





Abstract
Comparative genomics has revealed a class of non-protein-coding genomic sequences that display an extraordinary degree of conservation between two or more organisms, regularly exceeding that found within protein-coding exons. These elements, collectively referred to as conserved non-coding elements (CNEs), are non-randomly distributed across chromosomes and tend to cluster in the vicinity of genes with regulatory roles in multicellular development and differentiation. CNEs are organized into functional ensembles called genomic regulatory blocks–dense clusters of elements that collectively coordinate the expression of shared target genes, and whose span in many cases coincides with topologically associated domains. CNEs display sequence properties that set them apart from other sequences under constraint, and have recently been proposed as useful markers for the reconstruction of the evolutionary history of organisms. Disruption of several of these elements is known to contribute to diseases linked with development, and cancer. The emergence, evolutionary dynamics and functions of CNEs still remain poorly understood, and new approaches are required to enable comprehensive CNE identification and characterization. Here, we review current knowledge and identify challenges that need to be tackled to resolve the impasse in understanding extreme non-coding conservation.
INTRODUCTION
Extremely conserved sequences within the non-coding portion of metazoan genomes were initially identified more than three decades ago by comparing the introns and UTRs of mammalian and avian mRNAs (1–5). These pioneering studies identified elements that had maintained >70% sequence identity over hundreds of millions of years of evolution, far greater than that expected for neutrally evolving DNA. Progress in DNA sequencing technologies aided the further identification of numerous individual examples of non-coding conservation (6–9). The prevalence of these elements was only truly appreciated when multiple groups published the systematic, genome-wide identification of conserved non-coding elements (CNEs) (10–12). This established that there are hundreds to thousands of extremely conserved non-coding elements identifiable across more than 400 million years of evolution that, in many cases, exhibit levels of conservation well beyond those seen in protein-coding genes (Figure 1).

The phenomenon of extreme non-coding conservation. A conserved CNE (Human–Tetraodon CNE, on the left) shown here is more conserved than a protein-coding sequence (HIST1H4D, on the right). The multiple sequence alignment of 46 vertebrate species and the corresponding phyloP scores illustrate the evolutionary conservation of the CNE and protein-coding sequence. PhyloP scores range from negative to positive scores (red to blue) and indicate positive and negative selective pressure respectively. The 46-way alignment was downloaded from the UCSC genome browser and spans ∼600 million years of evolution since the last common ancestor of humans and lampreys.
Since then, numerous studies have defined sets of evolutionarily conserved CNEs, each using different conservation criteria, species comparisons and nomenclature (summarized in Supplementary Table S1). Our current understanding is that these overlapping sets represent the same elements, maintained by similar, poorly understood processes. Therefore, we collectively refer to those elements as CNEs. As conservation is dependent on the species compared, elements can be lineage-specific. For example, not all CNEs identified by comparing mammalian genomes appear conserved when the same conservation criteria are used on more distant genome comparisons. Additionally, all CNEs detected among closely related species (e.g. human and mouse) may not be functional elements, whereas an overwhelming majority of the CNEs conserved between distant species are likely to be functional.
A handful of resources, mainly databases, exist which contain pre-computed sets of CNEs (13–19) (Table 1). These databases contain many of the CNE sets studied so far; their disadvantage is that they are static and seldom updated.
Conserved non-coding elements (CNE) resources
| Abbreviation | Description | Identification | Reference |
|---|---|---|---|
| ANCORA | Atlas of non-coding conserved regions in animals | ≥70% seq. id. over 30 or 50 nt in different metazoa | (13) |
| CEGA | Conserved elements from genomic alignments | threshold-free phylogenetic modeling | (14) |
| cneViewer | Conserved non-encoding element viewer | user-specified | (15) |
| CONDOR | COnserved Non-coDing Orthologous Regions database | multiple and multi-pairwise alignments of orthologous regions between Fugu and human, mouse, rat, and dog; coding and repetitive regions are removed | (16) |
| UCbase | Ultraconserved elements database | 100% seq. id. over 200 nt between human and mouse | (17) |
| UCNEbase | Ultraconserved CNEs | ≥95% seq. id. over 200 nt in the human and chicken genomes; coding regions are removed | (18) |
| VISTA | ViSualization tool for alignment | extremely conserved sequences between human and rodents that have been tested in vivo for enhancer activity | (19) |
| Abbreviation | Description | Identification | Reference |
|---|---|---|---|
| ANCORA | Atlas of non-coding conserved regions in animals | ≥70% seq. id. over 30 or 50 nt in different metazoa | (13) |
| CEGA | Conserved elements from genomic alignments | threshold-free phylogenetic modeling | (14) |
| cneViewer | Conserved non-encoding element viewer | user-specified | (15) |
| CONDOR | COnserved Non-coDing Orthologous Regions database | multiple and multi-pairwise alignments of orthologous regions between Fugu and human, mouse, rat, and dog; coding and repetitive regions are removed | (16) |
| UCbase | Ultraconserved elements database | 100% seq. id. over 200 nt between human and mouse | (17) |
| UCNEbase | Ultraconserved CNEs | ≥95% seq. id. over 200 nt in the human and chicken genomes; coding regions are removed | (18) |
| VISTA | ViSualization tool for alignment | extremely conserved sequences between human and rodents that have been tested in vivo for enhancer activity | (19) |
| Abbreviation | Description | Identification | Reference |
|---|---|---|---|
| ANCORA | Atlas of non-coding conserved regions in animals | ≥70% seq. id. over 30 or 50 nt in different metazoa | (13) |
| CEGA | Conserved elements from genomic alignments | threshold-free phylogenetic modeling | (14) |
| cneViewer | Conserved non-encoding element viewer | user-specified | (15) |
| CONDOR | COnserved Non-coDing Orthologous Regions database | multiple and multi-pairwise alignments of orthologous regions between Fugu and human, mouse, rat, and dog; coding and repetitive regions are removed | (16) |
| UCbase | Ultraconserved elements database | 100% seq. id. over 200 nt between human and mouse | (17) |
| UCNEbase | Ultraconserved CNEs | ≥95% seq. id. over 200 nt in the human and chicken genomes; coding regions are removed | (18) |
| VISTA | ViSualization tool for alignment | extremely conserved sequences between human and rodents that have been tested in vivo for enhancer activity | (19) |
| Abbreviation | Description | Identification | Reference |
|---|---|---|---|
| ANCORA | Atlas of non-coding conserved regions in animals | ≥70% seq. id. over 30 or 50 nt in different metazoa | (13) |
| CEGA | Conserved elements from genomic alignments | threshold-free phylogenetic modeling | (14) |
| cneViewer | Conserved non-encoding element viewer | user-specified | (15) |
| CONDOR | COnserved Non-coDing Orthologous Regions database | multiple and multi-pairwise alignments of orthologous regions between Fugu and human, mouse, rat, and dog; coding and repetitive regions are removed | (16) |
| UCbase | Ultraconserved elements database | 100% seq. id. over 200 nt between human and mouse | (17) |
| UCNEbase | Ultraconserved CNEs | ≥95% seq. id. over 200 nt in the human and chicken genomes; coding regions are removed | (18) |
| VISTA | ViSualization tool for alignment | extremely conserved sequences between human and rodents that have been tested in vivo for enhancer activity | (19) |
In this review, we provide a comprehensive account of the genomic organization of CNEs and their intriguing sequence properties. We discuss CNE functions, their roles in disease aetiology and hypotheses regarding their emergence and evolutionary dynamics. We conclude with unaddressed questions important for our progress in understanding these elements in the future.
KNOWN FUNCTIONS OF CONSERVED NON-CODING ELEMENTS
Many CNEs function as developmental enhancers
A pioneering study by Aparicio et al. published more than two decades ago identified one of the first CNEs, and at the same time demonstrated that it exhibited enhancer activity in a transgenic mouse model (20). Since then, reporter gene assays throughout the vertebrate phylogeny, from mouse (21–24), chicken (25,26), frog (27) to zebrafish (28–30) have demonstrated that CNEs typically function as enhancers in various developmental contexts. This has led to the view of CNEs as cis-regulatory elements coordinating spatial-temporal gene expression, especially during embryonic development (11,31,32). While the majority of CNEs act as enhancers, it should be noted that not all functional enhancers display such extreme levels of conservation as CNEs (33,34), including many enhancers found within dense genomic clusters of CNEs.
In line with CNEs predominantly being developmental enhancers, detectable phenotypic changes have been associated with alterations in CNEs. A particularly well-characterized case is the SHH ZRS enhancer, in which point mutations result in preaxial polydactyly in both human and mouse (35–38). Mutations in a CNE proximal to the HMX1 gene cause aberrant external ear development in wild populations of rats and highland cattle (39). A mouse sequence called M280, which contains a CNE identical between human, mouse and rat, is indispensable for body growth in mice (40). Many more cases linking CNEs to both human disease and lineage-specific traits are discussed in more depth in the ‘Diseases associated with non-coding conservation’ and ‘CNE Modifications and Losses’ sections. They highlight the important role of CNEs as developmental enhancers.
GENERAL FEATURES OF NON-CODING CONSERVATION
Genomic organization of conserved non-coding elements
One of the most striking features of CNEs is their non-random distribution across genomes (10,11,41–43). CNEs reside in clusters (12) that often span regions with low gene density, including gene deserts (44). These clusters tend to coincide with key developmental regulatory target genes, such as SHH and HMX1 mentioned above, with CNEs driving the expression of these target genes without affecting unrelated bystander genes within the cluster (11,30,45,46). CNEs, their target genes, and associated bystanders are maintained in syntenic blocks due to the requirement for regulatory elements to remain in cis with their target genes. This has constrained the evolution of metazoan genomes, resulting in arrays of syntenic CNEs that form functional, long-range, gene regulatory modules. The regions spanned by these arrays are named genomic regulatory blocks (GRBs) and, in addition to the array of CNEs, always contain a target gene regulated by the CNEs. Some, but not all, GRBs also contain bystander genes. Bystander genes frequently contain CNEs within their introns; however, they are unresponsive to CNE regulation due to differences in their promoter architecture (45,46). GRB target genes share defining properties that distinguish them from bystander genes. In vertebrate genomes, these include: (i) longer CpG islands, often several of them bound by Polycomb proteins, (ii) distinct patterns of histone modifications, (iii) differences in the distribution of alternative transcription start sites and (iv) a characteristic spatial organization of transcription factor binding sites (TFBS) in their core promoters (46). In Drosophila, GRB target genes are also characterized by broad Polycomb binding, and longer than average introns that often contain CNEs (45). The GRB model is depicted schematically in Figure 2A. The MEIS2 target gene is presented in Figure 2B as part of a GRB with a well-characterized regulatory landscape.
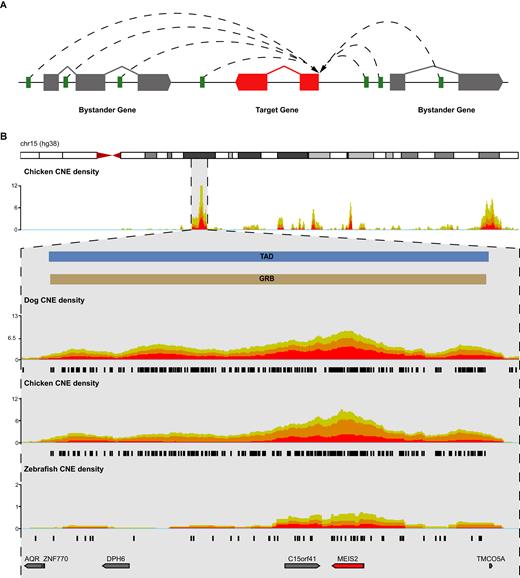
(A) The GRB model. The regulatory input for one or more target genes (red) is provided by long-range interactions (dashed lines) between CNEs (green) and the target gene’s promoter. Bystander genes (gray) often contain CNEs in their introns but remain unresponsive to CNE-mediated regulation. For more details, see text, (B) CNE clustering across chromosome 15 and at the MEIS2 locus (shown zoomed in below the whole chromosome track). The human MEIS2 GRB (brown) is a 3.3 Mb region defined by an array of conserved non-coding elements. MEIS2 (red) encodes a transcription factor involved in lens development through regulation of PAX6. Regardless of species used in the pairwise comparison against human, CNEs (black) clearly mark the boundaries of this GRB. The boundaries of the topologically associated domain (TAD) covering MEIS2 (TAD in blue; H1-ESC TAD calls are generated using HMM_calls) and the GRB spanning this locus are highly concordant.
Relationship with TADs
Recent studies in bilaterian genomes have led to the identification of genomic regions within which frequent chromatin interactions occur. These regions, known as TADs, form distinct genomic boundaries within which preferentially self-interacting regions are enriched (47–49). TAD boundaries are largely invariant across cell types (48,50,51) and between species (52). This robustness and prevalence of TADs prompted Harmston et al. to investigate whether TADs and GRBs reflect the same underlying phenomenon (53); see also Figure 2B. They demonstrated that GRB boundaries are resilient to CNE identification thresholds and the evolutionary distance of the species comparison used (53). Further, GRB boundaries coincide with TAD boundaries around developmental genes in both vertebrates and invertebrates, suggesting that TADs associated with GRBs display unique genomic features. TADs which closely correspond with GRBs are termed ‘GRB–TADs’, and those that show no evidence of non-coding conservation ‘nonGRB–TADs’. Several features distinguish GRB–TADs from nonGRB–TADs; GRB–TADs are larger than nonGRB–TADs, gene-sparse and their target genes are expressed in a cell-type and tissue-specific manner. In contrast, nonGRB–TADs more often span regions of high gene density, and the strength of within-TAD interactions in them is consistently lower than in GRB–TADs (53). This may indicate a less defined or less consistent 3D organization across Hi-C experiments. Since strong and stable GRB–TADs are interspersed with less strongly interacting nonGRB–TADs there is an open possibility that the weaker TADs are simply regions between stronger TADs, which would mean that a stable 3D arrangement is not required in the absence of long-range regulation. At present, this is still a hypothesis (53), whose testing will require more high-resolution Hi-C data across different cell types and different species.
The observation that GRBs coincide with TADs around developmental genes puts an interesting twist on the question of co-regulation of genes within TADs. Since the GRB model predicts different expression profiles of target and bystander genes, with bystanders typically broadly expressed, the co-regulation of target and bystander genes is not expected. Harmston et al. examined several GRB loci for co-regulation and showed that the dynamic range of target gene expression is much wider than that of bystander genes (53). Moreover, on the limited number of loci, they show that GRB–TADs in different cell types switch between the two compartments identified by Hi-C: the A compartment (reported to be dominated by actively transcribing chromatin) and the B compartment (enriched for heterochromatin and other transcriptionally inactive regions). Remarkably, the activity state of the GRB target gene is the only one that predicts whether the GRB will be in the A or B compartment: the expression of bystander genes appears to change little between the two. While still a preliminary observation, this pattern is consistent with the GRB model of long-range regulation.
CNEs are an ancient feature of metazoan genomes
While the bulk of CNE research has focused on the identification and functional characterization of CNEs in human and other vertebrate genomes, it is clear that CNEs are not a vertebrate innovation as equivalent elements are detected in multiple metazoan lineages (32,54–58). Despite a lack of CNEs identifiable between each clade, some general features of CNEs are recapitulated in each case:
CNEs occur in clusters around crucial regulators of early development such as MEIS1 (59), SHH (60), IRX3 (27) and around the orthologs of these genes in other lineages.
CNEs have constrained genome organization and thus occur in GRBs in other metazoa (30,45).
Finally, genes proposed as targets of CNE regulation are marked by broad polycomb repression in an inactive state (45).
The observation that CNEs are so prevalent and functionally similar within metazoan genomes indicates that they are an ancient and crucial feature of metazoan gene regulation.
Plant CNEs
Clusters of non-coding conservation also occur in genomes of higher plants (61–64), although their equivalence to metazoan CNEs is unclear. In plants, CNEs were found to cluster around genes involved in responses to hormonal stimuli, regulation of organ development (64–67) and flowering-time control (68). However, plant CNEs have so far not been shown to form GRB-like clusters, and much more work is needed to understand their distribution and roles.
Sequence features of CNEs
Walter et al. analyzed the nucleotide composition of human and fugu CNEs, showing that they are AT-rich and often contain runs of identical nucleotides (69). This is in contrast to the flanking regions just outside their boundaries which exhibit a marked drop in AT content, forming a distinct composition pattern. In line with this, Chiang et al. have shown that vertebrate CNEs are enriched in TAATTA, which contains the core recognition motif (TAAT) for homeodomain DNA-binding factors (70). In Figure 3, we order vertebrate CNEs (identified using human/chicken whole-genome alignments) by width, and plot the enrichment of AT, GC, WW and SS dinucleotides. This clearly illustrates this boundary effect. As regions outside boundaries of CNEs are by definition mismatched, i.e. mutated sites in the alignment, the WW depletion at the boundaries of CNEs might be due to the higher mutability of CpG nucleotides. However, the latter does not explain why we find the same pattern in Drosophila where CpG methylation is absent. Importantly also, such GC-content transitions are known to occur at transcription boundaries and serve as genomic punctuation marks (71). In summary, it is still unclear why we find this pattern in CNEs.
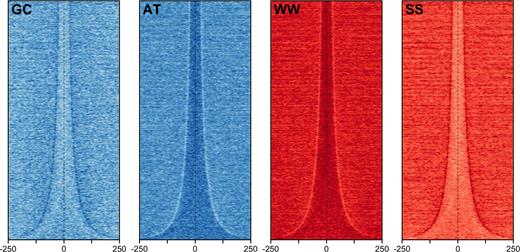
Sequence heatmaps showing dinucleotide content within and outside vertebrate CNEs. Plots are generated using heatmaps package (https://bioconductor.org/packages/release/bioc/html/heatmaps.html). CNEs which show sequence identity >98% for >50 nt between human and chicken are identified using CNEr (https://bioconductor.org/packages/release/bioc/html/CNEr.html). Sequences are ordered from shortest to longest on the Y-axis (aligned on the center) and X-axis shows distance in nucleotides from the center of each CNE.
CNEs overlap transcription factor binding sites (TFBS)
One of the suggested explanations for extreme non-coding conservation would be that CNEs constitute an ordered combination of overlapping TFBS (72,73). While there is clear evidence that CNEs are strongly enriched for overlapping TFBS (74,75), there is no evidence to suggest that this enrichment is higher for CNEs than for enhancers in general. Furthermore, it is unclear whether overlapping binding sites would be sufficient to explain extreme non-coding conservation, given promiscuity of binding sites and binding site divergence between species (76). A recent paper also proposed that CNEs are not under selective pressure as a whole DNA segment but are under various evolutionary constraints on the single nucleotide level (77), suggesting that overlapping TFBS likely do not account for the degree of conservation characteristic of CNEs.
Mining the distinguishing sequence features of CNEs
Two approaches have been presented to classify CNEs and distinguish them from other constrained elements within and between genomes: (i) N-gram graphs which combine neighborhood information (co-occurrence of substrings) with sequence compositional motifs (78) and (ii) logic alignment free which attempts to infer logic rules based on the underlying lexicon of sequences (79). Both approaches concluded that the most extremely conserved CNEs form a unique category bearing sequence features distinct from protein-coding exons. More sophisticated methods could be applied to CNE classification and deep learning, a variation of multi-layered artificial neural networks, is a promising candidate to elucidate potentially complex patterns within CNEs (80–82).
CNEs AS PHYLOGENETIC MARKERS
The deep sequence conservation of CNEs across phylogenies makes them particularly useful for the elucidation of evolutionary relationships. Using CNEs as the anchor points for targeted DNA enrichment and sequencing, Faircloth et al. recovered the established primate phylogeny (83) and McCormack et al. resolved the placental mammal phylogeny (84). As a further proof of concept, this approach was applied to 32 arachnids, again producing a highly resolved arachnid phylogeny consistent with transcriptome-based phylogenetic analyses. Due to the increasing variability of the sequence flanking the ‘core’ CNE region, the authors were also able to generate accurate phylogenies of the spider, scorpion and harvestman orders, demonstrating the utility of this method for shallower taxonomic scales (85,86). CNEs as probes have also proven useful in the case of ancient and degraded DNA (87). An overview of the workflow for using CNEs as phylogenomics markers is provided in Figure 4.
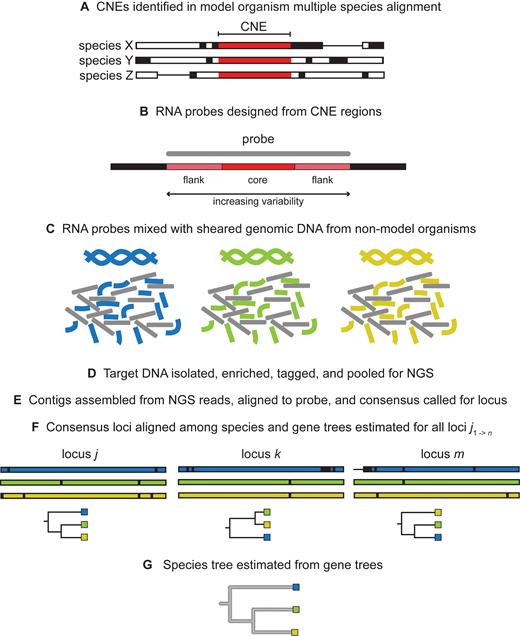
Methodology describing how CNEs are utilized as markers for constructing phylogenies (adopted and modified with permission from Brant Faircloth and John McCormack). Panels (A-G) describe the steps for constructing phylogenies starting from CNE sequences to generating species trees.
This method is implemented in the software package PHYLUCE (88). It is particularly useful for phylogenomic analysis of non-model organisms, as the extreme conservation of CNEs allows for targeted sequencing of informative loci without a complete reference genome. The ‘core’ regions of CNEs alone are sufficient to recapitulate gene trees, as demonstrated by Davies et al. (89), and have recently been considered for resolving nodes that are difficult to place in the eutherian tree (90). For a comprehensive review on CNEs as a tool in phylogenomics, see Edwards et al. (91).
DISEASES ASSOCIATED WITH NON-CODING CONSERVATION
Mutations in CNEs have been established as causal for a number of diseases. Single point mutations are associated with malformations, including Pierre Robin syndrome (92), cleft lip (93), holoprosencephaly (36,94), preaxial polydactyly (35,37,95) and Hirschsprung disease (96). A single nucleotide variant associated with IRX1, IRX2 and IRX4, located within a CNE, was also recently found to be involved in the pathogenesis of kyphoscoliosis (97). Beyond malformations, variations within CNEs can even be linked to complex behavioral or neurological disorders, evident from research linking cases of attention-deficit/hyperactivity disorder (98), autism (99) and restless leg syndrome (100) to single point mutations within CNEs.
Several diseases have been linked with duplications of CNEs, e.g. brachydactyly A2 (101) and brachydactyly-anonychia (102). Copy-number variations of the Indian Hedgehog region involving CNEs are related to syndactyly and craniosynostosis (103). Translocation of a CNE has been implicated in the aetiology of aniridia (104). Deafness (105), Leri-Weill dyschondrosteosis (25), blepharophimosis syndrome (106,107) and Waardenburg syndrome type 4 (108) are well-known cases of pathologies that are associated with deletions of CNEs. These findings further highlight and establish the role of CNEs in neurodevelopmental diseases. For a comprehensive review on this subject, see Amiel et al. (109).
Large-scale CNE deletions without visible phenotype
Despite strong evidence that the majority of CNEs play crucial roles in development, two early studies found that deletions of entire CNE-rich loci produced no detectable phenotypic changes in mouse models (110,111). However, it may be the case that either the phenotypes were difficult to detect in the tested experimental context, or that the tested elements were not deeply conserved. More elaborate methodologies are required to study CNE loss in greater detail. Towards this direction, Dickel and colleagues recently used CRISPR to knockout four CNEs near ARX, a gene with roles in sexual and brain development (112). At first, removing these elements individually or in pairs resulted in seemingly unaffected mice. A closer inspection of the brains of the knockout mice, however, revealed an atypical number of neurons or a diminished hippocampus, both of which were more pronounced in the double knockout mice. This supports the idea that a CNE phenotype might be context-specific, having little effect on mice in laboratory conditions but potentially detrimental in the wild.
Transcribed ultraconserved regions (T-UCRs)
Other than congenital abnormalities associated mainly with development, there are confirmed roles for CNEs in cancer. Calin et al. compared the transcription levels of the 481 ultraconserved regions from Bejerano et al. (10), and found that 93% of of those regions were transcribed over background in at least one of the tested normal human tissues. They named those elements transcribed ultraconserved regions (T-UCRs), and demonstrated that CNE transcriptional profiles could be utilized in order to differentiate carcinomas from leukemias (113). Since then, roles of T-UCRs have been investigated in hepatocellular carcinoma (114), prostate cancer (115,116), colorectal carcinoma (113,117), neuroblastoma (118,119), Barrett’s esophagus (120) and bladder cancer (121). In addition, reducing the overexpression of a T-UCR (uc.261) has been suggested as a therapeutic intervention for patients with Crohn’s disease (122).
At the level of GRBs, highly conserved elements that serve as long-range regulatory input for the TF genes HHEX, SOX4 and IRX3 were found to be associated with type 2 diabetes and obesity (123–125). Additional cases where GRBs are implicated in human diseases may be found in a review by Navratilova and Becker (126).
Summarizing this section, disruption of CNEs can contribute to the onset of severe diseases mainly associated with development and cancer. We anticipate more examples will be discovered in the future, especially now that it has been established that the majority of GWAS SNPs lie in the non-coding part of our genomes (127,128). It is possible that loss of even the most conserved CNEs is not guaranteed to result in visible phenotypes, emphasizing how much we still have to learn about extreme non-coding conservation.
EMERGENCE AND EVOLUTIONARY DYNAMICS OF CONSERVED NON-CODING ELEMENTS
The structural and implied functional equivalence of CNEs across vastly different realms of life, along with their extreme levels of sequence conservation, suggest that CNEs perform an ancient and irreplaceable function in genomes of multicellular eukaryotes. The emergence and maintenance of CNEs however remain poorly understood and no current theories can satisfactorily explain the source of selective pressure capable of maintaining such extreme levels of conservation (129).
Initial speculations that CNEs were not a product of negative selection, but simply genomic loci with a low local mutation rate, were dispelled by evidence that CNEs exhibit traits associated with sequences under purifying selection–namely very low derived allele frequencies, indicating evolutionary suppression of variation within these elements (130–132). This finding was corroborated in 2014 using 1000 Genomes human variation data (133).
Emergence
CNEs are readily recruited de novo from a diverse range of genomic sequences, an observation which is reflected by the general lack of similarity between CNEs on a sequence level. There is evidence for CNE recruitment from introns (10,54,56), transposable elements (TE) (134,135) and ancient repeats (136). There also exist several examples of CNEs which have been recruited from parts of exons (137,138). While GRBs are depleted of TEs, it seems that TEs that have been retained have significantly contributed to lineage-specific CNE evolution (139). Of the CNEs arising since split of eutheria and marsupials, 16% contain recognizable TEs. This is in contrast to 0.7% of the CNEs which had an orthologous marsupial CNE. These TE-derived, lineage-specific CNEs may underlie some of the innovative features responsible for eutherian-specific morphology and neural development.
CNE recruitment varies across lineages, with primates appearing to gain CNEs at particularly high rates (140). This recruitment tends to be enriched around different genes in different lineages, although CNEs are also gained around a conserved core of developmental genes (141). Furthermore, Lowe et al. (142) have proposed that there have been three distinct periods of regulatory innovation during vertebrate evolution: CNEs appear to have been preferentially recruited around genes that encode for TFs and key developmental regulators during early vertebrate evolution, then to cell signaling genes, and then to genes involved in post-translational modifications during placental mammal evolution. These patterns underline the importance of CNE recruitment in shaping vertebrate evolution. Further, it has been suggested that following the initial recruitment of a CNE, it’s flanking sequences could be co-opted to regulatory function over time in a lineage-specific manner (143). Under this model, the recruited flanking sequence would increase the modularity and complexity of the overall regulation by the element. This could explain that the core conserved regions of vertebrate enhancers are often sufficient to drive gene expression in reporter assays (144). However, these observations could also be explained by flanking sequences being under a relatively lower selection pressure than the core CNE region.
Taken together, the literature suggests that CNEs are recruited from any existing genomic sequence within the reach of the target gene if it contributes an advantageous alteration in gene expression. Furthermore, the alteration of key developmental genes’ expression can and does underlie the basis of many lineage-specific traits. Still, however, the mechanism by which CNE recruitment occurs remains unknown.
CNE modifications and losses
Despite the high degree of sequence and functional maintenance typical of CNEs, substitutions and deletions still occur, albeit at a much reduced rate. Such changes can be without apparent phenotype, but equally can be disruptive, and likely underlie many lineage-specific traits.
Losses of individual CNEs can be sufficient for alterations to anatomical structures. Deletions are thought to underlie penile spine loss (145) and foot digit shortening in humans (146). More extensive changes have been observed in other animals: snake limblessness, for example, is associated with partial and complete deletions of CNEs that regulate limb development genes (147–150). For example, a vertebrate conserved SHH enhancer has a few substitutions in snakes with vestigial hindlimbs, but a short deletion and multiple changes in snakes that have undergone complete limb loss (148). Interestingly, this ZRS enhancer is the same element implicated in human polydactyly. In stickleback, the deletion of a CNE, regulating the developmental gene PITX1, leads to pelvic reduction. The loss of this element, which may be beneficial due to reduced predation and calcium availability in freshwater environments, has occurred multiple times in independent freshwater populations, with strong evidence that PITX1 regulatory mutations are under positive selection in these populations (151). CNEs are even recurrently lost across species, with Hiller et al. discovering hundreds of CNEs independently lost in more than one mammal (152). A recent study demonstrated that many such losses could be linked to common anatomical changes. Those included an independent loss of an element proximal to EGR2, a TF linked to forelimb morphology, in manatees and dolphins. The deletions are postulated to play a role in elbow structure modifications common to both species (153). Additionally, the vast phenotypic diversity of teleost fish has been hypothesized to be related to large-scale loss of and increased substitution rates in ancestral vertebrate CNEs (154). The recent whole genome sequencing of one of the more morphologically distinct teleosts, the seahorse (Hippocampus comes), revealed a high degree of CNE loss compared to other teleost fish. Many of these CNEs cluster around key retained developmental genes, and may have contributed to the extensive morphological changes in seahorses (155).
As new elements are regularly recruited, a subset will have near-equivalent functions to extant CNEs, and thus could potentially replace them. This appears to be the case for nPE1 and nPE2, mammalian enhancers of POMC derived from TE (156). These elements have likely replaced ancestral enhancers, which are lost in mammals but still maintained in other vertebrates (157). The CNE turnover model posits that in the long term no sequences are indispensable, and that such turnover of elements may be quite common (129). This would explain why expression patterns are often preserved across species despite large changes in cis-regulation (158).
Several thousand CNEs have undergone bursts of lineage-specific positive selection in humans (159–164). These elements, referred to as human accelerated regions, are highly conserved in most mammals and often in other vertebrates, but have rapidly accumulated substitutions in humans. A number of these regions have been tested using transgenic mouse models, comparing how the human sequence drives gene expression compared to the equivalent in chimpanzee. This has demonstrated that some, often very highly conserved, CNEs have divergent function in humans, seemingly contributing to several human-specific traits, including bipedalism and increased brain size (165,166).
PERSPECTIVES
Most CNEs act as developmental enhancers, however, this does not explain the extreme levels of their conservation. Methodological advances for studying those elements in vivo are necessary: recently proposed genome-wide editing techniques for large-scale interrogation of regulatory elements (167,168) could prove promising towards addressing the role of CNEs on an individual or on a per-GRB basis.
We conclude by outlining questions, the answers to which may further our understanding of CNEs and their function:
What is the origin of selection pressure on CNEs? CNEs that form clusters around developmental genes share the unknown source of purifying selection. According to this criterion, they are not a set of elements with different function. However, we still lack understanding on what is being selected for in CNEs and how.
How ancient is the mode of regulation that includes CNEs? There are CNE-like elements in plants and some fungi, which cluster around genes involved in morphogenesis. We still do not know where in evolution the phenomenon of extreme non-coding conservation emerged, but the available evidence points to early emergence in multicellular eukaryotes.
Are CNEs that belong to the same GRB more similar to each other compared to CNEs found in other GRBs? Most CNEs are single copy elements (10) although there is a subset of non-unique CNEs in the human genome that appear to be the result of whole genome duplication events (169). We currently do not know what are the sequence properties of CNEs within different GRBs and whether different sequence features within CNEs play fundamental roles in the regulation of corresponding target gene(s).
What is the sequence grammar of extreme non-coding conservation? Our grasp of the importance of spacing, orientation and copy number of TFBS for CNEs function remains rudimentary. It is currently unknown whether the underlying organization of consensus motifs within CNEs could have potential impact on their functionalities.
What is the role (if any) for CNEs in the folding, establishment or maintenance of TADs? TAD structures are lost during mitosis, indicating that TADs must consistently refold after each cell division. The principles that guide TAD folding and maintenance are still largely unknown. Given the correspondence between GRBs and TADs, it would be tempting to investigate the possible role of CNEs in TAD formation.
What is the role of CNEs in disease pathogenesis? Recent studies have demonstrated that when elements that define TAD boundaries are disrupted, distinct developmental disorders and cancer arise (170–173). Given the role of CNEs in diseases and the correspondence between GRBs and TADs, it will be interesting to explore how enhancer–promoter interactions are perturbed in various diseases, in the context of GRBs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We are indebted to Drs Ya Guo, Nathan Harmston and Matthias Merkenschlager for comments on the manuscript. Thanks to Drs Brant Faircloth and John McCormack for help with the figure for CNEs and phylogenomics. We apologize to our colleagues whose work has not been discussed due to space limitations.
FUNDING
Medical Research Council [MC_UP_1102/1 to D.P., B.L., 1584095 to A.N., 1584144 to J.K.]. EU project ZF-Health [FP7/2010–2015, 242048 to G.T., B.L.]; Wellcome Trust [P55504_WCMA]. The open access publication charge for this paper has been waived by Oxford University Press - NAR.
Conflict of interest statement. None declared.
Present addresses:
Dimitris Polychronopoulos, Genomics England, Charterhouse Square, London EC1M 6BQ, UK.
Ge Tan, Functional Genomics Center Zurich, ETH Zurich and University of Zurich, Winterthurerstrasse 190, Zurich 8057, Switzerland.
REFERENCES
Author notes
These authors contributed equally to the paper as second authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments