





Abstract
Cpf1 nucleases were recently reported to be highly specific and programmable nucleases with efficiencies comparable to those of SpCas9. AsCpf1 and LbCpf1 require a single crRNA and recognize a 5′-TTTN-3′ protospacer adjacent motif (PAM) at the 5′ end of the protospacer for genome editing. For widespread application in precision site-specific human genome editing, the range of sequences that AsCpf1 and LbCpf1 can recognize is limited due to the size of this PAM. To address this limitation, we sought to identify a novel Cpf1 nuclease with simpler PAM requirements. Specifically, here we sought to test and engineer FnCpf1, one reported Cpf1 nuclease (FnCpf1) only requires 5′-TTN-3′ as a PAM but does not exhibit detectable levels of nuclease-induced indels at certain locus in human cells. Surprisingly, we found that FnCpf1 possesses DNA cleavage activity in human cells at multiple loci. We also comprehensively and quantitatively examined various FnCpf1 parameters in human cells, including spacer sequence, direct repeat sequence and the PAM sequence. Our study identifies FnCpf1 as a new member of the Cpf1 family for human genome editing with distinctive characteristics, which shows promise as a genome editing tool with the potential for both research and therapeutic applications.
INTRODUCTION
The ability to introduce targeted genomic sequence changes into living cells and organisms provides a powerful tool for biological research as well as a potential avenue for therapy of genetic diseases (1–3). CRISPR–Cas genome editing as a fledgling technology has redefined genetic and molecular biology research due to its simplicity and ease of design (2,3). According to the configuration of their effector modules, CRISPR–Cas systems can be classified into two groups: class 1 effectors, which utilize multi-protein complexes, and class 2 effectors which rely on single-component effector proteins such as the well-characterized Cas9 nuclease. CRISPR–Cas9 systems consist of a multi-domain endonuclease, along with CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA), with the ability to cleave both strands of the target DNA sequence. A guanine-rich protospacer adjacent motif (PAM) sequence at the 3′ end adjacent to the target site is essential for the cleavage process of Streptococcus pyogenes Cas9 (SpCas9), the most commonly used Cas9 at present.
Cpf1 is a class 2 CRISPR–Cas family of nucleases which was reported recently to be highly specific programmable nucleases with efficiencies comparable to those of the SpCas9 nuclease (4,5). It differs from Cas9 in several ways (4,6–8). It requires only a single crRNA without a tracrRNA and recognizes thymine-rich PAM sequence at the 5′ end of the protospacer, which increases the selection of CRISPR-endonuclease-editable genomic sites, compared with the only choice of CRISPR-SpCas9. crRNA is composed of a 19 nucleotide (nt) direct repeat followed by a 23–25 nt spacer sequence. Unlike Cas9 which creates blunt-ended cleavage products proximal to the PAM site, Cpf1 generates a staggered double-strand break (DSB) resulting in 5′-overhangs distal to the PAM site. Based on sequence analysis, Cpf1 contains only one RuvC endonuclease domain, which has led to the initial hypothesis that Cpf1 may form a dimer to cleave the two strands of the target DNA. However, recent structural and functional studies show that Cpf1 acts as a monomer and contains a second putative novel nuclease (NUC) domain (9). In contrast, Cas9 uses the RuvC and HNH endonuclease domains to cleave the non-target and target DNA strands, respectively.
GUIDE-seq analysis and targeted deep sequencing using a large number of different crRNAs with two types of Cpf1 (Acidaminococcus sp. BV3L6, AsCpf1 and Lachnospiraceae bacterium ND 2006, LbCpf1) suggest that Cpf1 nucleases are highly specific in human cells (10,11). Therefore, for multiple reasons, Cpf1 may be even better suited for accurate genome editing than Cas9. Among the 16 known Cpf1 family members, AsCpf1 and LbCpf1 have been reported to have DNA mutation inducing activity in mammalian cells (4,12–14). However, the application of Cpf1 may be dramatically limited because AsCpf1 and LbCpf1 require a 5′-TTTN-3′ PAM sequence which is not present in many potential targets. To address this drawback, we sought to identify a novel Cpf1 enzyme for genome editing in human cells. Here, we characterize FnCpf1 (Francisellanovicida U112. Cpf1), which recognizes a more suitable PAM (5′-TTN-3′) in human cells (4), and it may have great potential for further development as a genome editing tool.
MATERIALS AND METHODS
Plasmids encoding Cpf1 and crRNA
Plasmids for the expression of FnCpf1, AsCpf1 and LbCpf1 were obtained from Addgene (Addgene plasmids # 69976, # 69988 and # 69982, respectively). LSL–Cas9–Rosa26TV was also obtained from Addgene (#61408). The crRNA expression cassette (Figure 1A and B) was generated by insertion of PCR products into vector pJET1.2 (CloneJET PCR Cloning Kit, Thermo Fisher Scientific).The direction of crRNA expression cassette was selected by primer specific PCR. The oligonucleotide sequences used are summarized Supplementary data: Tables S1–S4. Plasmid DNA and genomic DNA were isolated by standard techniques. DNA sequencing confirmed the desired specific sequence in the constructs.
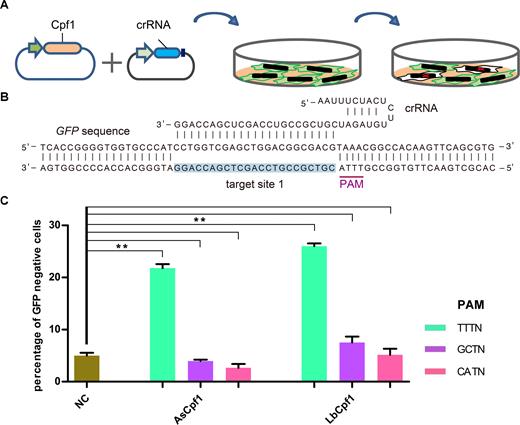
AsCpf1 and LbCpf1-mediated gene editing in human cells. (A) Illustration of the GFP-reporter system for measuring Cpf1-mediated DNA cleavage in human cells. Cpf1-family proteins were generated with the corresponding plasmids expressed in 293-SC1 cells. The corresponding crRNA was expressed droved with U6 promoter. Knock-out efficiency was analyzed using flow cytometry. (B) Schematic diagram of the AsCpf1 crRNA-DNA-targeting complex. The target sequence is shown in blue and the PAM sequence is shown in pink. (C) AsCpf1 and LbCpf1 recognized 5′-TTTN-3′ PAMs. AsCpf1 and LbCpf1 failed to cleave target sites with a 5′-GCTN-3′ PAM or 5′-CATN-3′ PAM, respectively. Error bars, S.E.M.; n = 3; NC, negative control. **P < 0.01.
Cells and cell culture
HEK-293 cells were obtained from ATCC (CAT#CRL-1573), and grown at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (Life Technologies, Carlsbad, CA, USA), 10% heat-inactivated fetal bovine serum, 1% PS (penicillin and streptomycin). HEK-293 cells expressing GFP were generated by lentiviral transduction as previously described (15). Drug-resistant single colonies of transduced HEK-293 cells were isolated and named 293-SC1. To maintain GFP expression, the medium for 293-SC1 culture included puromycin.
Flow cytometry analysis
The flow cytometry protocol was described previously (15). Briefly, 1.8 × 105 293-SC1 cells/well were seeded in 12-well plates on day 1, and transfected with Cpf1 and crRNA expression plasmids by the Transfection Reagent (TurboFect, Thermo Scientific) on day 2. Fresh medium was added to the transfected 293-SC1 cells on day 3. Cells were harvested for flow cytometry or genomic DNAs isolation on day 4. loxP-STOP-loxP-mG/FnCpf1 study was performed as described previously (16).
CAPS analysis and T7E1 nuclease assay for genome editing
Fragments harboring the indel mutation were amplified by PCR using the primer sets listed in Supplemental Table S3. For cleaved amplified polymorphic sequences (CAPS) analysis, 500 ng total of purified PCR products were digested by the appropriate restriction enzymes, and then analyzed on agarose gel. Quantification was based on relative band intensities. Indel percentage was determined by the formula,100 × (1 − sqrt(b + c)/(a + b + c)), where a is the integrated intensity of the undigested PCR product; b and c are the integrated intensities of the cleavage product. For T7 Endonuclease I(T7E1) assay, 500 ng purified PCR products were mixed with 1 ul 10 × NEB#2 buffer and ultrapure water to a final volume of 9.5 ul and were subjected to re-annealing process to enable heteroduplex formation. After re-annealing, products were treated with T7 Endonuclease I. Indel percentage was determined by the formula, 100 × (1 − sqrt(1 − (b + c)/(a + b + c))).
Off-target analysis for FnCpf1
We examined the possibility that FnCpf1 induced off-target mutations. The potential off-target sites were predicted using online software (http://www.rgenome.net/cas-offinder/). The fragments harboring potential off-target sites have been amplified (primer information in Supplementary Table S3) and tested with T7E1.
Statistics
All data were expressed as mean ± s.e.m. Differences were determined by 2-tailed Student's t-test between two groups, or one-way ANOVA followed by post-hoc Bonferroni test for multiple groups. The criterion for statistical significance was *P < 0.05, or **P < 0.01.
RESULTS
Genome editing with AsCpf1 and LbCpf1
Cpf1 orthologs can be programmed to induce DNA double strand breaks (DSBs) at specific genomic loci through a specific crRNA consisting of a 23–25 nt guide segment and a 19 nt scaffold (Supplementary Figure S1). When this sequence-specific RNA-guided endonuclease is targeted to coding regions of the GFP gene in 293-SC1 cells (HEK-293 cells harboring the GFP gene) (15), it can generate frameshift indel mutations, thus leading to loss of fluorescence (Figure 1A). It has been reported that two types of Cpf1 nuclease (AsCpf1 and LbCpf1) could be harnessed to edit genes in human cells with a 5′-TTTN-3′ PAM [4]. We designed a Cpf1 target site in the GFP gene (GFP site 1) with a 5′-TTTN-3′ PAM sequence (Figure 1B), and generated the corresponding crRNA expression plasmids. To test their genome editing efficiency, either AsCpf1 or LbCpf1 expression plasmids (500 ng) plus their crRNA expression plasmids (500 ng) were transfected into 293-SC1 cells. We found that both AsCpf1 and LbCpf1 could successfully trigger GFP gene inactivation in human cells with efficiencies of 22% and 26%, respectively (Figure 1C), which is consistent with the literature (4). While, only two target sequences (including one near the stop codon) in GFP satisfy the PAM requirement, thus we sought to test whether non-canonical PAMs could lead to DNA cleavage. We therefore tested two crRNAs with non-canonical PAMs (GCTN-3′ and 5′-CATN-3′). Our results showed that both AsCpf1 and LbCpf1 failed to cleave a GFP sites containing these non-canonical PAMs. The limited choice of targeting sites for AsCpf1 and LbCpf1 to a 5′-TTTN-3′ PAM may be a bottleneck for the application of Cpf1-mediated genome editing. Thus, we sought to identify additional Cpf1-family proteins for human genome editing, which requires a simpler PAM for their nuclease activities.
FnCpf1 has genome editing activity in human cells
An additional member of the Cpf1 family, FnCpf1, has been well investigated (4,17,18): Its canonical PAM sequence is 5′-TTN-3′. In the GFP gene, we only identified two 5′-TTTN-3′ sequences, while, there were fifty-three 5′-TTN-3′ sequences. Thus, if FnCpf1 or a variant form could be harnessed for gene editing in human cells, it could permit greater flexibility in the selection of target sequence. It has been reported that FnCpf1 cannot introduce detectable levels of nuclease-induced indels in the DNMT1 locus with target sequence of CTGATGGTCCATGTCTGTTACTC (labeled as DNMT1–3) in human cells, while with the recombinant FnCpf1 protein, crRNA and target sequence, cleavage activity could be detected in vitro (4,17). To investigate whether there is any locus bias in FnCpf1 activity, we designed and generated a crRNA expression cassette to target the GFP gene with a 5′-TTN-3′ PAM (site 2) in human cells (Figure 2A). Surprisingly, we found that FnCpf1 has robust DNA cleavage activity to target GFP gene, which is not consistent with the previous studies at the DNMT1 locus (4).
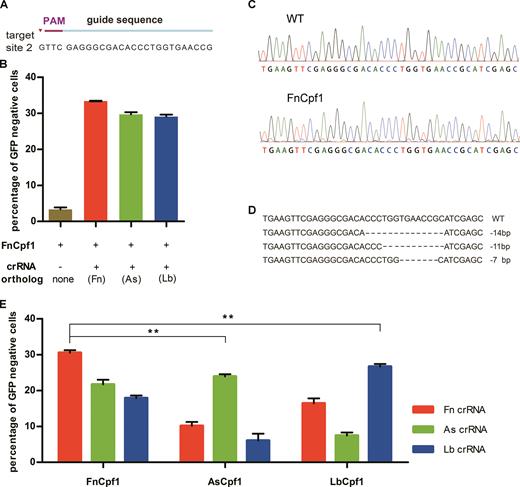
FnCpf1 possesses DNA cleavage activity in human cells. (A) Sequence of GFP target site 2. (B) Cells were co-transfected with plasmids encoding FnCpf1 and plasmids for crRNA expression target site 2. (C) DNA sequence chromatograms of the fragments harboring the target site obtained from the cells treated with FnCpf1 are in a mass, compared with controls. (D) DNA sequence analysis of single individual GFP-negative colonies. Dashes represent the DNA deletions. The number at the right side of each sequence is the length of indel (−, deletion). (E) Comparison of the activity of FnCpf1 with that of AsCpf1 and LbCpf1. Cells was co-transfected with plasmids encoding Cpf1 orthologs (FnCpf1, AsCpf1 and LbCpf1) and plasmids encoding crRNAs target site 1 in various combinations. Error bars, S.E.M.; n = 3; **P < 0.01.
We next asked whether the direct repeat needed for FnCpf1 activity could be altered. To address this, we used the direct repeat from AsCpf1 and LbCpf1 to target site 2 in GFP and found that the cleavage activity was still maintained (Figure 2B). Then we asked whether there were any dose-dependent effects. We examined different amounts of FnCpf1 and crRNA expression plasmids and observed dose-dependent effects on DNA cleavage (Supplementary Figure S2). This showed that co-transfection of 750 ng plasmids coding for FnCpf1 and 250 ng plasmids for crRNA per well of a 12-well plate was the lowest amount which could lead to the maximum cleavage efficiency of 34% (Supplementary Figure S2). Thus, all further experiments were performed using this condition.
We then investigated the cleavage effects of different Cpf1 family members (FnCpf1, AsCpf1 and LbCpf1) with these three different direct repeats. Because the 5′-TTN-3′ PAM from site 2 cannot be used for AsCpf1 and LbCpf1, which requires a 5′-TTTN-3′ PAM, we next targeted site 1, as it is compatible with all these enzymes. Not surprisingly, under conditions when each Cpf1 ortholog was used with its own direct repeat, the cleavage was the highest (Figure 2E and Supplementary Figure S3), with FnCpf1 having the highest cleavage efficiency. However, FnCpf1 could still lead to robust DNA cleavage with the direct repeats matching AsCpf1 and LbCpf1. Taken together, we conclude that, using exogenous gene (GFP) as an example, FnCpf1 possesses DNA cleavage activity in human cells and its majority of activity could be maintained using direct repeats from some of other Cpf1 family members.
FnCpf1-mediated genome editing at endogenous genes in human cells
Because our results showed that FnCpf1 can target the GFP gene in human cells, we sought to address whether it has targeting activity against endogenous genes in human cells. The original study for testing activity of FnCpf1 was at the DNMT1 locus and EMX1 locus (4). Thus, we sought to validate the activity of the exact target site from the literature (DNMT1–3 and EMX1–2) using FnCpf1 (Supplementary Figure S4). Not surprisingly, only 7% activity and no activity were detected at these two sites, respectively; therefore we examined more sites. We selected additional three genes (DNMT1, RS1 and NRL) with a 5′-TTTN-3′ PAM using CAPS analysis, thus allowing us to study FnCpf1, AsCpf1 and LbCpf1 simultaneously. Our results demonstrated that all these sites could be successfully targeted in human cells with these three Cpf1 proteins (Figure 3 and Supplementary Figures S4 and S5). These results were further confirmed with T7E1 assay at EMX1 and with additional genes (HBB, CCR5, VEGFA and GRIN2b, Supplementary Figure S6). Taken together, these data revealed that FnCpf1 possesses DNA cleavage activity for genome editing in human cells.
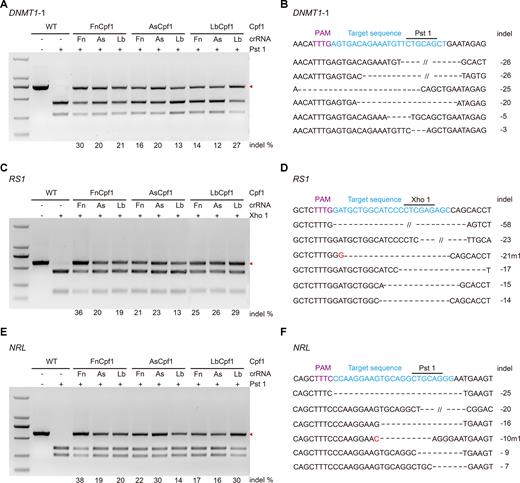
FnCpf1 induced indel mutations on endogenous genes. (A, C, E) Cleaved amplified polymorphic sequences (CAPS) analysis of DNMT1–1, RS1 and NRL loci. Arrow indicated the position of digestion-resistant PCR products. Digestion-resistant bands are the fragment with the indel mutation. Molecular markers in A, C and E are DL2000 (TaKaRa). (B, D, F) Restriction Enzyme cutting site (Pst1, Xho1) are shown in the target DNA sequences of each loci. Sequencing reads show representative mutations of Fncpf1 mediated gene editing with its own crRNA in DNMT1–1, RS1 and NRL loci. Dashes represent the DNA deletions. The number at the right side of each sequence is the length of indel (−, deletion).
The effects of spacer length and direct repeats on FnCpf1-mediated genome editing in human cells
crRNA is one of the two components of the CRISPR-Cpf1 genome-editing system. Compared with the sgRNA for Cas9, the guide RNA for FnCpf1 is notably simpler and only consists of a spacer sequence after the direct repeat sequence. It has been reported that optimizing sgRNA structure could improve CRISPR–Cas9 knockout efficiency (2). Here, in a similar fashion, we systematically investigated the parameters necessary in crRNA for FnCpf1-mediated gene editing in human cells. To investigate the length requirements, we constructed and tested a series of crRNAs with different spacer sequence lengths (14, 17, 20, 21, 22, 23, 24, 25 and 30nt) to target site 1 in GFP (Figure 4A). Surprisingly, crRNA with a 21 nt, but not 23–25 nt spacer sequence, which is commonly used as spacer sequence for Cpf1, achieved the highest GFP disruption at an efficiency 40%. This observation was further confirmed at site 2 (Figure 4B). These results showed that a spacer sequence for FnCpf1 with a length of 21 nt could lead to maximum gene knockout.
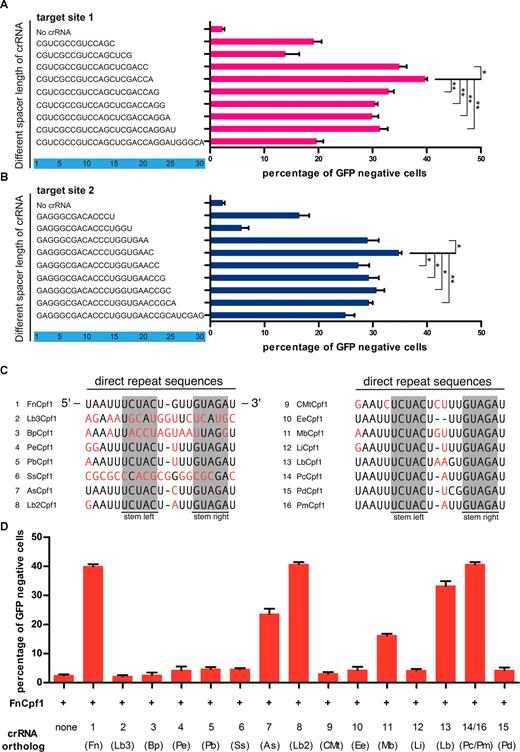
On-target gene editing activities of crRNA with different length of spacer sequence and direct repeat in human cells. (A, B) Lengths and sequences of crRNA spacer regions are shown. Indel frequencies were measured by flow cytometry. The two target sites used are named as previously described. The spacer sequence for FnCpf1 at 21nt could lead to maximum gene editing efficiency. (C) Alignment of direct repeat sequences from the 16 member Cpf1 family. The stem structure is highlighted in gray. Non-conserved sequences are in red. The direct repeats sequence of PcCpf1 (14) is identical with PmCpf1(P.macacae.Cpf1). (D) Cells were co-transfected with plasmids encoding FnCpf1 and plasmids for crRNA expression, of which direct repeats are from 16 Cpf1 members. Error bars, S.E.N.; n = 3; *P < 0.05, **P < 0.01.
The expressed crRNAs ended in a series of U’s to terminate transcription from the U6 promoter. We do not know whether the number of these U’s plays a role for its activity. To answer it, we generated the plasmids with different T (at RNA level, it would be U) and tested them. The results revealed that with different length U, no significant difference has been observed (Supplementary Figure S7A and B).
Because crRNA is typically composed of 19 nt of the direct repeat followed by 23–25 nt of spacer sequence, we were interested in the effects of mutations in the direct repeat sequence. To do this, we combined the direct repeats from 15 additional Cpf1 members with FnCpf1 and investigated the corresponding DNA cleavage efficiency. This revealed that the cleavage efficiencies of direct repeats from AsCpf1 and LbCpf1, Lb2Cpf1 (Lachnospiraceae bacterium MA2020.Cpf1), PcCpf1 (Porphyromonas crevioricanis.Cpf1) with FnCpf1 are all reasonably high (Figure 4C and D). In particular, direct repeats from Lb2Cpf1, PcCpf1 had the same DNA cleavage efficiency as FnCpf1 direct repeats when they were used with FnCpf1, which were further confirmed with additional sites (Supplementary Figure S7C and D). While, under this condition (FnCpf1 plus direct repeats from Lb2Cpf1 or PcCpf1), we don’t know whether the fidelity of FnCpf1would be maintained.
PAMs for FnCpf1-mediated genome editing in human cells
As previously mentioned, the canonical PAM for FnCpf1 is 5′-TT(N)-3′. To investigate the effects of the first two nucleotides (TT) on cleavage specificity, we tested the activity of crRNAs with all possible 5′-NN(N)-3′ combinations (Figure 5A). The results showed that the efficiency of FnCpf1-mediated DNA cleavage of CT(C) PAM was much higher than that of other PAMs except 5′-TT(N)-3′ (Figure 5B), which is consistent with results from recombinant protein-based in vitro assay and Escherichia coli based assay (4,17,18). Specifically, the average efficiency of FnCpf1-mediated DNA cleavage of 5′-CTC-3′ PAM sequence was 21%, compared with 40% cleavage efficiency for 5′-TTN-3′. With the addition of three pairs of targeting sequences with a 5′-CTN-3′ PAM at different locations in the GFP gene, we observed similar results (Figure 5C). Surprisingly, we found that we could obtain 40% of cells with GFP inactivation using crRNAs with a 5′-CTA-3′ PAM sequence.
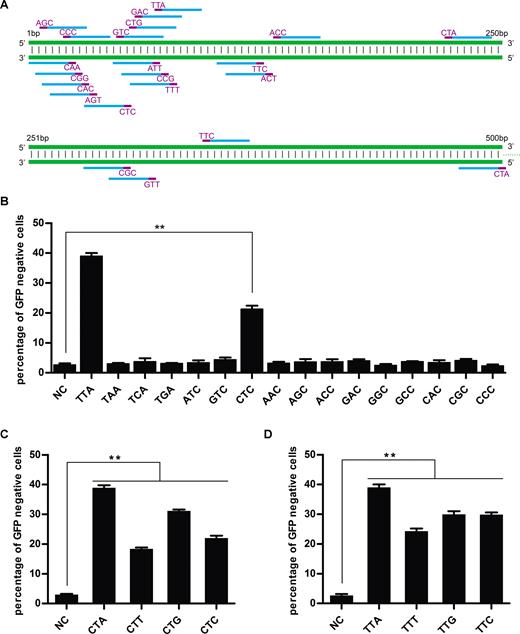
PAMs for FnCpf1-mediated gene editing in human cells. (A) Schematic diagram of targeted sites with different PAMs in GFP. Target sites and PAM sequences are in blue and purple, respectively. The target sequences were show in Supplementary Table S2. (B) Among these 16 PAMs (NNn), CTC PAM has the relative highest level of FnCpf1 mediated DNA cleavage expect TTN(TTA). (C, D) FnCpf1 recognizes a PAM, defined as 5′-YTV-3′. NC, negative control. Error bars, S.E.M.; n = 3; *P < 0.05, **P < 0.01.
We next sought to test the effect of changing the 3′ nucleotide of the PAM sequence. We designed, assembled and tested crRNAs with 5′-TTA-3′, 5′-TTC-3′, 5′-TTG-3′ and 5′-TTT-3′ PAM sequences (Figure 5A). The results showed that 5′-TTT-3′ has low activity, while TTA has higher activity than other PAM sequences (Figure 5D). Interestingly, both 5′-TTT-3′ and 5′-CTT-3′ had a relatively lower cleavage activity compared with 5′-TTV(A/C/G)-3′ or 5′-CTV(A/C/G)-3′, which suggests that 5′-TTT-3′ and 5′-CTT-3′ are not a good PAM sequence choice. We also noticed it was reported that 5′-CTG-3′could be used as non-classical PAM for FnCpf1, specifically, the reaction of FnCpf1 recombinant protein plus single crRNA and the DNA target sequence could lead to the DNA cleavage in vitro (17), which is consistent with our results. Taken together, our study clearly demonstrates that to achieve high cleavage efficiency, FnCpf1 requires a PAM defined as 5′-YTV-3′, where Y represents C or T, and V represents A or C or G. These results update the requirements for FnCpf1 PAM sequence in human cells.
Off-target effects of FnCpf1-mediated genome editing in human cells
The above results demonstrate that FnCpf1 can mediate efficient genome editing in human cells. Next, we wanted to determine the fidelity of FnCpf1 nuclease. To do this, we designed single-nucleotide mismatches within the spacer region of crRNAs (using 21 nt and 23 nt spacers, respectively) targeting site 1 in the GFP gene (Figure 6A and B). Not surprisingly, we found if the mismatches are located distal to the PAM sequence, FnCpf1 could trigger DNA cleavage at imperfectly matched target guide sequences (off-target genomic sites), similar to the off-target effects of CRISPR–Cas9, as CRISPR–Cas9 exhibits increased tolerance of mismatches at sites distal to the PAM. We also found that the 21 nt spacer has higher fidelity, compared with the 23 nt spacer. Specifically, single-nucleotide mismatches (G5A, C10U) in the 21 nt spacer only have 81–84% activity of the completely matched spacer (Figure 6C). While, the same single-nucleotide mismatches in a 23 nt spacer have 98–100% activity. These results also show that FnCpf1 is unable to distinguish the single-nucleotide mismatches between the spacer and targeting sequence, especially single-nucleotide mismatches at the 3′ end of the crRNA. These data collectively suggest that the crRNA with a 21 nt spacer has decreased off-target effects but increased efficiency. Then we sought to know the off-target effects of endogenous genes in human cells with 21 nt spacer. With the prediction of off-target with online software, we investigated it on these five genes and found there are detectable off-target effects of FnCpf1 at endogenous genes (HBB, Off-target 6; CCR5, Off-target 8; EMX1, Off-target 6, Supplementary Figure S8). While, these data revealed the off-target effects were limited, because the cleavage efficiency of off-target sites is far below that of the on-target sites. Further studies are needed to characterize additional potential off-target with genome analysis tool, i.e. whole genome sequencing. Taken together, these data provides a platform for the further development of FnCpf1 as a tool for genome editing in human cells.
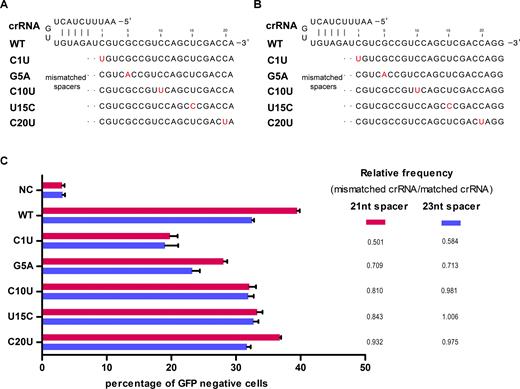
Specificities of FnCpf1 with different crRNAs. (A, B) The activity of single-nucleotide mismatched crRNAs. (C) Effect of crRNA-target DNA match or mismatch on FnCpf1 cleavage activity. NC, negative control. Error bars, S.E.M.; n = 3;
DISCUSSION
Schunder and his colleagues showed that the presence of spacers in the Cpf1 associated CRISPR arrays are similar to prophages, which indicated Cpf1 may be functionally analogous to Cas protein (19). Then AsCpf1 and LbCpf1 but not FnCpf1 were developed as human genome editing tool (4). Surprisingly, we found that FnCpf1 does possess DNA cleavage activity in human cells. Detectable levels of nuclease-induced indels haven’t be observed by a SURVEYOR nuclease assay at the human DNMT1 and EMX1 locus (4), indicating that these particular sites may not be a good choice. While, with CAPS analysis, a method for evaluation the activity of CRISPR/Cas (20,21), we could clearly detect FnCpf1 mediated genome editing activity at multiple endogenous loci (Figure 3, Supplementary Figure S6). Indeed, our further studies indicated that locus bias may explain the inconsistency (Supplementary Figure S6), which highlights the importance of the selection of target sequences.
crRNA used with Cpf1 consists of the spacer sequence (guide sequence) and the direct repeat sequence. We investigated the effect of different lengths of the spacer and direct repeat sequences for FnCpf1. We found that with a 21 nt spacer sequence FnCpf1 has better activity and higher fidelity. Structural studies revealed that the 24 nt spacer sequence and the target DNA strand form a 20 bp, rather than a 24 bp, RNA–DNA heteroduplex (6,9). We speculated that the 21 nt spacer sequence, the target DNA strand and FnCpf1 may form a complex which endows FnCpf1 with improved activity and higher fidelity. Further structural studies will dissect its molecular mechanism. Similar to previous study, our results also highlight the off-target effects of FnCpf1, with single-nucleotide mismatch especially distal to the PAM sequence, although CRISPR–Cpf1 systems has been reported that the genome-wide specificities is reasonably high in human cells (10,11). The off-target effects may be due to bacterial self-protection, because bacteria such as Acidaminococcus sp. BV3L6 (AsCpf1 originates from this species) may facilitate broader immunity from phages with similar genetic backgrounds. While these off-target effects represent a major bottleneck for safe Cpf1-mediated reliable genome editing. The fidelity of FnCpf1 at the genome-wide level remains an open question and parallel comparison of FnCpf1, AsCpf1 and LbCpf1 should be carried out. Also, to minimize off-target effects, special high-fidelity variants of FnCpf1 could be developed to achieve catalytically efficient, highly accurate genome editing.
In addition to target DNA interference activity, Cpf1 was also found to cleave precursor crRNA (pre-crRNA), leading to the generation of mature crRNAs (17). The active site for RNA cleavage is located in the OBD domain, which contributes to the cleavage of phosphodiester bond between A (–19) and U (–20) of the crRNA in vitro (Supplementary Figure S6). However, whether the RNase activity domain can enable structural maturation of the crRNA to guide Cpf1 in mammalian cells is still unknown. We compared the activities between the pre-crRNA with U (-20) and the mature crRNA in three GFP target sites (sites 1, 2, 3, Supplementary Table S2). The results showed there was no difference between these groups (P > 0.05) (Supplementary Figure S9).
The fourth position of the FnCpf1 PAM (the 5′ N in 5′-NTTN-3′) has been reported as preferring T and G (21). However, with the GFP system, we could not verify this preference because there were not enough sites to satisfy the requirement of 5′-ATTN-3′, 5′-TTTN-3′, 5′-GTTN-3′, and 5′-CTTN-3′ in the GFP gene as the PAM for FnCpf1. Furthermore, the selection of the target sequence, i.e. coding at N-terminus or C- N-terminus of GFP, may affect the editing efficiency. To address this lack, we generated another system, loxP-STOP-loxP-mG/FnCpf1 (genome editing tool), which is similar to the previously reported loxP-mT-loxP-mG (16). After the FnCpf1-mediated DNA double strands break via targeting loxP flanking STOP-cassette, the expression of the EGFP gene would be directly driven by the CAG promoter. With this system, we could investigate the PAM effects for the cleavage with an identical target sequence. Specifically, we designed crRNA to target the loxP, which has different adjacent sequences from PAM. Our study showed, with two different target sequences in the fourth position of the PAM (the 5′ N in 5′-NTTN-3′) for FnCpf1, T and G are the preferred nucleotides (Supplementary Figure S10), which is consistent with the literature (21). Our results showed, to achieve high efficiency targeting in the human genome, 5′-KYTV-3′ PAM for FnCpf1 may be preferred.
Recently, it was reported that using a single customized AsCpf1 array, four genes in mammalian cells and three in the mouse brain could be edited simultaneously (14). Here, FnCpf1 uses 5′-KYTV-3′ as the PAM; it presents a greater choice of the target sequence, compared with AsCpf1 and LbCpf1. Therefore FnCpf1 may be more suitable as a versatile genome editing tool for editing several genes simultaneously.
In summary, we found that FnCpf1 can be harnessed for genome editing in human cells. We comprehensively and quantitatively examined FnCpf1 genome editing parameters in human cells, including the spacer sequence, the direct repeat sequence and the PAM. In addition, our data also showed that FnCpf1 still has flaws; it may be possible to further improve its specificity by engineering. Our study provides evidence that FnCpf1, a new member of the Cpf1 family with distinctive characteristics, can act as an effective human genome editing tool for research applications and therapeutics.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Xianglian Ge, Fayu Yang, Liu Zhang, Lianchao Tang and Lulu Cheng for technical assistance.
FUNDING
Chinese National Program on Key Basic Research Project [973 Program, 2013CB967502 to F.G.]; Natural Science Foundation of China [81201181 to F.G., 81473295, 81670882 to Z.M.S., 81670840 to J.L.]; Natural Science Foundation of Zhejiang Province, China [LY13H040013 to J.Z.Z., Q16H040014 to H.T.X.]; Zhejiang Provincial & Ministry of Health Research Fund for Medical Sciences [WKJ2013-2-023 to F.G., 2016KYA146 to D.C.]; Science Technology Project of Zhejiang Province [2014C33260 to Z.M.S., 2017C37176 to F.G.]; Wenzhou city [Y20140633 to F.G., Y20150071 to D.C.]; Wenzhou Medical University [QTJ12011 to F.G.]; Eye Hospital at Wenzhou Medical University [YNZD201602 to F.G.]. Funding for open access charge: Natural Science Foundation of China [81201181 to F.G].
Conflict of interest statement. None declared.
REFERENCE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments