





Abstract
Cancer stem cells (CSCs) have been identified in several solid malignancies and are now emerging as a plausible target for drug discovery. Beside the questionable existence of CSCs specific markers, the expression of CD133 was reported to be responsible for conferring CSC aggressiveness. Here, we identified two G-rich sequences localized within the introns 3 and 7 of the CD133 gene able to form G-quadruplex (G4) structures, bound and stabilized by small molecules. We further showed that treatment of patient-derived colon CSCs with G4-interacting agents triggers alternative splicing that dramatically impairs the expression of CD133. Interestingly, this is strongly associated with a loss of CSC properties, including self-renewing, motility, tumor initiation and metastases dissemination. Notably, the effects of G4 stabilization on some of these CSC properties are uncoupled from DNA damage response and are fully recapitulated by the selective interference of the CD133 expression.
In conclusion, we provided the first proof of the existence of G4 structures within the CD133 gene that can be pharmacologically targeted to impair CSC aggressiveness. This discloses a class of potential antitumoral agents capable of targeting the CSC subpopulation within the tumoral bulk.
INTRODUCTION
Colorectal cancer (CRC) is one of the main causes of tumor-related mortality worldwide. Although surgical resection represents the elective therapeutic approach for the treatment of the tumor at its early stage, more than 50% of patients develop a metastatic disease, which is incurable with the available therapies (1). Therefore, the development of innovative therapeutic strategies is of crucial importance. In the last few years, growing evidence supported the hypothesis of the existence, within primary tumors, of cell subpopulations, referred as cancer stem cells (CSCs), characterized by self-renewal capacity and resistance to ‘conventional’ anticancer therapies. Although CSCs represent a small fraction of the overall tumor, these cells possess the potential to initiate and sustain tumor growth and metastasis (2). Identification of CSCs is mainly based on the surface marker expression, including CD133, CD166 and CD44 (3,4). Interestingly, CD133 is associated with tumorigenicity and progression of the disease, since its up-regulation in CRC strongly correlates with poor prognosis and metastatic dissemination (5–7). CD44 is a membrane adhesion molecule that binds hyaluronic acid and contributes to cell–cell and cell–matrix adhesion, growth and tumor progression (8). Finally, CD166 belongs to the immunoglobulin superfamily of adhesion molecules involved in cell migration and growth (9), and its expression has been correlated with bad prognosis and shortened survival (10). Altogether, these studies strongly indicate that the cell-surface phenotype of CSC can be mechanistically linked with metastatic process, suggesting that the surface molecules could be clinically exploited both as a biomarker and therapeutic targets.
G-quadruplexes (G4s) are non-canonical DNA secondary structures widely described at the chromosome ends (11,12) and in the promoters of a wide range of genes important in cell signaling, recognized as hallmarks of cancer (13–16). Interest in the more general therapeutic significance of G4s is expanding to include G4 structures in 5′-UTR regions and introns. Indeed, G4 sequences localized in promoter region (within 1000 nt from the starting codon) or at the 5′-UTR, can affect the expression of the target gene by regulating the transcriptional or the translational processes, respectively (17,18). On the other hand, G4s formed in the introns (if localized nearby to the splice acceptor sites of the adjacent exons) can affect the activity of the splicing machinery causing intron retention events (19,20) that could be lastly responsible for the synthesis of immature RNA transcripts.
Over the past two decades, several G4-interacting small molecules, designed to act as G4 stabilizers, have been developed with the aim of identifying potential anticancer therapeutics (21,22). Notably, many G4s have physicochemical properties and structural characteristics that make them druggable, and their structural diversity suggests that a high degree of selectivity might be possible (23). More interestingly, G4 DNA structures have been detected in human cells, corroborating the application of stabilizing ligands as a new class of anticancer agents (24,25). In this area, our group and others have made important contributions in identifying new G4-stabilizing agents. The profiling of the biological properties of several G4-binders demonstrated that they are outstandingly potent in inducing selective DNA-damage at telomeres of cancer versus normal cells (21,26,27). In addition, these drugs can also affect the expression of a number of cancer related genes (15–17,28,29), showing a potent multimodal antitumoral activity, either alone or in combination with antineoplastic drugs (21,26,30–33).
Here, we provide evidence that G4 ligands can dramatically impair the tumor-promoting activity of colon CSCs through triggering an alternative splicing of the CD133 mRNA, highlighting how G4 stabilization represents a valuable therapeutic option against CSCs.
MATERIALS AND METHODS
Cell culture, treatments and transfections
Patient derived colon CSC-enriched cell lines (CSC_1 and CSC_2) were obtained as previously described (34).
CSC-LUC, shCD133_1 and shCD133_2 cells were generated by transfecting the cells with the JetPEI® (Polyplus-transfection, Illkirch, France), according to the manufacturer's instructions. The following plasmids were used: PGL2 vector containing the firefly luciferase gene under the control of the SV promoter (Promega Madison, WI, USA), MISSION® CD133 shRNAs TRCN0000424799 and TRCN0000416340 (Sigma-Aldrich, St. Louis, MO, USA).
Cells were grown in ultra-low attachment plates (Corning, Lowell, MA, USA) and maintained in stemness medium (DMEM-F12, Euroclone, Milan, Italy) supplemented as reported in (34). When indicated, cells were plated under adherent condition in complete medium: DMEM (Euroclone) supplemented with 10% FCS (HyClone, Logan, UT, USA).
RHPS4, synthesized as already reported (35), was used at 1 μM concentration for different times. ATM-inhibitor KU-55933 (Sigma-Aldrich) was used at 5 μM for 96 h.
Immunofluorescence (IF)
For FACS analysis, untreated or treated cells were incubated with the following antibodies: anti human CD133 (mouse mAb AC133-Pur, Miltenyi Biotec, Bisley, UK), anti human CD44 (mouse mAb, BD Biosciences, San Diego, CA, USA) and anti human CD166 (mouse mAb, BD Biosciences); stained with FITC-conjugated Goat anti Mouse antibody (Jackson Immunoresearch, Suffolk, UK) and analyzed by flow cytometry using FACScalibur (BD Immunocytometry System-BDIS, San Jose, CA, US).
For microscopy analysis, untreated or treated cells were grown in complete medium and the IF analysis was performed (14), using the anti-CD133 antibody (AC133-PE, Miltenyi). Stained cells were analyzed with a Leica DMIRE2 microscope equipped with a Leica DFC 350FX camera and elaborated by a Leica FW4000 deconvolution software (Leica, Solms, Germany). For quantitative analysis of CD133 expression, 200 cells on triplicate slices were analyzed, and scored on the basis of the CD133 staining. In detail, the cells with isolated CD133 spots were considered CD133low, while cells with a marked and continuous staining were considered CD133high.
For interphase nuclei telomere-induced foci (TIFs) analysis, cells were fixed in 2% formaldehyde and permeabilized in 0.25% Triton X100 in phosphate buffered saline (PBS) for 5 min at room temperature (RT). For immunolabeling, cells were incubated with primary antibody (RT, 2 h), washed twice in PBS, and finally incubated with the secondary antibodies (RT, 1 h). The following primary antibodies were used: rabbit polyclonal anti-TRF1 antibody (Abcam Ltd., Cambridge, U.K.); mouse monoclonal anti-γH2AX antibody (Upstate, Lake Placid, NY, USA). The following secondary antibodies were used: TRITC-conjugated Goat anti Rabbit and FITC-conjugated Goat anti Mouse (Jackson Immunoresearch). Nuclei were counterstained with DAPI (Sigma-Aldrich).
Western Blotting
Western blot and detection were performed as previously reported (36). The activation state of DNA damage response proteins was analyzed by using the following antidodies: rabbit mAb anti-Ser1981 p-ATM (Abcam Ltd.); mouse mAb anti-p53 DO-1 and rabbit pAb anti-Thr68 p-CHK2 (Cell Signaling, Beverly, MA, USA). As loading control, levels of β-ACTIN were evaluated by using the mouse mAb anti-β-actin (Sigma-Aldrich).
RT–PCR
RNA, extracted with Trizol reagent (Invitrogen, Carlsbad, CA, USA), was converted to cDNA with the SuperScript ®VILOTM cDNA synthesis kit (Invitrogen). Evaluation of CD133 expression and analysis of the splice variants were performed using the primers reported in Figure 3G. Densitometry was performed with ImageJ software version 1.40.
Polymerase stop assay
Polymerase Stop Assay was performed as already reported (14). Of note, the 77-mer DNA templates were generated starting from a scaffold sequence 5′-CTG GAG ATC CCC GCC GGG TAC CCG GGT GAG - insert -TAT AGT GAG TCG TAT TA-3′, where the insert corresponds to the putative G4 sequences reported in Figure 2A.
Biophysical analyses
The sequences d(TGGGAAGGATGTGGGAGGAGAGGGCAGCGGGA) (intron 3(b)) and d(TGGGGATTGCCGGGCAGGGCTAAGCGTGGGT) (intron 7) were synthesized as already reported (37). Oligonucleotide samples were prepared by dissolving the lyophilized compound in potassium phosphate buffer (70 mM KCl, 10 mM KH2PO4, 0.2 mM EDTA, pH 7.0). The solutions were heated at 90°C for 5 min and slowly cooled to room temperature. The annealing procedure was followed by overnight storage at 4°C prior to use. The concentration of the oligonucleotides was evaluated by UV measurements at 260 nm, at a temperature of 90°C, using molar extinction coefficient values calculated by the nearest-neighbor model (38). Stock solutions of RHPS4 were prepared at 1–2 mM concentration in the same potassium phosphate buffer. Appropriate amounts of ligand were added to the oligonucleotide solutions to obtain the desired 2:1 and 4:1 ligand/DNA ratio, followed by mix at 25°C for 1 h, before data acquisition. CD spectra and melting experiments were recorded using a Jasco J-815 spectropolarimeter equipped with a thermoelectrically controlled cell holder (Jasco PTC-423S Peltier). A quartz cell of 1 mm optical path length was used. CD spectra at 25°C were the averages of three scans collected between 220 and 360 nm with a scanning rate of 100 nm/min, and a response time of 1 s. Buffer baseline was subtracted from each spectrum. CD melting were carried out in the 20–100°C temperature range, at 1°C/min heating rate by following changes of CD signal at the wavelengths of the maximum CD intensity. UV absorbance spectra were recorded using an Evolution 300 UV-Vis spectrophotometer (Thermo Scientific) equipped with a Peltier temperature controller. The 15 μM DNA solutions were placed in a quartz cuvette of 1 mm path length. Thermal difference spectra (TDS) were obtained by recording the absorbance spectra in the range 220–320 nm at 20 and 90°C and subsequently taking the difference between the two spectra (39).
Colony formation assay
Cells were grown in complete medium in absence or in presence of RHPS4 for different times. At the end of the treatment, 500 cells for each condition were seeded into 60-mm plates and after 10 days colonies were stained with 2% methylene blue in 95% ethanol and counted.
Sphere formation assay
Untreated or treated cells were grown for 96 h in stemness medium. At the end of the treatment, the primary tumorspheres were mechanically dissociated and the resulting cells were re-plated in stemness medium. After 5 days of growth, the secondary tumorspheres were analyzed and quantified by phase-contrast microscopy.
Cell migration
Chemotaxis was performed in a 48-wells Boyden chamber-slide using 8 μm pore-size polycarbonate filters (NeuroProbe Inc, MD, US) (14). Briefly, the lower compartment of each well was filled with serum free medium with 0.1% BSA, as negative control, or 50 ng/ml SDF1α (Sigma-Aldrich). After 6 h, cells were fixed in EtOH 70% and stained using Crystal violet. Each point was run in triplicate. Cells on five random fields on the lower face of the filter were counted at 40X magnification.
In vivo experiments
Six to eight weeks old male mice CD-1 nude (nu/nu) and NOD.SCID (Harlan Laboratories, S. Pietro al Natisone, Italy) were used. All animal procedures were approved by the ethics committee of the Regina Elena National Cancer Institute (CE/534/12) and were in compliance with the national and international directives (D.L. March 4, 2014, no. 26; directive 2010/63/EU of the European Parliament and of the council; Guide for the Care and Use of Laboratory Animals, United States National Research Council, 2011).
CSC-LUC cells (106 cells/mouse) pre-treated for 96 h with 1 μM RHPS4 or PBS (Vehicle) were intramuscularly injected into immunosuppressed CD-1 nu/nu mice (5 mice/group). Tumor appearance and growth were followed by caliper measurement and bioluminescence imaging analysis. CSC-LUC cells (6 mice/group) treated as before, were also injected in the spleen of anesthetized NOD.SCID mice and after 30 min the spleen was removed. Seven days post-injection, the mice were sacrificed, liver and gastrointestinal organs harvested and imaged. Real time tumor dissemination in the whole mice and in organs was imaged using the IVIS imaging system 200 series (Caliper Life Sciences, Hopkinton, MA, USA). Data were acquired and analyzed using the living image software version 3.0 (Caliper Life Sciences).
Statistical analysis
The Student's t-test (unpaired, two-tailed) was used for comparing statistical differences. Survival curves of mice were generated by Kaplan–Maier product-limit estimate, and statistical differences between the various groups were evaluated by log-rank analysis with Yates correction (software Primer of Biostatistics, McGraw-Hill, New York, NY, USA). Differences were considered statistically significant when P < 0.05.
RESULTS
G4 ligands can inhibit CD133 expression in colon CSCs
CSCs have been recently recognized as the main cause of treatment failure in several human malignancies (40). In spite of the continuous efforts in developing new molecules able to target CSCs, identification of really efficacious drugs has not been reported so far. Here, based on data describing G4 ligands as a new promising class of anticancer agents (28,41), we evaluated the effects of these molecules on patient-derived colon CSCs.
First, the typical properties of CSCs, including the expression of surface markers (CD44, CD166 and CD133), resistance to Oxaliplatin and 5-Fluorouracil, the two drugs routinely used in treatment of colorectal cancers, and the tumor-initiating potential were evaluated in two patient-derived colon CSCs (CSC_1 and CSC_2), and in two stabilized colon cancer cell lines (HCT116 and HT29) used as control (Supplementary Figure S1A–C). Interestingly, immunohistochemistry analyses revealed that the tumors originating from the CSCs are endowed with the ability to self-renew and faithfully reproduce the original tumor in immunocompromised mice, recapitulating some key points to be considered for the translatability of experimental findings (Supplementary Figure S1D).
Then, we investigated the ability of G4 ligands (Supplementary Figure S2) to affect CSC marker expression. Strikingly, despite a slight reduction of CD44 and CD166 levels, RHPS4, an effective and well-characterized G4 ligand (26,33,35,42–45), almost completely impaired the expression of CD133 (Figure 1A and Supplementary Figure S3A), and this potent inhibitory effect was also observed with two novel RHPS4-derivative molecules (Supplementary Figures S4A and S4B), characterized by improved on- and off-target profile (46,47), and, even if at a different extent, with Emicoron (Supplementary Figure S4C), a chemically unrelated G4 ligand, showing high selectivity for quadruplex versus duplex DNA and triggering telomere damage and antitumoral activity in mice (48,49).
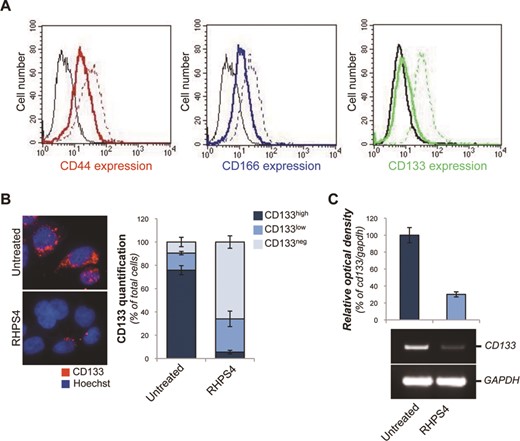
Expression of CD133 is impaired by G4 ligand treatment. (A) Colon CSCs (CSC_1), untreated or treated with 1 μM RHPS4 for 96 h, were immunostained with the antibodies against CD44, CD166 or CD133 and processed for FACS analysis. Histograms represent the fluorescence intensities in the negative controls (black lines), untreated (colored dashed lines) and treated (colored solid lines) samples. (B) CSC_1 cells were treated as in A and the expression of CD133 was analyzed by IF microscopy. Left panel: representative Immunofluorescence (IF) pictures are shown (63X magnification). Right panel: quantification of IF reporting the percentage of cells not expressing (CD133neg) or expressing high (CD133high) and low (CD133low) CD133 levels. (C) RT–PCR analysis of CD133 gene. The histogram shows the relative optical density of CD133. A representative picture of PCR products is shown. Histograms show the mean values ±SD of at least three independent experiments.
Moreover, a single-cell morphological analysis performed by IF microscopy (Figure 1B and Supplementary Figure S3B) revealed that, treatment with RHPS4 induced a robust decrease in the amount of CD133high cells with a concomitant increases in both CD133low and CD133neg cell subpopulations. Finally, to assess whether RHPS4 exerts its inhibitory effect by acting at transcriptional level, expression of the CD133 gene was evaluated. RT-PCR showed that treatment with the G4 ligand decreases the level of CD133 (Figure 1C and Supplementary Figure S3C), without substantially affecting the CD44 and CD166 mRNAs (Supplementary Figure S3D), suggesting the presence of at least one RHPS4-sensitive G-rich sequence within the CD133 gene.
Down-regulation of CD133 expression is dependent on the stabilization of intragenic G4 sequences
Following the initial identification of CD133 as RHPS4-sensitive CSC target, we investigated the mechanism(s) through which this G4 ligand regulates its expression. G4 structures are not exclusively located at the chromosome ends; indeed, these structures have been recently found along the entire genome (and on certain RNA transcripts) and their functions can range from transcriptional control to splicing regulation (20,41,50). Based on these data, we performed an in silico analysis on the entire CD133 gene looking for putative G4 sequences. Interestingly, the broad analysis carried by QGRS G4 structure prediction software (51) revealed the presence of five G-rich strands (one located within the promoter region and the other four within the introns 2, 3 and 7), whose probability to arrange into stable G4 structures (G-score) was very similar to that of the human telomeric repeats (TTAGGG)n (Figure 2A). The existence of these structures might explain the inhibitory effect of RHPS4 on the expression of CD133.
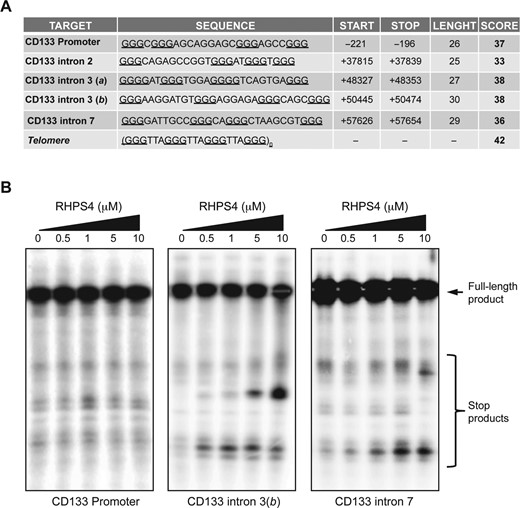
CD133 gene contains several putative G4-forming sequences. (A) The table shows the putative G4s identified within the CD133 gene. For each sequence is reported the score, calculated by QGRS mapper (G4 structure prediction software www.bioinformatics.ramapo.edu/QGRS/analyze.php). For comparison, the score of the human telomere repeat is reported. (B) Polymerase stop assay. Oligonucleotides reproducing the G4 sequences identified in the promoter and within the introns 3 and 7 of CD133 were amplified by PCR in presence of 5 mM KCl plus increasing concentrations of RHPS4. The stop products indicate the presence of stabilized G4 structures. Data shown are representative of three independent experiments.
Although all the identified target sequences had almost the same probability to form G4 structures, the G-rich motif located within the intron 2 and the first of the two identified in the intron 3(a), based on their huge distance from the proximal exon acceptor splicing sites (>1000 bp), were excluded from any additional investigation. The sequences found within the promoter and those found in the intron 3(b) and intron 7, were assayed for their capability to fold into stable G4 structures in vitro. First, we performed a polymerase stop assay (14,52) to exploit the capability of stable secondary DNA structures of arresting the activity of the DNA polymerase, allowing us to recognize those G-rich sequences that, under adequate conditions, fold into stable G4s. As showed in the Figure 2B, increasing concentrations of RHPS4 lead to a dose-dependent accumulation of stop products corresponding to the G-strands of the introns 3(b) and 7, whilst no effects were observed with the promoter candidate sequence.
Then, the two target sequences of the introns 3(b) and 7 were investigated by circular dichroism (CD) in order to determine the folding topology of G4s in solution (53), as well as to evaluate the ability of a G4 ligand to stabilize or alter these structures (54). Interestingly, the CD spectra indicated that the two oligonucleotides actually folded into G4 structures in the presence of K+ (Figure 3A and B), despite the presence of extended loops that could, in principle, compromise the stability of a G4 (55,56). Specifically, the spectrum of the intron 3(b) G-rich sequence was characterized by a negative band at about 240 nm and an intense positive band at 264 nm, typical of parallel-stranded G4s. While the spectrum of the sequence identified within the intron 7 showed a negative band at about 240 nm and a positive band at 265 with a shoulder at 288 nm, consistent with the presence of a hybrid G4 structure. Then, the formation of G4 motifs was confirmed by UV TDS analysis (Figure 3C), showing the characteristic G4 signature (39), i.e. the negative band at 295 nm and the positive ones at around 270 and 250 nm. The stability of these G4s was evaluated by CD melting experiments. In both cases, the melting experiments showed a sigmoidal transition curve (Figure 3D and E) from which the melting temperatures (Tm) were found to be 48.2 (±0.5) and 55.3 (±0.5)°C for intron 3(b) and 7 G4s, respectively. CD spectra and melting experiments were also performed to analyze the effect of RHPS4 on the structure and stability of G4s in solution. As shown in Figure 3A and B, no significant variations of CD spectra were observed for the investigated G4s upon RHPS4 addition (up to 4 equivalents), suggesting that the overall structure is preserved. On the other hand, increasing concentrations of RHPS4 induced a significant dose-dependent increase of thermal stability of the structures, indicating a tight interaction with the G4-forming sequences. Indeed, the CD melting curves (Figure 3D and E) showed that RHPS4 increased the Tm of 3(b) G4 of about 17 and 34°C at a 2:1 and 4:1 ligand/DNA ratio, respectively. While the ligand-induced increase in the Tm of the intron 7 G4 was of about 12 and 23°C at 2:1 and 4:1 ratio, respectively.
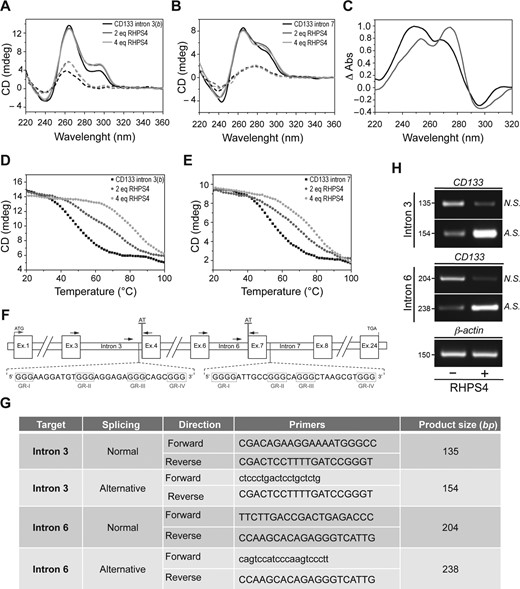
Stabilization of CD133 intragenic G4 sequences determines alternative splicing events. (A and B) CD spectra of the G4-forming DNA sequences found in the CD133 introns 3 and 7 at 20°C (black solid lines), 90°C (black dashed lines), and in presence of the indicated doses of RHPS4 at 20°C (gray solid lines) and 90°C (gray dashed lines). (C) Normalized thermal difference spectra of intron 3(b) (black solid line) and 7 G4s (gray solid line). (D and E) CD melting profiles of intron 3(b) and 7 G4-forming sequences in the absence (filled squares) and in the presence of the indicated doses of RHPS4 ligand (gray circles). (F) Scheme showing the positioning of the G4s within the introns 3 and 7 of CD133. For each sequence is indicated the proximal exon acceptor splicing site (AT). The arrows indicate the positioning and orientation of the primers designed for evaluating the splicing. (G) Nucleotide sequences of the primer used to discriminate between the normal and the alternative splicing of intron 3 and 6 of CD133. For each pair of primers, the size of the amplification product is indicated. (H) RT–PCR analysis of alternative splicing in CSC_1 cells untreated or treated with RHPS4 (1 μM for 96 h). Normal (N. S.) and alternative (A. S.) splicing of intron 3 and 6 were evaluated. Expression levels of β-ACTIN were measured as loading control.
Finally, we investigated whether the formation of G4 structures within the introns 3 and 7 may affect the splicing of CD133. Since the two G4s are located 118 bp upstream and 261 bp downstream of the acceptor sites of the exons 4 and 7, respectively (Figure 3F), we evaluated, using appropriately designed primers (Figure 3G), the effect of the G4 stabilization on the splicing of intron 3 and 6. We found that the RHPS4 treatment leads to a marked reduction of the mature transcripts counterbalanced by an increase in the splicing variants corresponding to an inhibition of the intron excision (Figure 3H). Overall these data indicate that the formation of stable intronic G4s could affect the proper positioning of the splicing machinery consequently interfering with the process of RNA maturation. Indeed, the inclusion of each of the two introns (3 and 6) leads to the formation of premature stop codons that can explain the reduction of CD133 expression.
G4 ligands can impair the tumorigenic potential of colon CSCs
The above results raise the interesting possibility that pharmacological G4 stabilization may affect the CSC activity. To address this question, the capability of RHPS4 to modulate the stemness properties of colon CSCs was assessed both in vitro and in vivo. Of note, these assays were performed by treating the cells for 96 h with 1 μM of RHPS4, an experimental setting that reduces the expression of CD133 without any overt effect on cell viability or apoptosis (Supplementary Figure S5A–C). Interestingly, both colony and sphere formation assay revealed that G4 ligands reduced cell survival up to about 50% (Figure 4A and B and Supplementary Figure S4D and E). Indeed G4 ligand treatments not only impaired self-renewal capability of the cells (Figure 4B, left panel and Supplementary Figure S4E, left panel) but, as revealed by qualitative microscopy analysis (Figure 4B, right panel and Supplementary Figure S4E, right panel), also significant reduced the size of the spheres. Then, cell migration, evaluated in presence of SDF1α, a potent chemoattractant cytokine, was almost completely abolished in RHPS4 pre-treated cells (Figure 4C). This effect was fully recapitulated by interference experiments performed by using two different short hairpin RNAs selective for CD133 (shCD133_1 and shCD133_2) (Figure 4C and D). Moreover, since RHPS4 is a potent inducer of DNA damage, including at the telomeres (Supplementary Figure S5D and E), the effect of RHPS4 was assessed also in presence of KU-55933, a specific ATM inhibitor (57). Notably, pharmacological inhibition of DDR pathway (Supplementary Figure S5D) did affect neither the expression of CD133 (Supplementary Figure S5F) or the cell migration (Figure 4C), indicating that impairment of certain stemness properties by G4 ligands can be independent from the activation of the DNA damage response.
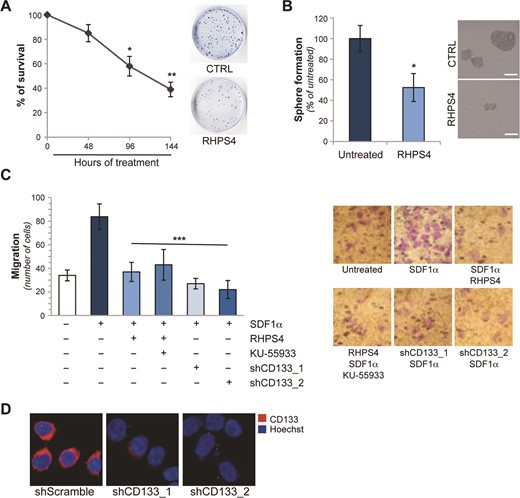
G4 stabilization impairs CSCs features in vitro. (A) Colon CSCs (CSC_1) were treated with 1 μM of RHPS4 for the indicated times and colony forming ability was evaluated. The graph shows the surviving fractions calculated as the ratio of absolute survival of the treated sample/absolute survival of the control sample. Representative images of colony assay for untreated and 96 h treated sample are shown. (B) Primary tumorspheres, untreated or treated with RHPS4 (1 μM for 96 h), were dissociated and the cell ability of originating secondary spheres was evaluated. The histogram shows the percentage of secondary tumorspheres obtained 5 days after dissociation. Representative images of secondary tumorspheres are shown in the right panel. (C) CSC_1 cells, treated as reported (RHPS4 1 μM for 96 h, KU-55933 5 μM for 96 h), or interfered for 48 h with two different shRNA constructs targeting CD133 (shCD133_1 and shCD133_2), were processed for chemotaxis assay in response to sdf-1α (50 ng/ml). Histograms represent the number of migrating cells. For each condition, representative pictures of cell migration (40X magnification) are reported. (D) Analysis of CD133 expression in colon CSC_1 cells 48 h after the transfection with a control shRNA (shScramble) and two different shRNA constructs targeting CD133. Representative IF images are shown. Graphs show the mean ±SD of three independent experiments (*P < 0.1 **P < 0.01; ***P < 0.001).
Finally, the effects of RHPS4 on the tumorigenic potential of CSCs were assessed in vivo. Pre-treatment with RHPS4 markedly impaired the aggressiveness of colon CSCs in immunosuppressed mice (Figure 5). Indeed, bioluminescence analysis performed at day 25 after cell injection showed that while in control group all the mice (5/5) developed tumors, in the RHPS4 group only one out of the five (1/5) injected mice developed tumor (Figure 5A). Afterwards, the tumor mass was detectable in the rest of mice (Figure 5B) but with a median time of tumor appearance of 29 days significantly reduced (P < 0.01) compared to that observed in untreated group (16 days). The delay in tumor development resulted in a different tumor growth rate as at day 67 the tumor weight in RHPS4-treated group was significantly decreased (660 ± 320) compared to untreated group (1747 ± 682, P = 0.012). Moreover, being the high metastatic potential one the main features of CSCs, we evaluated the efficacy of RHPS4 on an experimental model of disseminated disease established after intrasplenic injection of colon CSCs, stably transfected with a luciferase-expressing vector. Interestingly, RHPS4 pre-treatment limited the colon CSCs dissemination being the luminescence of treated animals significantly reduced (about 50%) in comparison to that of the untreated ones. This is evident in the analysis performed both on the whole mice (Figure 5C, P = 0.023) and on the organs (liver P = 0.026 and intestine, P = 0.038) excised after the euthanization of animals (Figure 5D).
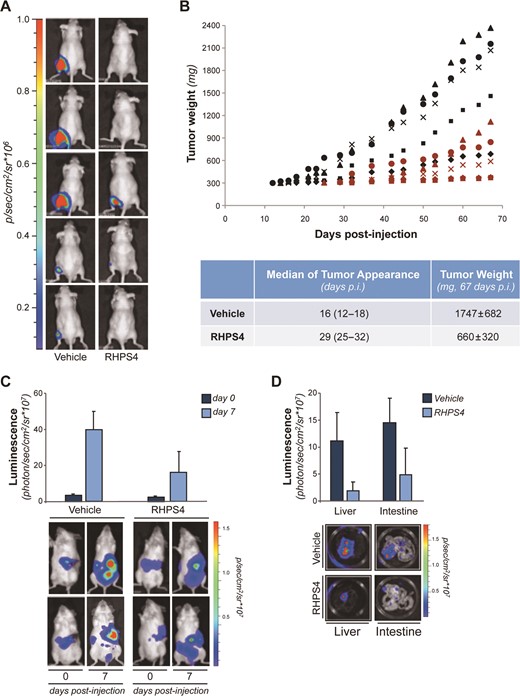
G4 stabilization impairs CSCs tumor promoting activity in vivo. (A and B) CSC-LUC cells, pre-treated for 96 h with 1 μM of RHPS4 or a vehicle, were intramuscularly injected in immunosuppressed mice. (A) Luminescence images (quantified as number of photons/s) acquired 25 days after cell injection are shown. (B) Tumor weights assessed in mice intramuscularly injected with CSC-LUC cells treated with RHPS4 (red symbols) or Vehicle (black symbol) are reported. Each symbol represents a different mouse. Median of tumor appearance and average tumor weight are reported in the table. (C and D) CSC-LUC cells treated as in A were injected in the spleen of NOD-SCID mice and the real time tumor dissemination was monitored by the imaging system. (C) Histograms report bioluminescence in the whole mice at time 0 (immediately after the spleen removal) and at day 7 after tumor cells injection. Representative images of 2/6 mice/group are shown. (D) At day 7, mice were sacrificed and organs were harvested and analyzed. Histograms report bioluminescence in liver and gastrointestinal organs. Representative images of liver and intestine are also shown. Bars indicate mean values ±SD.
DISCUSSION
Over the last decade, CSC biology has acquired a huge relevance both at basic and preclinical cancer research level. In this context, particular attention has been paid to the biological relevance of stemness markers. The significance of these surface proteins expression, initially considered as CSCs specific markers, has been extensively debated in the last few years (58). Indeed, a number of recent studies reported that these proteins would be detectable, even if at lower levels, also in more differentiated cell types, questioning their real CSCs specificity (59). However, beside this controversial issue, the expression of these surface proteins was reported to be responsible for conferring CSC peculiar properties, including chemoresistance, self-renewal capability and metastatic potential (60,61). Here, we found that G4 ligands, including the well-characterized RHPS4, two novel RHPS4-derivatives, and Emicoron, markedly affect the stemness properties of patient-derived colon CSCs by impairing the expression of CD133. Although the effect of RHPS4 in counteracting the growth of CSCs was already proposed by Burger et al. in 2007, the mechanism through which the drug acts was not deeply investigated (43).
G4 structures, first identified at the chromosome ends, were found dislocated along the entire genome (and in the RNAs) (41,62–64) and their stabilization has been demonstrated to affect, through modulation of transcription (50), translation (65) or splicing (20), the expression of various target genes (15,16,66). Based on these data, and with the intent of defining the mechanism through which the G4 ligands can affect the expression of CD133, we analyzed the entire gene sequence, looking for G-rich strands with features compatible with the formation of G4 structures. Among the putative G4 sequences identified by bioinformatic analysis, two motifs (located within introns 3 and 7, respectively) were experimentally validated by CD. Notably, the CD analysis evidenced that both G4s are stabilized by RHPS4, affecting the process of transcript maturation, which, in turn, generated two alternatively spliced mRNAs, each deriving from a distinct event of intron retention. To date a number of CD133 splice variants have been reported (67). However, the two splice variants induced by treatment with G4 ligand were never described, suggesting that these transcripts could generate aberrant CD133 isoforms that are then degraded. In line with these data, we found that intron retention determined the formation of premature stop codons leading to the production of truncated forms, probably removed by the nonsense-mediated decay pathway. In several cases, CD133 isoforms, derived from alternative splicing events, were not recognized by specific antibodies due to the lack of the AC133 epitope (68). At this level, we cannot completely exclude that the reduction observed in the CD133 protein expression could be due to the loss of the immunogenic epitope. However, data in the literature clearly indicate that AC133 epitope is, per se, sufficient to confer the aggressive phenotype to CSCs (68). Therefore, independently on the mechanism, our in vitro and in vivo functional studies clearly demonstrate that treatment of colon CSCs with the G4 ligand determines an almost complete impairment of their tumor-promoting activity both in terms of self-renewal and metastatic potential. Of note, other relevant stemness marker of colon CSCs (i.e. CD44 and CD166) were found slightly reduced by the drug treatment. Even though a cross-talk among different stemness markers has been described (4), we cannot completely exclude that G4 ligand might directly affect their expression by stabilizing G4 structures within their promoters as revealed by the QGRS G4 structure prediction software (51). Therefore, even if other genes can contribute to the biological effects observed with the G4 ligand on colon CSCs, the possibility of fully recapitulating the effects of the drug by specific interfering RNAs, indicates that treatment with G4 stabilizing agents inhibits the tumor-promoting signaling pathways regulated by CD133 (69,70).
CSCs for their aggressiveness and resistance to canonical therapeutics have been recognized as the main cause of failure in the treatment of several malignancies. Therefore, identification of a new class of antineoplastic drugs able to target these cells is of crucial importance. Here, we provide, for the first time, the evidence for the existence of intragenic G4 structure formed in the human CD133, corroborating the application of stabilizing ligands in a cellular context to target G4 and interfere with gene expression and function. Indeed, these molecules, initially designed to bind to and stabilize the telomeric G4 sequences, triggering telomere damage and cell death, can act, through a multimodal therapeutic strategy, on the expression of cancer-related genes, including the CD133, counteracting the tumor-promoting activity of colon CSCs. Notably, being CD133 relevant in the progression of tumors of different histotype, it will be possible to extend our results also to other malignancies. Moreover, since the folding of G4s localized within the genes can assume different shapes, depending on their specific DNA sequence, it might be possible to design and develop new and more potent drugs able to selectively hit certain, well-defined, drivers depending on the tumor histotype.
In conclusion, here, we provided an additional proof of the potential therapeutic use of G4 ligands as a novel multitargeting anticancer strategy. Moreover, the ability of these drugs to directly target CSCs derived from patients and the use of advanced preclinical colorectal cancer models, significantly increases the possibility to translate our results in clinical studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
The authors thank Prof. Ruggero De Maria for the critical reading of the manuscript and Prof. Armandodoriano Bianco for Emicoron synthesis. C.C. is recipient of a fellowship from the Italian Foundation for Cancer Research (FIRC).
FUNDING
Italian Association for Cancer Research [#11567, #9979 to A.B, #14337 to C.L. and #14150 to A.R.]. Funding for open access charge: Italian Association for Cancer Research [#11567, #9979 to A.B, #14337 to C.L. and #14150 to A.R.].
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments