan, A. N., P. R. Crane, and S. B. Hoot.
Edwards, D., and J. Feehan.
Elliott, R. C., A. S. Betzner, E. Huttner, M. P. Oakes, W. Q. J. Tucker, D. Gerentes, P. Perez, and D. R. Smyth.
Enright, A., S. Van Dongen, and C. Ouzounis.
Felsenstein, J.
Finkelstein, R. R., M. L. Wang, T. J. Lynch, S. Rao, and H. M. Goodman.
Floyd, S. K., and J. L. Bowman.
Fujimoto, S. Y., M. Ohta, A. Usui, H. Shinshi, and M. Ohme-Takagi.
Gifford, E., and A. Foster.
Giraudat, J., B. M. Hauge, C. Valon, J. Smalle, F. Parcy, and H. M. Goodman.
Goff, S. A., D. Ricke, T. H. Lan et al. (55 co-authors).
Huala, E., and I. M. Sussex.
Huelsenbeck, J., and F. Ronquist.
Irish, V. F., and I. M. Sussex.
Ito, T., and E. Meyerowitz.
Jack, T., L. L. Brockman, and E. M. Meyerowitz.
Jofuku, K. D., B. G. W. Denboer, M. Van Montagu, and J. K. Okamuro.
Kagaya, Y., K. Ohmiya, and T. Hattori.
Kasschau, K. D., Z. Xie, E. Allen, C. Llave, E. J. Chapman, K. A. Krizan, and J. C. Carrington.
Kidner, C., and R. Martienssen.
Kim, S., M.-J. Yoo, V. A. Albert, J. S. Farris, P. S. Soltis, and D. E. Soltis.
Klucher, K. M., H. Chow, L. Reiser, and R. L. Fischer.
Komaki, M. K., K. Okada, E. Nishino, and Y. Shimura.
Krizek, B.
Kunst, L., J. E. Klenz, J. Martinezzapater, and G. W. Haughn.
Lagos-Quintana, M., R. Rauhut, W. Lendeckel, and T. Tuschl.
Lang, W.
Lau, N., L. Lim, E. Weinstein, and D. Bartel.
Lee, R., and V. Ambros.
Li, H., and M. Chye.
Llave, C., Z. Xie, K. Kasschau, and J. Carrington.
Lynch, M., and J. Conery.
Maddison, W., and D. Maddison.
Maes, T., M. Van Montagu, and T. Gerats.
Maes, T., N. Van de Steene, J. Zethof, M. Karimi, M. D'Hauw, G. Mares, M. Van Montagu, and T. Gerats.
Magnani, E., K., Sjölander, and S. Hake.
Mandel, M. A., J. L. Bowman, S. A. Kempin, H. Ma, E. M. Meyerowitz, and M. F. Yanofsky.
Mapes, G., and G. W. Rothwell.
———.
Mathews, S., and M. J. Donoghue.
Moose, S. P., and P. H. Sisco.
Mourelatos, Z., J. Dostie, S. Paushkin, A. Sharma, B. Charroux, L. Abel, J. Rappsilber, M. Mann, and G. Dreyfuss.
Ohmetakagi, M., and H. Shinshi.
Okamuro, J. K., B. Caster, R. Villarroel, M. VanMontagu, and K. Jofuku.
Park, W., J. Li, R. Song, J. Messing, and X. Chen.
Posada, D., and K. Crandall.
Rambaut, A, and N. C. Grassly.
Reinhart, B., E. Weinstein, M. Rhoades, B. Bartel, and D. Bartel.
Riechmann, J. L., and E. Meyerowitz.
Riechmann, J. L., J. Heard, G. Martin et al. (17 co-authors).
Sakuma, Y., Q. Liu, J. G. Dubouzet, H. Abe, K. Shinozaki, and K. Yamaguchi-Shinozaki.
Schmid, M., N. H. Uhlenhaut, F. Godard, M. Demar, R. Bressan, D. Weigel, and J. U. Lohmann.
Schultz, E. A., and G. W. Haughn.
Shannon, S., and D. R. Meekswagner.
Shigyo, M., and M. Ito.
Soltis, D. E., P. S. Soltis, V. A. Albert, D. G. Oppenheimer, C. W. dePamphilis, H. Ma, M. W. Frohlich, and G. Theissen.
Soltis, P. S., D. E. Soltis, and M. W. Chase.
Soltis, P. S., D. E. Soltis, V. Savolainen, P. R. Crane, and T. G. Barraclough.
Stebbins, G. L.
Stockinger, E. J., S. J. Gilmour, and M. F. Thomashow.
Swofford, D.
Thompson, J., T. Gibson, F. Plewniak, F. Jeanmougin, and D. Higgins.
Vahala, T., B. Oxelman, and S. von Arnold.
Wessler, S. R.
Wilson, K., D. Long, J. Swinburne, and G. Coupland.
Won, H., and S. Renner.
Wuitschick, J. D., P. R. Lindstrom, A. E. Meyer, and K. M. Karrer.
Xue, G. P., and C. W. Loveridge.
Author notes
*Department of Botany, University of Florida, Gainesville; †Florida Museum of Natural History, University of Florida, Gainesville; and ‡Department of Biology, Pennsylvania State University

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}