





Abstract
More than 100 distinct chemical modifications to RNA have been characterized so far. However, the prevalence, mechanisms and functions of various RNA modifications remain largely unknown. To provide transcriptome-wide landscapes of RNA modifications, we developed the RMBase v2.0 (http://rna.sysu.edu.cn/rmbase/), which is a comprehensive database that integrates epitranscriptome sequencing data for the exploration of post-transcriptional modifications of RNAs and their relationships with miRNA binding events, disease-related single-nucleotide polymorphisms (SNPs) and RNA-binding proteins (RBPs). RMBase v2.0 was expanded with ∼600 datasets and ∼1 397 000 modification sites from 47 studies among 13 species, which represents an approximately 10-fold expansion when compared with the previous release. It contains ∼1 373 000 N6-methyladenosines (m6A), ∼5400 N1-methyladenosines (m1A), ∼9600 pseudouridine (Ψ) modifications, ∼1000 5-methylcytosine (m5C) modifications, ∼5100 2′-O-methylations (2′-O-Me), and ∼2800 modifications of other modification types. Moreover, we built a new module called ‘Motif’ that provides the visualized logos and position weight matrices (PWMs) of the modification motifs. We also constructed a novel module termed ‘modRBP’ to study the relationships between RNA modifications and RBPs. Additionally, we developed a novel web-based tool named ‘modMetagene’ to plot the metagenes of RNA modification along a transcript model. This database will help researchers investigate the potential functions and mechanisms of RNA modifications.
INTRODUCTION
More than 100 different types of RNA modifications have been characterized across all living organisms (1–5). RNA modifications occur in diverse RNA molecules, including mRNAs, tRNAs, rRNAs, lncRNAs and snoRNAs (1–8). Increasing numbers of studies have demonstrated that RNA modifications play important roles in RNA splicing, protein localization and translation, stem cell pluripotency and human diseases (1–12).
To determine the global landscape of RNA modifications, many studies have recently developed transcriptome-wide sequencing technologies (e.g, Pseudo-seq, Ψ-seq, CeU-seq, Aza-IP, MeRIP-seq, m6A-seq, miCLIP, m6A-CLIP, RiboMeth-seq, Nm-seq and m1A-seq) to identify distinct epitranscriptomic marks (4,5,9,12–19). These new methods have helped researchers to identify the genomic locations of RNA modifications and reveal the distinct distributions of various modification types (e.g. Ψ, m6A, m5C, 2′-O-Me and m1A) throughout the transcriptome. Moreover, in combination with other emerging technologies and tools (e.g. CLIP-seq) (20), researchers have used these technologies to identify novel RNA-modifying enzymes and their targets, and have revealed the spatial-temporal dynamics of distinct RNA modifications under different physiological and pathological conditions (1–12). Although these sequencing technologies have provided comprehensive profiling of valuable data for functional epitranscriptomic investigations, the integration of these large-scale data sets for the exploration of the prevalence, mechanisms and functions of various modifications remains a daunting challenge.
In this study, we developed RMBase v2.0 to perform a large-scale integration of RNA modification sites derived from high-throughput epitranscriptome sequencing data that covered 13 species including humans, mice, zebrafish, yeast, etc. (Figure 1, Table 1). RMBase provides web interfaces that display the maps of RNA modifications for various cell types. Moreover, by integrating miRNA targets, RNA-binding protein (RBP) binding sites, single-nucleotide variations (SNVs) and genome-wide association study (GWAS) data, RMBase can be used to explore the relationships between these data and RNA modification sites. Additionally, we also developed novel web-based tools that include the ‘modMetagene’, ‘Motif’ module and genome browser to visualize metagene profiles, logos of modification motifs and various types of genomic features. RMBase v2.0 is expected to help researchers investigate the potential functions and mechanisms of RNA modifications.
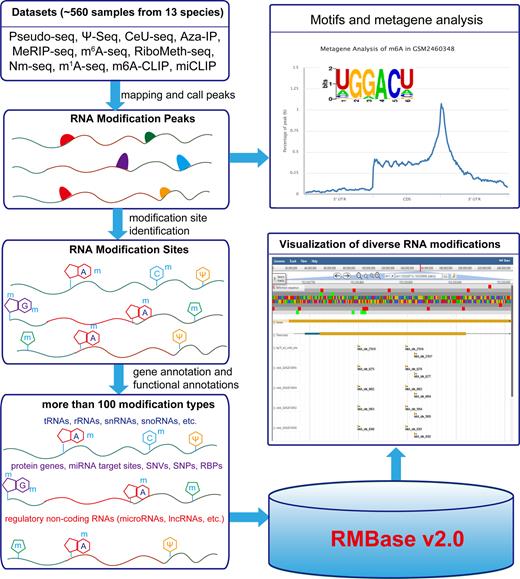
The scheme of the RMBase v2.0 workflow. RMBase v2.0 provides the comprehensive transcriptome-wide landscape of more than 100 types of RNA modifications. All results generated by RMBase v2.0 are deposited in MySQL database and displayed in the visual browser and web pages.
The modification site statistics in RMBase v2.0. The statistical data indicating the number of each RNA modification type for 13 species. m6A is N6-methyladenosine methylation, m1A is N1-methyladenosine methylation, m5C is 5-methylcytosine methylation, Ψ is pseudouridine modification and 2′-O-Me is 2′-O-methylation and ‘other types’ contains diverse rare modification types
| Species | m6A | m1A | m5C | Ψ | 2′-O-Me | Other types |
|---|---|---|---|---|---|---|
| Human | 477 452 | 2574 | 680 | 4128 | 4795 | 525 |
| Mouse | 490 704 | 1052 | 97 | 3320 | 59 | 435 |
| Rhesus | 38 838 | / | / | / | / | / |
| Chimpanzee | 38 369 | / | / | / | / | / |
| Rat | 60 769 | / | / | / | / | / |
| Pig | 121 409 | / | / | / | / | / |
| Zebrafish | 43 027 | / | / | / | / | / |
| S. cerevisiae | 67 671 | 1220 | 211 | 2122 | 242 | 1864 |
| Fly | 6798 | / | / | / | / | / |
| A. thaliana | 20331 | / | / | / | / | / |
| S. pombe | / | 565 | / | / | / | / |
| E. coli | 2173 | / | / | / | / | / |
| P. aetuginosa | 5814 | / | / | / | / | / |
| Species | m6A | m1A | m5C | Ψ | 2′-O-Me | Other types |
|---|---|---|---|---|---|---|
| Human | 477 452 | 2574 | 680 | 4128 | 4795 | 525 |
| Mouse | 490 704 | 1052 | 97 | 3320 | 59 | 435 |
| Rhesus | 38 838 | / | / | / | / | / |
| Chimpanzee | 38 369 | / | / | / | / | / |
| Rat | 60 769 | / | / | / | / | / |
| Pig | 121 409 | / | / | / | / | / |
| Zebrafish | 43 027 | / | / | / | / | / |
| S. cerevisiae | 67 671 | 1220 | 211 | 2122 | 242 | 1864 |
| Fly | 6798 | / | / | / | / | / |
| A. thaliana | 20331 | / | / | / | / | / |
| S. pombe | / | 565 | / | / | / | / |
| E. coli | 2173 | / | / | / | / | / |
| P. aetuginosa | 5814 | / | / | / | / | / |
| Species | m6A | m1A | m5C | Ψ | 2′-O-Me | Other types |
|---|---|---|---|---|---|---|
| Human | 477 452 | 2574 | 680 | 4128 | 4795 | 525 |
| Mouse | 490 704 | 1052 | 97 | 3320 | 59 | 435 |
| Rhesus | 38 838 | / | / | / | / | / |
| Chimpanzee | 38 369 | / | / | / | / | / |
| Rat | 60 769 | / | / | / | / | / |
| Pig | 121 409 | / | / | / | / | / |
| Zebrafish | 43 027 | / | / | / | / | / |
| S. cerevisiae | 67 671 | 1220 | 211 | 2122 | 242 | 1864 |
| Fly | 6798 | / | / | / | / | / |
| A. thaliana | 20331 | / | / | / | / | / |
| S. pombe | / | 565 | / | / | / | / |
| E. coli | 2173 | / | / | / | / | / |
| P. aetuginosa | 5814 | / | / | / | / | / |
| Species | m6A | m1A | m5C | Ψ | 2′-O-Me | Other types |
|---|---|---|---|---|---|---|
| Human | 477 452 | 2574 | 680 | 4128 | 4795 | 525 |
| Mouse | 490 704 | 1052 | 97 | 3320 | 59 | 435 |
| Rhesus | 38 838 | / | / | / | / | / |
| Chimpanzee | 38 369 | / | / | / | / | / |
| Rat | 60 769 | / | / | / | / | / |
| Pig | 121 409 | / | / | / | / | / |
| Zebrafish | 43 027 | / | / | / | / | / |
| S. cerevisiae | 67 671 | 1220 | 211 | 2122 | 242 | 1864 |
| Fly | 6798 | / | / | / | / | / |
| A. thaliana | 20331 | / | / | / | / | / |
| S. pombe | / | 565 | / | / | / | / |
| E. coli | 2173 | / | / | / | / | / |
| P. aetuginosa | 5814 | / | / | / | / | / |
MATERIALS AND METHODS
Integration of the public epitranscriptome sequencing data sets and genome sets
We manually collected high-throughput Pseudo-seq, Ψ-seq, CeU-seq, Aza-IP, MeRIP-seq, m6A-seq, m1A-seq, miCLIP, m6A-CLIP, RiboMeth-seq and Nm-seq data from the Gene Expression Omnibus (GEO) and Sequence Read Archive (SRA) databases (21). The barcodes and 3′-adapters of the raw sequencing data were clipped using cutadapt software (22). All trimmed reads were aligned to the genomes using hisat2 (23) and the mapping results were then converted into BAM format for display in the genome browser and peak calling using samtools (24). Other known RNA modification sites were integrated and curated as described in our RMBase v1.0 (25). The genome sequences and annotations of all 13 species were downloaded from the UCSC genome browser (26), GENCODE (27) and Ensembl databases (28) (Supplementary Table S1).
Identification and annotation of modification sites
The m6A modification peaks were called with the exomePeak program (29) with strict criteria (false discovery rate (FDR) <0.05, P-value <0.01 and fold change (FC) >2). To locate the m6A modification sites in a genome, we predicted the exact m6A positions from the m6A-seq or MeRIP-Seq peaks by searching for consensus RRACH motifs (where R denotes A or G and H denotes A, C or U) (18,30) among the 13 species. Similarly, the m1A modification sites of four species were identified from the m1A-seq peaks by searching for the GAAGAAG motif (14,19). We performed de novo motif identifications of the m6A and m1A peak data by using the HOMER software (31) to obtain their position weight matrices (PWMs) and accurate motif regions. We used these PWMs to score the m6A and m1A modification sites.
We assigned all modification sites to multiple types of genes, including tRNAs, rRNAs, Mt-tRNAs, Mt-rRNAs, scRNAs, snRNAs, snoRNAs, microRNAs (miRNAs), lincRNAs, misc_RNAs, protein-coding genes, processed_transcripts, pseudogenes and gene regions covering CDS, 3′ UTR, 5′ UTR, intron, exon and intergenic region.
Association analysis of the RBP and miRNA binding sites with the RNA modifications
The RBP-RNA and miRNA-target interactions that were supported by the CLIP-seq data were downloaded from our starBase platform for humans and mice (32,33) (Supplementary Table S2). To study the relationships between the RBPs and RNA modifications, we intersected their binding sites with curated and identified RNA modification sites to obtain regulatory pairs. We also annotated the RBPs that were putatively affected by the m6A modifications as readers, writers and erasers to facilitate the queries and use by researchers. A similar workflow was applied to the interactions between the miRNA targets and RNA modifications.
Identification of disease-related SNVs and SNPs associated with RNA modification sites
To investigate the relationships of disease-related SNVs and SNPs with RNA modifications, we collected cancer somatic mutations from the COSMIC database (34) and previous publications (35,36). As described in our previous study (37), the human disease/trait-associated SNPs were curated from published GWAS data provided by the NHGRI GWAS Catalog (38), Johnson and O’Donnell (39), dbGAP (40) and GAD (41), as well as the SNPs in linkage disequilibrium (LD, r2 ≥ 0.5; Supplementary Table S2) with reported disease-related loci that were selected in at least one of the four populations genotype data (CEU, CHB, JPT and YRI) from the HapMap project (release 28) (42). All SNVs and SNPs were intersected with the RNA modification regions that were extended an additional 10nt in both the 5′- and 3′-directions for each modification site to identify the SNVs and SNPs that might interact with the RNA modifications. The modification regions were defined as described in our previous RMBase (25).
Construction of a web-based function tool and the RMBase genome browser
We developed ‘modMetagene’ to present a metagene plot of the RNA modifications along a transcript model from uploaded user data. We also improved the web-based tool ‘modAnnotation’ to identify and annotate the modification sites that were based on all of the RNA modification sites recorded in our database.
We used JBrowse (43), which is a fast and embeddable genome browser that was built completely with JavaScript and HTML5, to construct the improved RMBase Genome Browser that is used to integrate and display reference sequences, RNA modification sites, protein-coding genes, transcripts, modification sequencing data and aligned sequencing reads.
Database and web interface implementation
All data sets were processed and stored in a MySQL Database Management System. The database query and user interface were developed using PHP and JavaScript. The query result table is based on jQueryUI and DataTables and is a highly flexible tool for sorting and filtering search results.
DATABASE CONTENT AND WEB INTERFACE
Comprehensive atlas of various types of RNA modifications
By manually collecting and identifying RNA modification sites from high-throughput epitranscriptome sequencing data, we obtained a total collection of 1,397,244 RNA modification sites, of which covered more than 100 types of chemical modifications among the 13 species (Table 1) including ∼1 373 000, ∼5400, ∼9600, ∼1000 and ∼5100 m6A, m1A, Ψ, m5C and 2′-O-Me with sites, respectively. To study the distributions of RNA modifications on the transcript products, we mapped their sites onto the genomic coordinates of the genes with comprehensive annotated information such as gene types and regions. Our results demonstrated consistencies with previous studies in that the m6A, Ψ and 2′-O-Me modifications tended to occur in protein-coding genes, whereas the m5C modifications often appeared among tRNA molecules.
Exploring the associations between RNA modifications and RBPs
To investigate whether RNA modifications influence the function of RBPs or vice versa, the relationships between CLIP-Seq experimentally supported RBP binding sites (32,33) and all RNA modification sites from RMBase were constructed. We located all of the identified modification sites in the binding regions of the RBPs and found that thousands of RNA modifications were associated with RBPs. In human cells, RMBase characterized regulatory relationships between ∼30 RNA modification types that included m1A, m6A, 2′-O-Me, m5C and 120 RBPs (Supplementary Table S3). In RMBase, many RBPs bound to thousands of RNA modification sites, such as AGO, and m6A writers (e.g. METTL3 and METTL14) and readers (e.g. YTHDF1 and YTHDF2; Supplementary Table S3).
Selection of RNA modifications associated with disease-related SNPs or SNVs
Some RNA modifications have also been found to be correlated with a variety of human diseases and cancers, such as leukemia, glioblastoma and colon cancer (44,45). To investigate the clinical connections between RNA modifications and SNPs as well as SNVs, we systematically drew the distributions of the SNP and SNV coordinates around the RNA modification sites. We identified 192 283 and 1862 sites of RNA modifications assembled around disease-associated SNVs and SNPs, which included somatic point mutations, such as substitutions and deletions. Additionally, we obtained an overview that at least 10 RNA modification types were potentially correlated with dozens of diseases or cancers, such as melanoma (MEL), lung adenocarcinoma (LUAD) and uterine corpus endometrial carcinoma (UCEC) (Supplementary Table S4).
Web interfaces for web-based modules that were developed to explore different types of RNA modification sites
In RMBase v2.0, we provide multiple user-friendly web interfaces to help users explore RNA modification sites that have been identified from high-throughput epitrancriptome sequencing data. In most of the modules deposited on the web interface, the users are allowed to choose their favored organism to browse the corresponding relationships between RNA modifications and multiple interacting factors, such as RBPs, microRNA targets, SNPs and SNVs.
The interface provides users with six basic web-based modules (m6A, m1A, m5C, 2′-O-Me, Pseudouridine/Ψ and otherType) to quickly retrieve the RNA modification sites from various modification types. In the result pages of these modules, users can browse genomic coordinates, associated genes, located gene regions, sequence contexts, motif scores and supporting evidence of RNA modifications. The motif scores provided by RMBase in m6A and m1A web pages will improve the accuracies of analyses of modification prediction.
To display the consensus sequence preferences of the modification sites of m6A and m1A under different cell conditions, we provided the ‘Motif’ module to visualize the PWMs and the distribution patterns of the transcripts (Figure 2). The identified PWMs can be evaluated in many aspects, including P-values, percentages of targets and backgrounds.
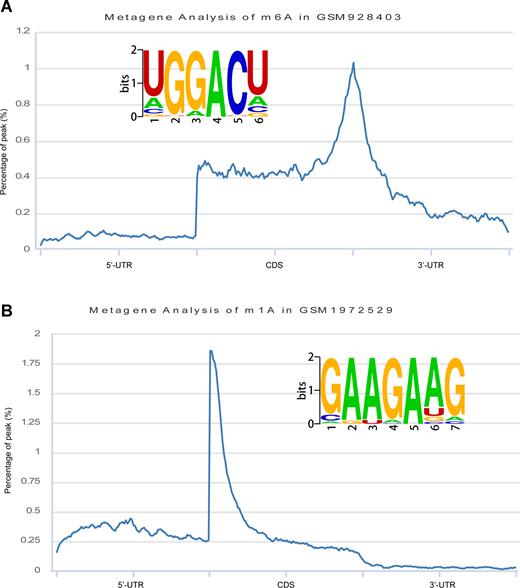
De novo motif logos identified and metagene analyses of RNA modifications. (A) The m6A modification motif and metagene. (B) The m1A modification motif and metagene.
The constructed clinical connections between RNA modifications and SNPs as well as SNVs were placed in the ‘modVar’ module. In the result pages, RNA modification sites were connected to SNPs or SNVs with information about disease types, mutation types, mutation regions and the relative distances between these features.
Additionally, the identified regulatory relationships between RNA modifications and post-transcriptional regulation-associated factors, including RBPs and miRNAs were deposited in ‘modRBP’ and ‘modMirTar’. In these modules, users can inspect the RBPs or miRNAs of interest in terms of whether they have interactions with any RNA modifications.
Web-based analysis tools developed for epitranscriptome sequencing data
To facilitate the research on RNA modifications, we developed a web-based tool suite termed ‘modTool’ that consists of ‘modMetagene’ and ‘modAnnotation’ by employing bin strategies and gene annotation files. The tool suite allows users to upload their identified RNA modification sites, which are formatted into the BED6 format and take up less than 2MB disk volume. After a few seconds to minutes of running, the ‘modMetagene’ sub-module displays the analysis results with metagene plots, and the ‘modAnnotation’ sub-module was displays its results with a data table that is similar to that of the m6A web-based module. Additionally, users can export or download the analysis results for use in downstream analyses.
RMBase genome browser
To facilitate the comparative analyses of the RNA modification sites and exploration epitranscriptome sequencing datasets, we developed an improved genome browser in RMBase v2.0. We provide users with an integrated view of various genomic features including genomic coordinates and genome-wide experiment densities of RNA modification sites, and information about genome and gene annotations downloaded from GECODE or Ensembl. For example, Figure 3 illustrates the visualization of the ‘2-O-Me_site_3709’ modification site located within rRNA.
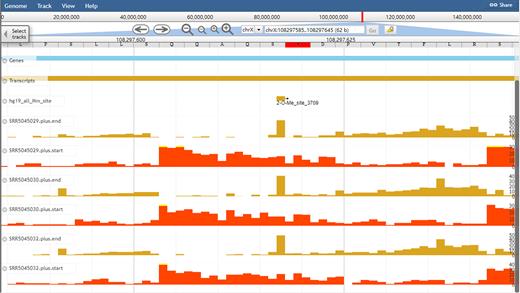
An example of the display of RNA modification sites and epitranscriptome data in the improved RMBase genome browser. Visualization of the genomic context of a ‘2-O-Me_site_3709’ modification site located within rRNA using the RMBase browser.
EXAMPLE APPLICATIONS
M6A modifications occurred along long-noncoding RNAs
While m6A modification is well characterized in mRNA, there are fewer reports on m6A in long-noncoding RNAs (lncRNAs). Thus, we filtered the ‘lincRNA’ in the ‘modGene’ web page, and found that 15 lincRNAs were associated with >50 RNA modification sites (Supplementary Figure S1). We were able to recapitulate the highly methylated lincRNA XIST that has been reported in a recent study (46). The results page sorted by the number of modification sites revealed that the XIST contains 89 m6A modification sites. Additionally, a recent study (46) revealed that m6A RNA methylation promotes XIST-mediated transcriptional repression. Thus, we speculate that other highly methylated lincRNAs identified by RMBase may play important roles in various biological processes.
M6A modification sites bound by RNA-binding proteins
The function of m6A is heavily dependent on m6A associated proteins that are characterized as readers, writers and erasers. Consequently, we used the ‘modRBP’ web page to explore the connection between m6A modification sites and related RNA binding proteins (RBPs). We found that many RBPs, as well as known m6A-related RBPs (e.g. YTHDC1, YTHDF1, METTL3/14, WTAP and ALKBH5), interacted with a large number of m6A sites (Supplementary Table S3). Interestingly, consistent with a recent report about FMR1 as a sequence-context-dependent m6A reader (47,48), we also discovered FMR1 bound to 74,460 m6A modification sites (Supplementary Figure S2).
DISCUSSION AND CONCLUSIONS
By integrating and analyzing numerous high-throughput epitranscriptome sequencing data and collecting data from public resources, RMBase v2.0 provides a gallery of RNA modification marks that cover more than 100 types of RNA modifications on transcript products and revealed that RNA modifications are involved in complex post-transcription regulatory networks.
In comparison to other databases and our previous release version (RMBase v1.0) (25), the advances and improvements of RMBase v2.0 are listed as follows: (i) RMBase v2.0 expanded the previous version by up to 10-fold with ∼600 datasets from 47 studies in 13 species. (ii) N1-methyladenosine (m1A) is one of the most prevalent post-transcriptional modifications in ncRNAs and mRNAs. We first integrated ∼5000 m1A modification sites identified from m1A-seq data into RMBase v2.0. (iii) RMBase v2.0 integrated over 4000 2′-O-Me sites identified from Nm-seq data (16) and provides the first map of the 2′-O-Me sites in mRNAs and other regulatory ncRNAs. (iv) We performed de novo motif identifications of modification sites of m6A and m1A and identified thousands of motif matrices and sequences. We also constructed a module called ‘Motif’ that presents de novo identified PWMs and visualized logos of modification motifs. (v) We built a web-based tool called ‘modMetagene’ for plotting the metagenes of RNA modification sites uploaded by the user. (vi) We added disease-related SNV data to our database and built a disease link between RNA modification sites and SNVs sites. (vii) RMBase v2.0 includes a newly developed functional module called ‘modRBP’, which displays the relationships between post-transcriptional RNA modification sites and RNA-binding proteins (RBPs).
RMBase v2.0 allows for the global investigation of more than 100 RNA modification types and reveals extensive and complex post-transcriptional modifications of RNA (Figure 1, Table 1). RMBase v2.0 provides a variety of interfaces and graphic visualizations to facilitate analyses of the massive modification sites in normal tissues and cancer cells. Overall, RMBase v2.0 provides researchers with a comprehensive and powerful platform to discover potential functional roles of RNA modifications hidden in these data.
FUTURE DIRECTIONS
Recent advances in high-throughput epitranscriptome sequencing technology have produced large amounts of single-nucleotide-resolution modification sequencing data. We developed an automatic pipeline that is used to map, annotate, analyze and merge all high-throughput epitranscriptome sequencing data sets, and integrate these data into our local MySQL database. RMBase will continue to improve the computer server performance for storing and analyzing these new incoming data. We also developed new tools to decode the maps of RNA modifications from epitranscriptome sequencing data. We will maintain RMBase to ensure that it remains a useful resource for the research community.
AVAILABILITY
RMBase v2.0 is freely available at http://rna.sysu.edu.cn/rmbase/. All of the data files can be downloaded and used in accordance with the GNU Public License and the licenses of the primary data sources.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
National Key R&D Program of China [2017YFA0504400]; National Natural Science Foundation of China [91440110, 31770879, 31370791, 30900820, 31230042, 31471223, 31771459, 31401975]; Ministry of Science and Technology of China, National Basic Research Program [2011CB811300]; Guangdong Province [2017A030313106, S2013010012457]; The project of Science and Technology New Star in ZhuJiang Guangzhou city [2012J2200025]; Fundamental Research Funds for the Central Universities [2011330003161070, 14lgjc18]; China Postdoctoral Science Foundation [200902348]; Guangdong Province Key Laboratory of Computational Science and the Guangdong Province Computational Science Innovative Research Team. Funding for open access charge: National Key R&D Program of China [2017YFA0504400].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
Comments