





Abstract
We present a new likelihood method for detecting constrained evolution at synonymous sites and other forms of nonneutral evolution in putative pseudogenes. The model is applicable whenever the DNA sequence is available from a protein-coding functional gene, a pseudogene derived from the protein-coding gene, and an orthologous functional copy of the gene. Two nested likelihood ratio tests are developed to test the hypotheses that (1) the putative pseudogene has equal rates of silent and replacement substitutions; and (2) the rate of synonymous substitution in the functional gene equals the rate of substitution in the pseudogene. The method is applied to a data set containing 74 human processed-pseudogene loci, 25 mouse processed-pseudogene loci, and 22 rat processed-pseudogene loci. Using the informatics resources of the Human Genome Project, we localized 67 of the human-pseudogene pairs in the genome and estimated the GC content of a large surrounding genomic region for each. We find that, for pseudogenes deposited in GC regions similar to those of their paralogs, the assumption of equal rates of silent and replacement site evolution in the pseudogene is upheld; in these cases, the rate of silent site evolution in the functional genes is ∼70% the rate of evolution in the pseudogene. On the other hand, for pseudogenes located in genomic regions of much lower GC than their functional gene, we see a sharp increase in the rate of silent site substitutions, leading to a large rate of rejection for the pseudogene equality likelihood ratio test.
Introduction
It has long been held that pseudogenes provide the molecular evolutionist with an important tool for studying the rate and pattern of neutral evolution (Li, Gojobori, and Nei 1981 ). To the extent that pseudogenes evolve without selective constraint, the rate and pattern of substitutions in pseudogenes will faithfully reflect the underlying mutational process. Pseudogenes have, therefore, been used to infer the mutational process for nucleotide changes within species (Gojobori, Li, and Graur 1982 ; Li, Wu, and Luo 1984 ) and to compare this process between species (Petrov and Hartl 1999 ), as well as to study deletion rates among taxa (Graur, Shuali, and Li, 1989 ; Petrov, Lozovskaya, and Hartl 1996 ; Ophir and Graur 1997) and the effect that rates of DNA loss have on genome size (Petrov et al. 2000 ; Bensasson et al. 2001) .
The question of whether silent sites are evolving at the neutral mutation rate is far from resolved. The analysis of polymorphism data and codon frequencies in Escherichia coli and Salmonella enterica suggest that there is considerable weak selection operating on silent sites (Sharp and Li 1986 ; Andersson and Kurland 1990 ; Hartl, Moriyama, and Sawyer 1994 ; Hartl et al. 2000 ). Recent work in the comparative analysis of Drosophila genes has also revealed evidence for constraint on synonymous site evolution, presumably because of codon bias (Akashi 1996, 1997 ; Akashi and Schaeffer 1997 ; McVean and Vieira 2001 ). In rodent genes, however, there seems to be no relationship between the rate of synonymous substitution and codon bias (Smith and Hurst 1999 ). It has been suggested that a balance between mutation and selection may account for genomic variation in GC content; this would affect both the apparent codon bias and rate of substitution at silent sites (for a review see Bernardi 2000) .
Pseudogenes provide the molecular evolutionist with a direct opportunity to infer the strength of selection on changes at synonymous sites. Ophir et al. (1999) employed a distance method to analyze a set of 12 human and murid (rat and mouse) pseudogene gene trees; they found that, on average, murid and human third-position sites evolve at 40% the rate of pseudogene third-position sites. Because not all changes at third-position sites are synonymous, it is difficult to extrapolate from this result as to how much selection is acting on synonymous changes. The issue of selection on synonymous sites is of considerable practical importance in the study of molecular evolution because the ratio (ω) of the number of replacement substitutions per replacement site (dn) to the number of synonymous substitutions per synonymous site (ds) is widely used to detect adaptive evolution at the protein level.
The methods we present in this paper to study pseudogene evolution exploit recent statistical developments in molecular phylogenetics. In particular, we develop codon-based models for gene trees that contain a (processed) pseudogene, the functional gene from which the pseudogene was derived, and the ortholog of the functional gene from a closely related species (fig. 1 ). The models are implemented in a maximum likelihood framework and lead to two likelihood ratio tests of neutral evolution assuming a shared mutational process for all lineages—one to test the equality of silent and replacement substitution rates in a putative pseudogene, and another to test if the synonymous sites in the functional gene are evolving at the same rate as the pseudogene. We apply this method to a data set consisting of 121 processed pseudogenes (74 from human, 22 from rat, and 25 from mouse [Ophir and Graur 1997]) to test the assumption that processed pseudogenes evolve neutrally and to estimate the strength of selection on synonymous sites in the functional paralogs of pseudogenes.
Using the informatics resources of the Human Genome Project, we localized 67 of the gene-pseudogene pairs and estimated the GC content of the surrounding genomic areas for both. We used this data as well as the GC content of fourfold redundant sites (GC4) in the functional genes to assess whether rejection of pseudogene or silent site neutrality is correlated with the GC content.
It is important to note that the data sets presented here contain only retropseudogenes (i.e., pseudogenes generated by reverse transcription of mRNAs and insertion in the genome). These genes lack a promoter for expression and can, therefore, safely be termed “dead on arrival.” Furthermore, they generally insert far from their functional paralog and thus are expected to avoid the concerted evolution frequently observed for duplicated genes arranged in tandem.
Statistical Methods
The Model

The transition probabilities of the process can be calculated by exponentiating the rate matrix using standard numerical methods (e.g., numerical diagonalization of Q). The codon frequencies are estimated from the data using the method of moments to reduce the number of parameters in the model and to save computational time. The estimates are obtained from the observed base frequencies in the three codon positions (see Yang and Nielsen 1998 for details).
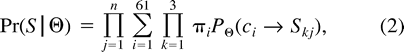
Likelihood Ratio Tests
The first hypothesis we will test is whether the putative pseudogene has been an untranscribed pseudogene or a neutrally evolving gene in the entire evolution of the pseudogene lineage. If a putative pseudogene is truly a pseudogene, then ωψ ought to equal 1; that is, the rate of synonymous substitution should equal the rate of replacement substitution in the pseudogene. To do so, we compare the log maximum likelihood of the general model with seven parameters (tf, to, tψ, ωf, ωo, ωψ, κ) to the log maximum likelihood of the nested six-parameter model (tf, to, tψ, ωf, ωo, κ), which assumes that ωψ = 1. Appealing to the usual large sample results for nested hypotheses (e.g., Stuart, Ord, and Arnold 1987 , p. 246), two times the logarithm of the maximum likelihood ratio of the two hypotheses (LRT[6,7]) is asymptotically distributed as a χ2 random variable with one degree of freedom (df). In particular, if the maximum log likelihood ratio under the seven-parameter model is more than 1.92 (3.84/2) log likelihood units larger than the maximum likelihood value under the six-parameter model, we reject the null hypothesis (ωψ = 1) at the 5% significance level. Throughout the rest of the paper, we will refer to LRT[6,7] as the pseudogene equality likelihood ratio test (PELRT).
Once we have established that the rates of silent and replacement substitutions are statistically similar in the pseudogene, suggesting that the pseudogene rate of substitution reflects the underlying neutral mutation rate, we will wish to test whether silent sites in the functional gene evolve at the same rate as the pseudogene rate. To do so, we will exploit the genealogical relationship between the pseudogene, the functional gene, and the functional ortholog. If the pseudogene arose after the two species diverged, the pseudogene sequence and the functional ortholog will be more closely related to each other than either of them would be to the outgroup sequence. Therefore, if the rate of evolution of the pseudogene equals the rate of synonymous evolution in the functional gene, then tf = tψ. To test this hypothesis, we will compare the maximum likelihood under the constrained model with six parameters {tf, to, tψ, ωf, ωo, κ} to the maximum likelihood under the nested model with five parameters {tf = tψ, to, ωf, ωo, κ}. Again, significance is tested by comparing twice the log likelihood ratio, LRT[5,6], to a χ21-distribution. Throughout the rest of this paper, we will refer to LRT[5,6] as the silent site equality likelihood ratio test (SSELRT).
Both these tests can be sensitive to the codon frequencies. Biased estimates of ω will be obtained if codon frequency biases are not properly accounted for (e.g., Yang and Nielsen 2000 ). This should cause little problem in the current context because the codon frequencies are similar. However, if the genomic context differs widely between pseudogenes and their functional paralog, the codon frequencies might be affected. In such cases, the result could potentially be an excess of significant results of the PELRT.
Pooled Likelihood Ratio Test
One issue of interest is whether we can reject the null hypotheses when combining the data across loci. If we assume that the genes are independent of one another, we can sum the log likelihoods under each model and perform the SSELRTs and the PELRTs on the pooled data. SSELRT applied to a combined data set would test whether all pseudogenes in the data set conform to ωψ = 1. PELRT applied to the whole data set would test whether the average rate of silent site evolution in each gene is the same rate as in the respective pseudogene for all genes. As models 6 and 7 differ by one df, the PELRT for n genes will be distributed as χ2n. Likewise, the combined SSELRT statistic will be χ2n − r distributed, where r is the number of genes that reject the PELRT; the PELRTs are relevant because the SSELRT on a gene is justified only if the gene fails to reject the PELRT.
Binomial Likelihood Ratio Test
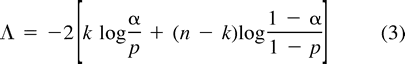
Logistic Regression
To explore whether the GC content correlates with the rate of rejection of either the PELRT or the SSELRT, we used the logistic regression framework. This is an appropriate analysis to employ because our response variable is dichotomous (whether a particular test rejects the null hypothesis or not), and our predictor variable is continuous (GC content). We ran four separate logistic regressions using the genomic GC content or the GC content of fourfold redundant sites as explanatory variables, and whether or not the SSELRT or the PELRT rejects the null hypothesis as the outcome variables. We use the notation βGenomic GC and βGC4 to refer to the maximum likelihood estimates of the slope parameter for regressions using the genomic GC content and the GC4 content as predictor variables, respectively, and α to refer to the intercept parameter for each regression. The P value reported for each regression is for a likelihood ratio test comparing the fit of the model with the maximum likelihood estimate of the slope parameter to a model with slope equal to 0 (Christensen 1997 ). The 95% confidence intervals for the predicted rate of rejection of each test as a function of GC content were found using nonparametric bootstrap sampling.
Bootstrap Test
To test whether the average number of replacement substitutions per replacement site in pseudogenes, d̅n̅, equaled the average number of silent substitutions per silent site in pseudogenes, d̅s̅, we used a paired bootstrapping procedure. Namely, we sampled (dnψ, dsψ) pairs as estimated from our data with replacement and calculated d̅n̅ and d̅s̅ for each sample of pseudogenes 100,000 times. The P value reported is twice the proportion of times d̅n̅ was less than d̅s̅ (because we were performing a two-sided test). Again, this test is not independent of the pooled likelihood ratio test.
Estimating Effective Level of Selection on Synonymous Sites
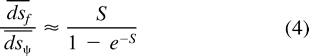
To generate confidence intervals for d̅s̅f̅ /d̅s̅, we used 100,000 nonparametric bootstrap samples generated by sampling with replacement (dsf,dsψ) pairs estimated from our data. Using equation (4) , we can transform the distribution of bootstrap estimates of d̅s̅f̅ /d̅s̅ into a distribution for estimates of S.
Materials and Methods
Data
For each pseudogene reported in Ophir and Graur (1997), we searched the nonredundant database (NR) of the NCBI server (http://www.ncbi.nlm.nih.gov/BLAST/) using the program (BLASTN) (Altschul et al. 1990 ) to find the closest available paralogous sequence from the same species. We then ran BLASTN on the paralog to find the single closest murid or human ortholog for the paralog-pseudogene pair (GenBank accession numbers available via the MBE website). Genes for which no functional ortholog could be found—as evidenced by the intercalation of phylogenetically incongruent sequences in the search results—were omitted (e.g., if a search using a functional human gene revealed a higher homology between the human gene and a crocodile gene rather than the human gene and the mouse gene, the pseudogene-ortholog pair was omitted).
Sequences were obtained, edited, and aligned by CLUSTALW using various versions (3.5–4.0.30) of the program DAMBE (Xia 2000 ). All stop codons and codons containing nucleotides with gaps in the alignment were removed and the reading frame was set to the reading frame of the functional gene. Pseudogenes that were identical to their functional paralog, except for gaps, were omitted from the analysis. Likewise, genes for which the estimated pseudogene, t, was longer than the outgroup, t, in model 7 were omitted because such a phylogeny indicated that the assumption of pseudogene emergence after the primate-rodent split was not met. We used BLASTN to identify the genomic position of 67 of the human pseudogene-functional gene pairs using the NCBI-curated sequences of the Human Genome Project. All genomic GC content measures are for the NCBI GenBank entry for the BAC (>100,000 bp) containing the functional gene.
Results and Discussion
In the Additional Information, which can be accessed via the Molecular Biology and Evolution web site, we summarize the maximum likelihood parameter estimates and the results of the individual likelihood ratio tests for each gene. The estimated number of silent substitutions per silent site between human and murid genes, as estimated from the full model, ranged from 0.143 to 2.427. Likewise, the estimated number of replacement substitutions per replacement site ranged from 0 to 0.447. The mean and standard deviation of these distances (d̅s̅f̅+̅d̅s̅o̅ = 0.596 ± 0.295; d̅n̅f̅+̅d̅n̅o̅ = 0.052 ± 0.064) are comparable to those previously cited for the human-mouse comparison (Matassi, Sharp, and Gautier 1999 ). The estimated distance between a pseudogene and its respective functional gene under the full model ranged from 0 to 0.684 silent substitutions per silent site (d̅s̅f̅+̅d̅n̅ψ̅ = 0.128 ± 0.106) with 0–0.257 replacement substitutions per replacement site (d̅n̅f̅+̅d̅n̅ψ̅ = 0.0645 ± 0.0513).
One key conclusion from our analysis is that, overall, the majority of pseudogene-gene pairs (>70%) do not reject either test. This suggests that, as a first approximation, the most constrained model (i.e., equal codon frequencies and mutation rates in all lineages accompanied by neutral evolution in both pseudogenes and silent sites in functional genes) seems to fit the majority of the data. We did find that the PELRT is significant at the 5% significance level for 13 of the 121 genes (11%). Among those genes for which the PELRT is not significant (n = 108), 25 genes (23%) have significant SSELRTs. Given the large number of tests, we would expect approximately 5% of the genes to reject either null hypothesis if the deviations were simply the result of chance. However, for both tests the proportion of genes that reject the null hypothesis is significantly greater than 5% by the BLRT (PELRT: P < 0.012; SSELRT: P ≪ 0.0000001). The source of these discrepancies is discussed in the following sections.
Implications of the Results for Pseudogene Evolution
To learn if the GC content correlates with an increase in the rejection rate of either test, we divided the data set into two equal groups based on the GC content of fourfold redundant sites in the functional paralogs of the pseudogenes (GC4). The rationale for using GC4 rather than overall GC content is that replacement sites are presumably constrained by purifying selection on amino acid changes. For 67 of the human genes, we were also able to estimate the GC content of the surrounding genomic region for the functional gene using the NCBI sequences of the Human Genome Project. Not surprisingly, GC4 was highly correlated with the surrounding genomic GC content (r = 0.72), and the conclusions are the same if one analyzes the flanking GC content of the functional gene rather than GC4.
In table 1 , we report the rejection rates, 95% confidence intervals for the rejection rates, and the results of the pooled likelihood ratio tests for gene pairs grouped on the basis of the GC content of the functional gene for both the PELRT and the SSELRT. We find, by applying Fisher's Exact Test of homogeneity, that regardless of whether we classify humans genes on the basis of the regional GC content or GC4 of the functional gene, the processed pseudogenes derived from GC-rich genes reject the PELRT more frequently than do pseudogenes derived from GC-poor genes (Genomic GC, P < 0.027; GC4, P < 0.011). Likewise, a pooled likelihood ratio test implies that pseudogenes derived from GC-rich human genes, in general, have unequal rates of substitution at silent and replacement sites (P < 0.0001). This result is unexpected, but the same phenomenon is also observed in the rodent pseudogenes (P < 0.0032).
The inequality in substitution rates between silent and replacement sites is not observed in pseudogenes derived from GC-poor regions. From table 1 , we see that the rejection rate of the PELRT is not significantly different from that expected by chance for human genes as classified by genomic GC (1/33; P ≈ 0.5766), for human genes classified by GC4 (1/39; P ≈ 0.4430), or for rodent genes (1/22; P ≈ 0.9209). Furthermore, we do not reject the pooled PELRT for GC-poor genes in either humans or rodents (P > 0.45 for all; see table 1 for details). This tentatively suggests that in the cases where pseudogenes land in genomic regions similar to those in which their respective functional genes have evolved, the equal codon frequencies model and pseudogene neutrality is supported.
To investigate whether these results are because of our dichotomous classification of GC groups based on GC4, we used a logistic regression with GC4 in the functional gene as a continuous predictor variable and whether the pseudogene rejected or failed to reject PELRT as the outcome variable. As illustrated in figure 2 , we found a strong positive association between GC4 in the functional gene and the probability of rejecting the PELRT (βGC4 = 6.374, α = −6.156; P < 0.0016). However, for pseudogenes derived from genes with GC4 < 0.60, the 95% confidence intervals for the probability of rejecting the PELRT contain 5%. A logistic regression for human pseudogenes based on the local GC content yielded comparable results (βGenomic GC = 21.118, α = −11.864; P < 0.0058).
Why should pseudogenes derived from high-GC functional genes tend to have unequal rates of silent and replacement substitutions, whereas pseudogenes derived from low-GC functional genes conform to the expectations of neutral theory? One logical hypothesis is that selection for regional GC content (or differential mutation rates away from G or C) accounts for the difference in substitution rates at silent and replacement sites in processed pseudogenes derived from high-GC functional genes. Because mammalian functional genes tend to be nonrandomly distributed with respect to the GC content, such that a majority of genes are found in GC-rich regions, although a majority of the genome is composed of AT-rich regions (Mouchiroud et al. 1991 ; Saccone et al. 1997 ), if processed pseudogenes are randomly incorporated into the genome, the majority of pseudogenes will move to areas of lower GC content. Consequently, pseudogenes derived from genes in relatively GC-poor coding regions will usually land in genomic regions that are similar to the region in which their functional gene has evolved, whereas pseudogenes derived from GC-rich regions will usually land in regions of much lower GC content.
A prediction of this hypothesis is that, in the pseudogene lineage, the substitution rate per silent site should be elevated relative to the substitution rate per replacement site. In other words, the silent sites in the pseudogenes that move from regions of high GC content to regions of low GC content should be changing faster than the neutral rate. The reason that silent sites in pseudogenes are disproportionately affected is that it is the silent sites in functional genes that are most affected by local GC content because replacement sites are predominantly under purifying selection. Therefore, processed pseudogenes that move from an area of high GC to one of low GC have an excess of G or C at their silent sites, relative to their new genomic neighborhoods. This will cause silent sites to be more strongly affected by either mutation or selection pressure for GC content, leading to an excess of silent substitutions vis à vis replacement substitutions. The same phenomenon would hold for genes that move from low- to high-GC regions, but given the high abundance of AT-rich regions and the fact that most genes are in GC-rich regions, these events would occur infrequently.
Our data afford strong support for this hypothesis. For pseudogenes derived from high-GC genes, the average number of replacement substitutions per replacement site, d̅n̅, relative to the average number of silent substitutions per silent site, d̅s̅, is 0.0537/0.0935 = 0.5743; this is significantly different from unity by the bootstrap test (P < 0.00001). For pseudogenes derived from low-GC genes, d̅n̅ = 0.0624 and d̅s̅ = 0.0619, yielding a ratio of 1.008, which is extremely close to the expected ratio of 1.0 (P ≈ 0.9900). Furthermore, a higher rate of silent substitution in pseudogenes derived from high-GC functional genes is evident from application of an unpaired t-test. The average replacement substitution rate does not vary between pseudogenes derived from high- and low-GC groups (t = −1.027; P ≈ 0.3062), whereas the average silent substitution rate for pseudogenes derived from high-GC genes is significantly higher than the average rate for pseudogenes derived from low-GC genes (t = 2.1808; P < 0.0312). Together, these results suggest that silent and replacement sites in pseudogenes derived from low-GC genes, as well as replacement sites in genes derived from high-GC genes, are evolving at or near the neutral mutation rate, whereas silent sites in pseudogenes derived from high-GC genes are evolving at a rate faster than the neutral mutation rate.
In addition, pseudogenes derived from genes found in high-GC regions tend to decrease in GC4 content (Wilcoxon signed rank test [WSRT]: Z = 3.561, P < 0.0005) relative to their respective functional genes, whereas pseudogenes derived from low-GC regions tend to maintain the same level of GC4 content (WSRT: Z = 0.950, P ≈ 0.3422). These findings corroborate the hypothesis that positive selection for GC content, or large differences in mutation rates among genomic regions, drives the observed difference in substitution rate between silent and replacement sites in a significant number of processed pseudogenes.
Implications of Results for Silent Site Evolution
We find that, for functional genes derived from both GC-rich and GC-poor regions, a significant number of pseudogenes evolve faster than silent sites in the genes from which they were derived. This comparison is statistically significant whether one considers the proportion of genes that reject the SSELRT or whether one considers the pooled SSELRT across genes (table 1 ). For functional genes in GC-rich regions, the high rate of rejection is, in part, attributable to the excess of silent site fixation events along the pseudogene branch in the gene tree because of selection or mutation bias in the novel genomic region. This explanation does not apply to relatively GC-poor genes because we have shown that pseudogenes derived from these genes do not reject the PELRT more than expected by chance. In this class of genes, the substitution rates of silent and replacement sites in the pseudogenes are roughly equal. Furthermore, a logistic regression with GC4 of the functional gene as a predictor variable and whether the gene rejects the SSELRT as the outcome variable is not significant (β = 1.471, α = −2.040; P ≈ 0.3434), suggesting that GC4 alone cannot account for the overall level (∼25%) of rejection for the SSELRT.
How does the detection of non-neutral evolution at silent sites in functional genes relate to the rate of substitution? To estimate the overall difference in the average rates of substitution between silent sites and pseudogene sites, we compare the ratio of the average rate of substitution per synonymous site in functional paralogs to the average rate of substitution per site in the pseudogenes (d̅s̅f̅ /d̅s̅). Figure 3 illustrates the distributions of 100,000 nonparametric bootstrap estimates of d̅s̅f̅ /d̅s̅ from model 6 (pseudogene neutrality model) and model 7 (free model) for gene-pseudogene pairs derived from GC-poor regions, and from model 7 only for gene-pseudogene pairs derived from GC-rich regions. We did not perform the bootstrap analysis for parameter estimates based on model 6 for gene-pseudogene pairs derived from GC-rich regions because this class of genes rejects model 6 as a group based on the pooled likelihood ratio test. Table 2 reports the mean and the 2.5% and 97.5% quantiles of these distributions. There is approximately a 30% reduction in the average substitution rate of silent sites in functional genes relative to silent sites in pseudogenes. We also note that the mean of the distribution of d̅s̅f̅ /d̅s̅ from GC-rich regions for model 7 (0.62) differs slightly from the mean of the distribution for models 6 and 7 for GC-poor regions (0.69–0.70), which reflects the accelerated substitution rate at silent sites in the pseudogene noted previously. The means of these distributions are all significantly different from unity because the interval between the 2.5% and 97.5% quantiles of these distributions does not contain 1 (table 2 ).
Using Kimura's result presented in the Statistical Methods section, one can transform the distributions of d̅s̅f̅ /d̅s̅ in figure 3 into a distribution for S. From table 2 , we see that the means of these distributions are in the interval [−0.6753, −0.8814]. This suggests that the average strength of selection at synonymous sites is in the range where genetic drift and weak negative selection are important in determining the rate of substitution. However, if there is considerable variation within genes in selection intensity at silent sites, or if the mutation rates at silent sites differ considerably between pseudogenes and functional genes, then the above result underestimates the effect of selection. To estimate the effect of selection in these more complex scenarios, comparative data within and between species will be needed.
Adam Eyre-Walker, Reviewing Editor
Keywords: pseudogene evolution synonymous site evolution codon-based models maximum likelihood neutral evolution
Abbreviations: BLRT, binomial likelihood ratio test; PELRT, pseudogene equality likelihood ratio test; SSELRT, silent site equality likelihood ratio test; WSRT, Wilcoxon signed rank test.
Address for correspondence and reprints: Daniel L. Hartl, 16 Divinity Avenue, Cambridge, Massachusetts 02138. dhartl@oeb.harvard.edu .
Table 1 Proportion of Genes that Reject Null Hypothesis for Pooled Data and Results of Pooled Likelihood Ratio Tests
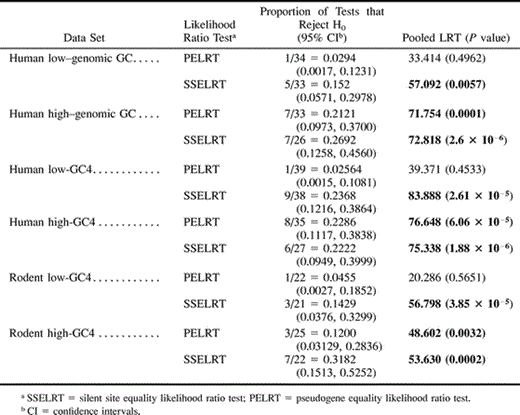
Table 1 Proportion of Genes that Reject Null Hypothesis for Pooled Data and Results of Pooled Likelihood Ratio Tests
Table 2 Average Ratio of Substitution Rates at Synonymous Sites in Functional Genes and in Pseudogenes and Corresponding values for 4Ns

Table 2 Average Ratio of Substitution Rates at Synonymous Sites in Functional Genes and in Pseudogenes and Corresponding values for 4Ns
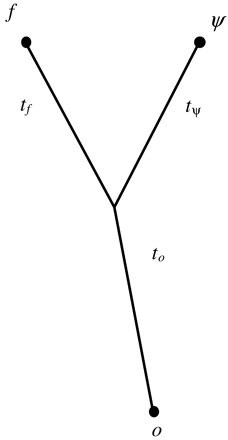
Fig. 1.—A graphical illustration of the model assumed in this paper. f is the codon sequence in the functional gene, ψ is the codon sequence in the pseudogene, o is the codon sequence in the outgroup and tf, tψ, and to are the respective branch lengths of the lineages leading to these sequences
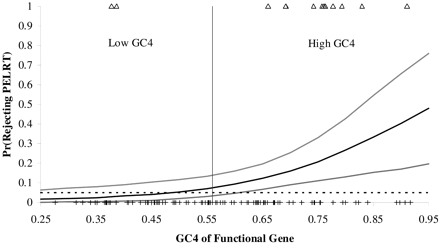
Fig. 2.—Probability of rejecting the PELRT as a function of GC4 content of the functional gene. The solid line is the predicted rate of rejection from the logistic regression analysis and the gray lines are the 95% confidence intervals. The locations of the genes that reject the test are denoted by open triangles, and the locations of the genes that do not reject the test are denoted by hatched marks. The dotted line traces out the expected 5% rejection rate
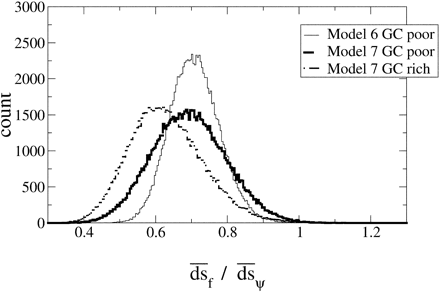
Fig. 3.—Distributions of d̅s̅f̅ /d̅s̅ estimated from 100,000 nonparametric bootstrap samples of estimates from model 6 (solid line) and model 7 (dotted line) from GC-poor gene-pseudogene pairs, and from model 7 (thick solid line) from the GC-rich pairs
The authors thank H. Akashi and D. Petrov as well as two anonymous reviewers for comments on earlier drafts of this manuscript. This work was supported by a Howard Hughes Medical Institute award to C.D.B.
References
Altschul S. F., W. Gish, W. Miller, E. W. Myers, D. J. Lipman,
Andersson S. G. E., C. G. Kurland,
Akashi H.,
———.
Akashi H., S. W. Schaeffer,
Bensasson D., D. A. Petrov, D. Zhang, D. L. Hartl, G. M. Hewitt,
Goldman N., Z. Yang,
Gojobori T., W. H. Li, D. Graur,
Graur D., Y. Shuali, W.-H. Li,
Hartl D. L., E. F. Boyd, C. D. Bustamante, S. A. Sawyer,
Li W. H., T. Gojobori, M. Nei,
Li W. H., C. I. Wu, C. C. Luo,
Matassi G., P. M. Sharp, C. Gautier,
McVean G. A., J. Vieira,
Mouchiroud D., G. D'Onofrio, B. Aissani, G. Macaya, C. Gautier, G. Bernardi,
Muse S. V., B. S. Gaut,
Nielsen R., Z. Yang,
Ophir R., D. Graur,
Ophir R., T. Itoh, D. Graur, T. Gojobori,
Petrov D. A., E. Lozovskaya, D. L. Hartl,
Petrov D. A., D. L. Hartl,
Petrov D. A., T. A. Sangster, J. S. Johnston, D. L. Hartl, K. L. Shaw,
Saccone S., S. Caccio, P. Perani, L. Andreozzi, A. Rapisarda, S. Motta, G. Bernardi,
Sharp P. M., W.-H. Li,
Smith N. G., L. D. Hurst,
Stuart A., J. K. Ord, S. Arnold,
Xia X.,
Yang Z.,
Yang Z., R. Nielsen,

{kind=link}
{kind=link}
{kind=link}