





Abstract
Biomedical literature is traditionally used as a way to inform scientists of the relevance of genes in relation to a research topic. However many genes, especially from poorly studied organisms, are not discussed in the literature. Moreover, a manual and comprehensive summarization of the literature attached to the genes of an organism is in general impossible due to the high number of genes and abstracts involved. We introduce the novel Génie algorithm that overcomes these problems by evaluating the literature attached to all genes in a genome and to their orthologs according to a selected topic. Génie showed high precision (up to 100%) and the best performance in comparison to other algorithms in most of the benchmarks, especially when high sensitivity was required. Moreover, the prioritization of zebrafish genes involved in heart development, using human and mouse orthologs, showed high enrichment in differentially expressed genes from microarray experiments. The Génie web server supports hundreds of species, millions of genes and offers novel functionalities. Common run times below a minute, even when analyzing the human genome with hundreds of thousands of literature records, allows the use of Génie in routine lab work. Availability: http://cbdm.mdc-berlin.de/tools/genie/ .
INTRODUCTION
Complete genome sequences give an overview of all the genes of an organism. Such collections allow researchers to screen for genes associated with particular properties, which can then be further used to design new experiments or to prioritize analysis ( 1 ). Classically, the literature dealing with genes, as stored in the MEDLINE database of biomedical references ( 2 ), has been used to do this prioritization ( 3 ). Although many genes whose sequences are obtained from complete genomes have never been experimentally characterized and accordingly have no related literature, especially when they are from poorly studied organisms, the literature from equivalent (orthologous) genes in related organisms is usually taken into consideration under the assumption that proteins bearing high sequence similarity also share similar functions ( 4 ). However, the large number of genes per organism and the increasing number of publications with associated genes makes it difficult to find the required information without computational assistance. This prompted the development of computational methods to assist researchers in evaluating gene function based on analysis of the literature ( 5 , 6 ). However, to date, there is no method that either ranks the complete set of genes of any given organism according to a particular gene function or takes advantage of all available orthology information to expand the related MEDLINE literature. Such a method should be fast and flexible despite working with a large amount of data, so that a user can try different queries to get the desired information.
With these objectives in mind, we developed the Génie algorithm and web server. Génie takes a biological topic as input (ideally related to a gene function), evaluates the entire MEDLINE for relevance to that subject, and then evaluates all the genes of a user’s requested organism according to the relevance of their associated MEDLINE records. Importantly, one can evaluate the genes of the desired organism using information from their orthologous counterparts, which could enhance the results for poorly studied organisms. We evaluated the performance of the algorithm in the identification of genes known to be involved in molecular pathways and diseases, and assessed its effectiveness in finding novel associations between genes and functions by comparison with experimental measurements of gene expression.
RESULTS
Algorithm and web server
The novel Génie algorithm was developed to prioritize all of the genes from a species according to their relation to a biomedical topic using all available scientific abstracts and orthology information. Génie takes advantage of literature, gene and homology information from the MEDLINE, NCBI Gene and HomoloGene databases ( 2 ) ( Figure 1 ).
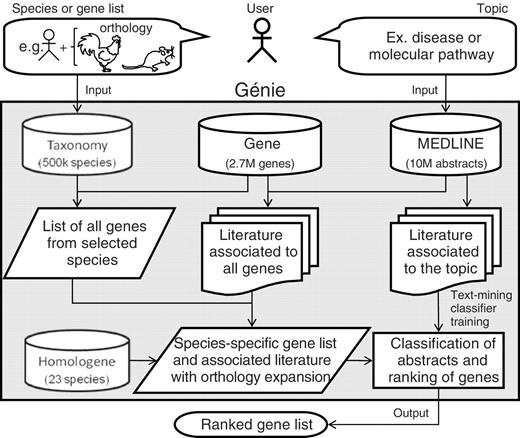
Flow chart of the Génie web tool and algorithm. As an example, a user could query human genes related to a disease or a molecular pathway using chicken and rat orthologs. Usage of orthology information is optional. Data are extracted from four NCBI databases: Taxonomy, Gene, MEDLINE and HomoloGene. As the retrieved literature associated to the topic may not be complete, it is used to train a text mining classifier that will select relevant gene literature. The output gene list (human genes in the given example) is ranked using Fisher’s statistics.
The system needs two basic inputs: a target species (e.g. Homo sapiens ) and a biomedical topic ideally related to a gene function (e.g. ‘Cardiovascular diseases’) according to which the genes of the target species have to be prioritized. The target species is most often defined by its scientific name or its taxonomic ID [in the NCBI taxonomy database ( 2 )], though an arbitrary list of Entrez Gene IDs can be used instead. The biomedical topic is ultimately defined by a set of biomedical references represented by MEDLINE records. In Génie’s current implementation, such a set can be defined directly by providing a list of PubMed identifiers (PMIDs), or indirectly via either a typical text query to PubMed or a set of Medical Subject Heading terms from the MeSH database ( 2 ). Notably, the query to PubMed handles free text and synonym resolution. Importantly, the most discriminative words for classification will be automatically defined by the algorithm. An optional, but powerful, third input is a list of species (e.g. Mus musculus and Danio rerio ), which is used to search for literature that is associated to the genes of the target species (e.g. Homo sapiens ) following orthology relationships. A wizard assistant can help in the selection of the most appropriate species by showing the total number of relevant abstracts for each species.
The first step in the algorithm is the retrieval of a sample set of MEDLINE abstracts (limited to 1000 abstracts) that are representative of the topic being studied, which is used to train a naïve linear Bayesian classifier. The training consists of automatically building a statistical model, which is a weighted list of discriminative words in the selected set of records in comparison to the rest of MEDLINE ( 7 ) (see Supplementary Methods ). Then, all of the abstracts associated to the genes from the target species in the Gene database (including manual links created by NCBIs curators using full-text information) are evaluated by the Bayesian classifier and assigned a P -value representing the confidence of the classification. If requested as an option, this set of bibliography can be extended with abstracts associated with orthologous genes from some or all available species as defined in HomoloGene. In this case, the genes of the target species are ranked using, in addition to the abstracts directly associated to them, the abstracts associated to their orthologs in other species. Finally, given a cutoff for abstract selection ( P < 0.01 by default) a one-sided Fisher's exact test is computed to define the significance of gene-to-topic relationship, comparing the number of selected abstracts to what is observed in a simulation using a set of ten thousand randomly selected abstracts. The genes are then presented in a list sorted by false discovery rate (FDR) with hyperlinks to the most significant abstracts, to Entrez Gene and to HomoloGene databases. A list of the words that were detected as relevant to the topic is provided to facilitate the interpretation of the results.
Ranking human and model organism genes
Currently, over half a million genes are associated to more than one abstract in the Gene database and a total of 4418 eukaryotic and prokaryotic species are available in Génie.
We tested Génie’s ability to provide an overview of an organism’s genes without using orthology information for five different topics and species ( Arabidopsis thaliana , Drosophila melanogaster , Homo sapiens , Mus musculus and Saccharomyces cerevisiae ). Following manual validations of the top 50 predicted genes ( Table 1 , Supplementary Tables S1–S5 and Supplementary Methods ), observed precision ranged from 92% ( Drosophila genes ranked for planar cell polarity) to 100% ( Arabidopsis genes ranked for host–pathogen interactions).
Manual evaluation of the top genes for five species and five topics
| Species | Topics | Evaluated genes | True positives | Precision (%) |
|---|---|---|---|---|
| Arabidopsis thaliana | Host–pathogen interactions | 50 | 50 | 100 |
| Saccharomyces cerevisiae | Cell cycle | 50 | 49 | 98 |
| Mus musculus | Pain measurement and knockout mice | 50 | 48 | 96 |
| Homo sapiens | Alzheimer's disease | 50 | 47 | 94 |
| Drosophila melanogaster | Planar cell polarity | 24 | 22 | 92 |
| Species | Topics | Evaluated genes | True positives | Precision (%) |
|---|---|---|---|---|
| Arabidopsis thaliana | Host–pathogen interactions | 50 | 50 | 100 |
| Saccharomyces cerevisiae | Cell cycle | 50 | 49 | 98 |
| Mus musculus | Pain measurement and knockout mice | 50 | 48 | 96 |
| Homo sapiens | Alzheimer's disease | 50 | 47 | 94 |
| Drosophila melanogaster | Planar cell polarity | 24 | 22 | 92 |
Manual evaluation of the top genes for five species and five topics
| Species | Topics | Evaluated genes | True positives | Precision (%) |
|---|---|---|---|---|
| Arabidopsis thaliana | Host–pathogen interactions | 50 | 50 | 100 |
| Saccharomyces cerevisiae | Cell cycle | 50 | 49 | 98 |
| Mus musculus | Pain measurement and knockout mice | 50 | 48 | 96 |
| Homo sapiens | Alzheimer's disease | 50 | 47 | 94 |
| Drosophila melanogaster | Planar cell polarity | 24 | 22 | 92 |
| Species | Topics | Evaluated genes | True positives | Precision (%) |
|---|---|---|---|---|
| Arabidopsis thaliana | Host–pathogen interactions | 50 | 50 | 100 |
| Saccharomyces cerevisiae | Cell cycle | 50 | 49 | 98 |
| Mus musculus | Pain measurement and knockout mice | 50 | 48 | 96 |
| Homo sapiens | Alzheimer's disease | 50 | 47 | 94 |
| Drosophila melanogaster | Planar cell polarity | 24 | 22 | 92 |
Noteworthy, human genes were ranked for Alzheimer’s disease with 94% precision, and among the three false positives, two genes were related to neurodegenerative diseases ( HTT and IAPP ), and one to macular degeneration ( ABCA4 ) ( Supplementary Table S4 ). Moreover, the Génie’s top 50 Arabidopsis genes were all true positives though the overlap with the existing Arabidopsis -related KEGG ( 8 ) plant–pathogen interaction pathway (138 genes in total) was only 16 genes (one-sided Fisher’s exact test: P < 2.2e-16) ( Supplementary Table S1 ). For example, NPR1 was selected by Génie but missing from the KEGG plant–pathogen interaction pathway. NPR1 is a known inducer of defense genes, and interacts with TGA2 and TGA3 ( 9 ), which bind the salicylic acid responsive element of the ‘pathogenesis-related’ (PR)-1 gene promoter. Therefore, NPR1 could be linked to the ‘defense-related gene induction’ in the KEGG plant–pathogen interaction pathway.
Zebrafish as a model for heart development
Zebrafish ( Danio rerio ) is rapidly gaining importance as a model organism to study cardiovascular development and disease ( 10 ); however, there are still comparatively few publications characterizing genes from this species. To demonstrate the usefulness of orthology expansion of relevant bibliography by our method, we evaluated zebrafish genes for their role in heart development and function using the literature directly associated with zebrafish genes as well as the literature associated with orthologous genes in human and mouse. The ranked gene lists were compared to differentially expressed genes in a microarray data set comparing whole heart of 3-day-old zebrafish embryos with rest of the embryo ( 11 ), under the assumption that these genes will include many with a role in heart development (see Supplementary Methods ).
The training data set for the topic of heart development was defined by an expert query to PubMed (see Supplementary Methods ) and resulted in 1408 abstracts. When the zebrafish genes were ranked using only the abstracts directly associated to them, only 55 genes were selected (FDR < 5%). In contrast, when extending the literature from zebrafish to human and mouse, via orthology relationships, a total of 1247 genes were returned (FDR < 5%). The latter gene list contained all the 55 genes from the previous ranking.
A comparison of the Génie confidence score, defined here as −log 10 (FDR), versus the gene-expression log 2 -fold change of overexpressed genes shows that the majority of genes that have very high changes in expression are also highly scored by Génie ( Figure 2 a and Supplementary Table S6 ). For instance, out of 14 genes with a log 2 -fold change greater than six (above the dashed horizontal line), only two were not scored by Génie. We also evaluated the precision of Génie in predicting differentially expressed genes ( Figure 2 b). Precision increased with Génie’s rank cutoff, showing that top ranked genes were enriched in differentially expressed genes. Results were better when considering both over and underexpressed genes (blue line), and overexpressed genes (red line) were better predicted than underexpressed genes (green line).
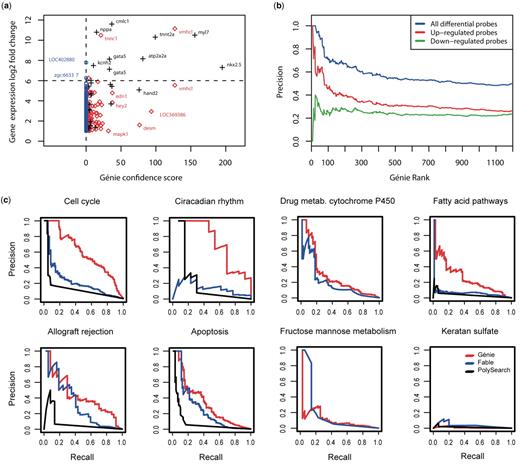
Benchmarks. ( a ) Génie confidence scores versus log 2 -fold expression changes for all up-regulated probes (at least 2-fold expression change) in a zebrafish microarray data set between hearts from 3-day-old zebrafish embryos and whole body tissue. All probes with a positive confidence score were selected by Génie using orthology to zebrafish, mice and humans (red diamonds and black crosses). Probes also selected by Génie using only zebrafish-related abstracts are plotted with black crosses. Genes not selected by Génie have a score equal to zero (blue circles). The scores and gene expression fold changes for each gene are available as Supplementary Table S6 . ( b ) Precision when predicting differentially expressed genes using gene ranks given by Génie. From the zebrafish microarray data analysis, differentially expressed genes are selected by a FDR < 0.01 and a minimum 2-fold expression change between heart and body samples. ( c ) These precision–recall plots show the performance of Génie (red curves), Fable (blue curves) and PolySearch (black curves) when ranking genes from eight randomly chosen KEGG pathways. The three tools were used with default parameters. PolySearch returned no results for two pathways: drug metabolism cytochrome P450 and fructose mannose metabolism (see Supplementary Methods ). Génie was run without using orthology expansion of the literature.
This comparison shows that the integration of Génie output with other types of gene information can be used as a powerful discovery tool. For instance, highly differentially expressed zebrafish genes such as nkx2.5 and vmhcl have very high Génie confidence score. The mouse and human orthologs of nkx2.5 are at the base of an ancestral cardiac specification program and hence belong to the best studied genes in heart development and disease ( 12 ). Zebrafish nkx2.5 is similarly involved in cardiac morphogenesis ( 13 , 14 ). Likewise, the human ortholog of vmhcl , named MYH6 , is associated with cardiomyopathy ( 15 ), and similar findings have been obtained from studies on murine Myh6 ( 16 ). These two genes could thus be considered as functional models of human cardiac genes.
Comparison to other methods
We have compared Génie against two text-mining tools, Fable ( 17 ) and PolySearch ( 18 ), both of which fulfill the following minimal requirements: the tools are regularly updated and publicly accessible, they use abstracts from PubMed or MEDLINE, and they do not limit queries to specific biomedical domain or controlled vocabulary ( Table 2 ).
Features comparison
| Feature | Génie | Fable | PolySearch |
|---|---|---|---|
| Abstracts database | Medline | Medline + ‘Publisher Status’ from PubMed | PubMed |
| Updates | Weekly | Several times a year | Direct access (web services) |
| Concepts to query | Unlimited | Unlimited | Unlimited |
| Query input | Text, PMIDs or MeSH terms | Keywords | Keywords |
| Running time a | 2–25 s | 2–10 s | 1 min to hours |
| Species | 4418 | 1 | 1 |
| Orthology species extension | Yes, 23 species | No | No |
| Gene-concept association method | Naïve Bayesian classifier | cooccurrence statistics | co-occurrence statistics |
| Gene ranking method | Fisher's exact test | Frequency | Z -score |
| Gene name extraction | Manual | Trained probabilistic model | Dictionary based |
| Synonymous resolution | Manual | Dictionary based | Dictionary based |
| Gene names ambiguity problem | no | Yes | Yes |
| Feature | Génie | Fable | PolySearch |
|---|---|---|---|
| Abstracts database | Medline | Medline + ‘Publisher Status’ from PubMed | PubMed |
| Updates | Weekly | Several times a year | Direct access (web services) |
| Concepts to query | Unlimited | Unlimited | Unlimited |
| Query input | Text, PMIDs or MeSH terms | Keywords | Keywords |
| Running time a | 2–25 s | 2–10 s | 1 min to hours |
| Species | 4418 | 1 | 1 |
| Orthology species extension | Yes, 23 species | No | No |
| Gene-concept association method | Naïve Bayesian classifier | cooccurrence statistics | co-occurrence statistics |
| Gene ranking method | Fisher's exact test | Frequency | Z -score |
| Gene name extraction | Manual | Trained probabilistic model | Dictionary based |
| Synonymous resolution | Manual | Dictionary based | Dictionary based |
| Gene names ambiguity problem | no | Yes | Yes |
a The run-time depends strongly on the parameters for Génie (here queries without orthology information) and PolySearch.
Features comparison
| Feature | Génie | Fable | PolySearch |
|---|---|---|---|
| Abstracts database | Medline | Medline + ‘Publisher Status’ from PubMed | PubMed |
| Updates | Weekly | Several times a year | Direct access (web services) |
| Concepts to query | Unlimited | Unlimited | Unlimited |
| Query input | Text, PMIDs or MeSH terms | Keywords | Keywords |
| Running time a | 2–25 s | 2–10 s | 1 min to hours |
| Species | 4418 | 1 | 1 |
| Orthology species extension | Yes, 23 species | No | No |
| Gene-concept association method | Naïve Bayesian classifier | cooccurrence statistics | co-occurrence statistics |
| Gene ranking method | Fisher's exact test | Frequency | Z -score |
| Gene name extraction | Manual | Trained probabilistic model | Dictionary based |
| Synonymous resolution | Manual | Dictionary based | Dictionary based |
| Gene names ambiguity problem | no | Yes | Yes |
| Feature | Génie | Fable | PolySearch |
|---|---|---|---|
| Abstracts database | Medline | Medline + ‘Publisher Status’ from PubMed | PubMed |
| Updates | Weekly | Several times a year | Direct access (web services) |
| Concepts to query | Unlimited | Unlimited | Unlimited |
| Query input | Text, PMIDs or MeSH terms | Keywords | Keywords |
| Running time a | 2–25 s | 2–10 s | 1 min to hours |
| Species | 4418 | 1 | 1 |
| Orthology species extension | Yes, 23 species | No | No |
| Gene-concept association method | Naïve Bayesian classifier | cooccurrence statistics | co-occurrence statistics |
| Gene ranking method | Fisher's exact test | Frequency | Z -score |
| Gene name extraction | Manual | Trained probabilistic model | Dictionary based |
| Synonymous resolution | Manual | Dictionary based | Dictionary based |
| Gene names ambiguity problem | no | Yes | Yes |
a The run-time depends strongly on the parameters for Génie (here queries without orthology information) and PolySearch.
Génie incorporates comprehensive orthology information and can be used on hundreds of species while Fable and PolySearch are limited to human genes. In contrast to the two other tools, Génie uses a naïve linear Bayesian classifier to select abstracts rather than simpler term co-occurrence statistics. Moreover, Génie avoids gene name ambiguity as it relies on curated links between genes and abstracts.
The performance of Génie (without orthology expansion of the literature), Fable and PolySearch were compared on eight randomly chosen human molecular pathways ( Figure 2 c) from the KEGG PATHWAY database ( 8 ), which contains a description of reference genes of molecular pathways. Each tool was asked to rank human genes with default parameters, and pathway names were used as input (see Supplementary Methods ). Génie showed the best performance in four (50%) pathways, where its precision–recall curves were above the others in the whole range (Cell cycle, Circadian rhythm, Drug metabolism cytochrome p450 and Fatty acid pathways). For Allograft rejection and Apoptosis pathways, Génie and Fable results were comparable to each other in the low sensitivity range (sensitivity < 0.41 and <0.25, respectively), but Génie’s precision was the best for higher sensitivity. Including the Fructose Mannose metabolism pathway, Génie performed better than the other tools for high sensitivity ranges in seven out of eight (87.5%) pathways.
DISCUSSION
We have created the novel Génie algorithm that ranks the genes from hundreds of genomes for different topics while taking advantage of resources provided by NCBI. The Génie web server is free and open to all users and it is not necessary to create an account before using the service. Its features are unique and more extensive than comparable resources ( Table 2 ), gene lists related to various species and functions are ranked with high precision ( Table 1 ) and it performs better than other tools in most benchmarks ( Figure 2 c).
The benefits of using orthology analysis to find human genes associated to disease using the phenotypes associated to their murine orthologs has already been shown by several data mining tools, e.g. ToppGene ( 19 ), GenSeeker ( 20 ) and G2D ( 21 ). The Génie orthology-based method applies this idea using all species in Homologene to increase the bibliography between a target organism and a set of other organisms, such as zebrafish, human and mouse. Moreover, the use of orthology information is helpful when ranking poorly studied genes. We evaluated this feature by comparing Génie’s results with gene-expression microarray data to show the ability of Génie to predict functional heart-related genes in zebrafish ( Figure 2 a and b). Such an analysis can reveal multiple types of information. First, the correlations found can tell us which zebrafish genes are homologous to human and mouse genes with known cardiac functions, and may be considered to generate models for human disease. Second, genes with high differential expression, whose human ortholog has no associated bibliography, could potentially indicate human genes with undiscovered functions in cardiac development and disease ( 10 ). Thus, besides selecting well-known genes, Génie also highlights new candidate genes lacking relevant bibliography for the selected topic, and may help in the characterization of the system under study.
We have also found informative mismatches between a gene’s microarray expression and Génie ranking ( Supplementary Table S6 ) due to various biological causes. For instance, cx43 , which encodes the ortholog of human heart malformation associated GJA1 protein ( 22 ) is highly ranked by Génie (FDR = 5.43e-105) but shows greatly reduced expression in the cardiac expression profile (log 2 -fold change = −4.33), consistent with its expression in only a defined subset of cardiac tissue ( 23 ). Expression of the shha gene is similarly correlated to its Génie FDR (log 2 -fold change = −3.22, and Génie FDR = 9e-12). Studies of the mouse ortholog Shh have identified it as a factor involved in heart development; however, it was shown to originate from an extra-cardiac source, thus explaining this discrepancy ( 24 , 25 ). Phylogenic differences between zebrafish and other vertebrate species become obvious in the case of isl1 . Isl1 in higher vertebrates is essential in forming the second heart field, which is absent in zebrafish ( 26 ). Expression of isl1 is, therefore, absent in the zebrafish data, despite a strong cardiac association via orthologs by Génie (log 2 -fold change: −4.91; Génie FDR = 1.23e-28).
Many methods for prioritizing genes using literature data are based on co-occurrence analysis of given keywords and automatically extracted gene names from scientific abstracts [e.g. Fable ( 17 ), PolySearch ( 18 ), Facta ( 27 )]. The main hypothesis of this type of analysis is that the more often two words co-occur in abstracts the more likely they are to be functionally linked. However, automatic gene name extraction and normalization methods wrongly identify a significant portion of gene mentions in text ( 28 , 29 ), and consequently bring noise and ambiguity into the text-mining results. Génie avoids this problem by relying on NCBIs curated associations between MEDLINE records and unambiguous gene identifiers. A possible drawback is that some abstracts may not yet be associated to genes. However, for our system, the benefit of having accurate associations outweighs the cost of missing some associations. The current subset of MEDLINE associated to genes (547 168 distinct MEDLINE records) seems to properly reflect all experimental knowledge on gene function. Via orthology relationships, any of these records, can potentially be used to rank all genes of hundreds of species.
An additional advantage of NCBIs annotations, some of which are manually assigned, is that abstracts can be associated to genes based on full-text evidence. For instance, the human gene presenilin 1 has been related to Alzheimer’s disease in the literature ( 30 ) and is mentioned in the full text of the corresponding manuscript by its synonym PS1 , but in the abstract we can only find a mention to the presenilin complex, which could refer to either of PS1 or presenilin 2 ( PS2 ). Accurately, the manuscript is associated to PS1 and not to PS2 , although PS2 is mentioned twice in the full text, because the manuscript describes experiments that involve research on PS1 and not on PS2 . A more extreme example is the association of the human dermatopontin gene ( DPT ) to a genetic study of longevity and age-related phenotypes ( 31 ): its name or synonyms are mentioned in neither the abstract nor in the full text of the corresponding manuscript, but DPT is listed in a table alongside P -values for significant association to morbidity-free survival at age 65 years. These examples illustrate that automated methods to associate text to genes cannot reach the sophisticated level of reasoning of a human curator.
CONCLUSION
Génie is a powerful and fast tool that prioritizes the whole gene set of hundreds of species for any biomedical topic, taking advantage of annotations-linking genes and bibliography. By using orthology relationships, Génie can transfer annotations between 23 species. Those annotations, including those that are manually assigned, may not be complete but we believe that the increase in the number of available genomes and the use of orthology produces a synergistic effect resulting in a coverage of genes and topics that compensates for the shortcomings inherent to manual curation. The bibliography increases continuously as new experimental and genetic data is generated by the scientific community. At the same time, the new sequences deposited in the databases have an increased tendency to be similar to already existing sequences ( 32 ), and therefore to be covered by bibliography of some ortholog. Both factors suggest that the results of Génie, which were shown to be already very precise, will continually improve over time.
FUNDING
This project is funded within the framework of the Medical Genome Research Programme NGFN-Plus by the German Ministry of Education and Research (BMBF) with the reference number 01GS08170, and by the Helmholtz Alliance in Systems Biology (Germany). Funding for open access charge: German Medical Genome Research Programme NGFN.
Conflict of interest statement . None declared.
ACKNOWLEDGEMENT
The authors thank Matthew Huska for critical reading of the text.

{kind=link}
{kind=link}
Comments