





Abstract
The performance of 14 different recombination detection methods was evaluated by analyzing several empirical data sets where the presence of recombination has been suggested or where recombination is assumed to be absent. In general, recombination methods seem to be more powerful with increasing levels of divergence, but different methods showed distinct performance. Substitution methods using summary statistics gave more accurate inferences than most phylogenetic methods. However, definitive conclusions about the presence of recombination should not be derived on the basis of a single method. Performance patterns observed from the analysis of real data sets coincided very well with previous computer simulation results. Previous recombination inferences from some of the data sets analyzed here should be reconsidered. In particular, recombination in HIV-1 seems to be much more widespread than previously thought. This finding might have serious implications on vaccine development and on the reliability of previous inferences of HIV-1 evolutionary history and dynamics.
Introduction
The detection of recombination from DNA sequences is relevant to the understanding of evolutionary and molecular genetics. Not surprisingly, a plethora of methods have been developed during the last 15 years to detect the presence of recombination in sequence alignments. However, the performance of these methods has been evaluated only recently (Maynard Smith 1999 ; Brown et al. 2001 ; Posada and Crandall 2001 ; Wiuf, Christensen, and Hein 2001 ).
Although computer simulations offer an unlimited range of possibilities for the evaluation of the statistical performance of a given method, results from simulation studies alone are always limited. Models used in simulation studies are simplifications of reality. Recombination among sequences has been simulated using the coalescent with recombination (Brown et al. 2001 ; Posada and Crandall 2001 ; Wiuf, Christensen, and Hein 2001 ) or using a more classical forward approach (Maynard Smith 1999 ). In these simulations, recombination fragments are defined from a single breakpoint to the end of the sequences, and the exchange is performed in a reciprocal way, so each sequence is a donor and a receptor of a fragment. However, this is not the way that recombination occurs in many organisms, including viruses and bacteria, where recombinational events often include more than one breakpoint, and nonreciprocal exchanges are the norm. In addition, nucleotides are evolved under simple reversible substitution models, which assume independence of sites and a stationary and homogeneous substitution process. Moreover, it is not easy to design a simulation study that does not favor some method(s) (Hillis, Mable, and Moritz 1996 ). Different methods make different assumptions on the processes that generate data. In a simulation study, methods that make the same assumptions as the model used to generate the data should be, in principle, favored. For example, Brown et al. (2001) use the performance of a likelihood ratio test of recombination based on the coalescent as the reference with which the performance of other recombination detection methods is compared.
Despite their limitations, computer simulations are useful. If a result holds in simulations across a wide variety of conditions, they can suggest that the result is general and should apply in real data sets (Swofford et al. 1996 ). One obvious way of checking this proposal is through the analysis of real data sets utilizing the same software implementations used in the computer simulations. By comparing the results obtained from computer simulations with the inferences obtained from real data, we simultaneously serve two purposes. First, similar results from both types of studies strengthen conclusions regarding performance of the methods evaluated. Second, the analysis of empirical data provides a check of simulation studies, validating (or not) the utility of the models used in the simulations to evaluate a method's performance.
The analysis of empirical data sets alone may be useful to study the performance of recombination methods, especially when the recombinational events are more or less known (Drouin et al. 1999 ). However, performance of a method is best evaluated through the combined analysis of empirical and simulated data. Indeed, the analysis of empirical data sets is a very desirable and rare complement to simulation studies (Hillis and Huelsenbeck 1994 ). Here, several empirical data sets are analyzed for the presence of recombination to compare the inferences with those obtained from previous computer simulations (Posada and Crandall 2001 ). The objectives of this study are threefold (1) to decipher the presence of recombination in several representative empirical data sets, (2) to obtain a better understanding of the performance of recombination detection methods, and (3) to evaluate the utility of models of reticulated sequence evolution.
Materials and Methods
The methodology employed here consists of the following steps: (1) select two groups of empirical data sets, where (a) recombination is assumed to be absent and (b) recombination has been suggested; (2) apply 14 different methods to detect the presence of recombination; and (3) for each data set, compare the inferences obtained among different recombination methods.
Methods for Detecting Recombination
For the last 20 years, a number of methods have been developed to detect recombination from a set of aligned DNA sequences. David Robertson (Zoology, University of Oxford) provides a web site with links to many of these methods at http://grinch.zoo.ox.ac.uk/RAP_links.html. In general, recombination methods could be tentatively classified as described in the subsequent paragraphs.
(a) Distance Methods
Distance methods search for inversions along the alignment of the distance patterns among sequences (Weiller 1998 ). In general, they use a sliding window approach and estimate statistics based on genetic distances among sequences. Because the phylogeny does not need to be known, these methods are typically fast.
(b) Phylogenetic Methods
Several methods infer recombination when phylogenies from different parts of the genome result in discordant topologies. When comparisons of adjacent sequences yield different branching patterns, there is reason to suspect recombinational events. If the consequence of such changes results in reconciling different sequence phylogenies to a single phylogeny, then the existence of such events becomes a reasonable hypothesis (Hein 1990, 1993 ; Grassly and Holmes 1997 ; Holmes, Worobey, and Rambaut 1999 ; Lole et al. 1999 ; Martin and Rybicki 2000 ). These are the methods most extensively used in the literature.
(c) Compatibility Methods
Compatibility methods are phylogenetic methods that are based on site-by-site analyses (Drouin et al. 1999 ). These methods define two sites as compatible when their evolutionary history is congruent with the same tree (Sneath, Sackin, and Ambler 1975 ), and they do not require the phylogeny of the sequences to be known (Jakobsen and Easteal 1996 ; Jakobsen et al. 1997 ).
(d) Substitution Distribution
Nucleotide substitution distribution methods examine the sequences either for a significant clustering of substitutions or for a fit to an expected statistical distribution (Stephens 1985 ; DuBose, Dykhuizen, and Hartl 1988 ; Sawyer 1989 ; Maynard Smith 1992 ; Takahata 1994 ; Sneath 1995 ; Maynard Smith and Smith 1998 ; Sawyer 1999 ; Worobey 2001 ).
Implementation of Recombination Detection Methods
Fourteen recombination methods were implemented (table 1 ). The details of each of these methods have been described elsewhere (see also Posada and Crandall 2001 ). Particular details of the implementations are described subsequently.
(1) Bootscanning (Salminen et al. 1996 )
The windows program Simplot (Lole et al. 1999 ) (http://www.med.jhu.edu/deptmed/sray/download/) was modified by Stuart Ray (Simplot's author) to implement the bootscanning of every sequence in the alignment against the rest. A sliding window size of 200 bp and step size of 10 nt were used. Neighbor-Joining trees were estimated using F84 distances (Felsenstein 1984, 1991 ), and bootstrap values were obtained from 100 replicates. Several bootstrapping thresholds for assignment of parenthood were explored (70%, 90%, and 95%), but because only the 95% threshold provided a false positive rate below 10% in computer simulations, this was the threshold used here.
(2) Geneconv (Sawyer 1999 )
The program Geneconv 1.81 (http://www.math.wustl.edu/∼sawyer/geneconv/index.html) was employed. The global permutation P-values based on BLAST-like global scores (10,000 replicates) smaller than 0.05 were considered as evidence of recombination. A multiple comparison correction is already built into these P-values. The default value of the parameter gscale (gscale = 0) was used.
(3) Homoplasy Test (Maynard Smith and Smith 1998 )
Two qbasic programs written by J. Maynard Smith (http://www.biols.susx.ac.uk/home/John_Maynard_Smith/) were translated into a single C program. No outgroup was used, and the number of effective sites, Se, was taken to be 0.6 × the total number of sites. For coding sequences, only third positions were included in the analysis.
(4) Informative Sites Test (Worobey 2001 )
The program Pist (Worobey 2001 ) (http://evolve.zoo.ox.ac.uk/software/PIST/PIST.html) was used. Maximum likelihood trees and substitution estimates were obtained in PAUP* (Swofford 2000 ) for the best-fit model selected by Modeltest 3.06 (Posada and Crandall 1998 ). In the case of coding sequences, only third positions were used. For the parametric simulation of the null distribution of the test statistic, 100 replicates were used.
(5) Maximum Chi-Square (Maynard Smith 1992 )
The computer program MaxChi2 was written in C, implementing a modification of the maximum chi-square method (Maynard Smith 1992 ) suggested by Wiuf, Christensen, and Hein (2001) . The statistic employed was the maximum chi-square in the original alignment. For each pair of sequences, this statistic was calculated on a sliding window that moved one nucleotide at a time and included only variable sites. The width of this window was arbitrarily set to the total number of variable sites divided by 1.5. The P-value for the null hypothesis of no recombination was estimated as the number of times the maximum chi-square was smaller than the maximum chi-square out of 1,000 permuted alignments (obtained by randomizing the position of the columns in the alignment).
(6) Maximum Match Chi-Square (Posada and Crandall 2001 )
The computer program Chimaera was written in C implementing the maximum mismatch chi-square method. This method is a modification of the maximum chi-square (see previously). The only differences are that a different test statistic is used (in this case the statistic is the maximum match chi-square), and that the test statistic is calculated for each possible triplet of sequences (instead of for each pair). For each triplet, each sequence was treated alternatively as the potential recombinant, and the maximum match chi-square statistic was calculated. The maximum match chi-square is the chi-square statistic from the contingency table built with the number of sites in the putative recombinant that match each one of the parental, before and after an arbitrary breakpoint.
(7) Phylogenetic Profile (Weiller 1998 )
The computer program PhyPro was written in C extending the Phylogenetic Profile method, which in its original form does not provide statistical significance. The test statistic employed was the minimum distance vector correlation in the original alignment. For each data set, this statistic was calculated on a sliding window that moved one nucleotide at a time and included only variable sites. The width of this window was arbitrarily set to the total number of variable sites divided by 1.5. The P-value for the null hypothesis of no recombination was estimated as the number of times the minimum distance vector correlation was smaller than the minimum distance vector correlation out of 1,000 permuted alignments (obtained by randomizing the position of the columns in the alignment).
(8) Partial Likelihood Assessed Through Optimization (Grassly and Holmes 1997 )
The program Plato (Grassly and Holmes 1997 ) (http://evolve.zoo.ox.ac.uk/software/Plato/Plato2.html) was used. Maximum likelihood trees and substitution model estimates were obtained in PAUP* (Swofford 2000 ) for the best-fit model selected by Modeltest 3.06 (Posada and Crandall 1998 ). For the simulation of the null distribution, 100 Monte Carlo replicates were used. Default window settings were used (minimum size = 5, step = 1).
(9) Recombination Detection Program (Martin and Rybicki 2000 )
The Windows program Rdp (Martin and Rybicki 2000 ) (ftp://ftp.uct.ac.za/pub/data/geminivirus/recomb.htm) was generously modified by D. Martin for the simulations. The reference sequences used were internal and external. The window size was set to 10 nt.
(10) Recombination Parsimony (Hein 1990, 1993 )
The C program RecPars (K. Fisker; ftp://ftp.daimi.aau.dk/pub/empl/kfisker/programs/RecPars) was used. In order to specify the substitution costs between nucleotides, a step matrix was estimated upon the maximum likelihood tree for each data set using MacClade4.0 (Maddison and Maddison 2000 ). A recombination cost of three times the maximum substitution cost (d = 3 × s) was used. This particular recombination cost was chosen because it performed the best in previous computer simulations. Recombination was inferred when more than one history was suggested for a given data set.
(11) Reticulate (Jakobsen and Easteal 1996 )
The C program Reticulate (Jakobsen and Easteal 1996 ) (http://jcsmr.anu.edu.au/dmm/humgen/ingrid/reticulate.htm) was modified for the simulations. The test statistic used was the neighbor similarity score, and the number of permutations was set to 1,000.
(12) Runs Test (Takahata 1994 )
A C program was written implementing the Runs Test proposed by Takahata (1994) .
(13) Sneath Test (Sneath 1995, 1998 )
A program written by P. Sneath in qbasic (ftp://ftp.ebi.ac.uk/pub/software/dos/) was translated into C for the simulations.
(14) Triple (Kuhner et al. 1991 )
A program written in Fortran by Mary Kuhner (Kuhner et al. 1991 ) that implements an extension to the method of Stephens (1985) was translated into C. On the basis of the results from computer simulations, the Bonferroni correction was applied to each individual test with a family alpha level of 0.01 to obtain an approximate false positive rate of 5%.
Performance Evaluation
The question addressed in this study is whether recombination methods detect the presence of recombination. Although some methods provide a qualitative answer for the presence of recombination (yes or no), most methods calculate a P-value (table 1 ). In the latter case, recombination was inferred when the provided P-value (calculated using some statistical distribution or using permutations) was smaller than 0.05.
Selection of Empirical Data Sets
A total of 24 nucleotide data sets were selected from the literature and public databases. In 12 of these data sets recombination is assumed to be absent, whereas in the other 12 data sets the presence of recombination has been suggested in the literature (table 2 ). Data sets were selected to represent different conditions (levels of genetic diversity, taxonomic level, number of sequences, number of sites, rate heterogeneity among sites) (table 3 ). Already aligned data sets (available from the author) were obtained. In some cases, regions with abundant missing data were removed from the alignment. Nucleotide substitution parameters (required by some recombination methods) for each data set were estimated in PAUP* (Swofford 2000 ) for the best-fit model of nucleotide substitution (Posada and Crandall 1998 ).
Results
Recombination Inference
Different methods for detecting recombination disagreed regarding the presence or absence of recombination for several data sets. In some cases, inference was not possible because of the requirements of a given method (e.g., more than three sequences). In only one case was recombination not inferred by all methods (DmelCytB). There were no data sets for which all methods detected recombination. Notice that in figure 1 the data sets are quite scrambled in terms of the preconceived notions of recombination (data sets in bold vs. not data sets not in bold). However, four of five data sets with the least recombination detected were previously thought to be recombination free and all five data sets with the strongest inference of recombination were previously thought to contain recombinants. The behavior of each recombination method is briefly summarized subsequently.
MaxChi2 and Rdp seem to identify quite well when recombination is present (or likely to be present) without claiming that recombination has occurred when it seems unlikely.
Chimaera could be inferring potential false positives (MammPGK) and missing some likely recombinant data sets (HIV(B)EnvNR).
Reticulate and Geneconv seem to wrongly infer recombination in some divergent data sets (DaphniaCO1 and InsectaCOII), but they do recognize well the occurrence of recombination where it is most likely.
PhyPro does not infer many potential false positives, but it seems to fail to identify some likely recombinant data sets (HIV(B)EnvNR and MaizACT).
The Homoplasy Test seems to incorrectly infer recombination when rate heterogeneity among sites is high (WolfCR and Armillaria-mtDNA). Also, it did not seem able to detect recombination in highly diverse data sets (HIV(B)EnvNR, HIVEnvNR, BoletalesATP6, and HIVEnv). Interestingly, this is exactly the same behavior observed in previous computer simulations (Posada and Crandall 2001 ).
Pist showed a tendency to wrongly detect recombination in divergent data sets (e.g., GymnND4, DaphniaCO1, and VertebCOI), although it did not clearly detect recombination in the most divergent data set (PetuniaS-RNase). In the simulations, however, the amount of diversity did not seem to induce false positives for Pist, except when high rate variation was present in the data.
Plato seems to erroneously infer recombination in two data sets with high rate variation (WolfCR and Armillaria-mtDNA), even though rate variation was included in the model used for the calculation of the likelihoods. In addition, when rate variation is included in the model, recombination was not detected in argF; this data set was used as an example of recombination in the original paper.
RecPars did not detect recombination in clear recombinant data sets, such as Fusarium3 or Candida-mtDNA. This could be because of the somehow low number of parsimony informative sites in these data sets (118 and 58, respectively). This may imply that RecPars needs a minimum sequence divergence (>1%) to detect recombination.
Runs Test performance does not fit an obvious pattern, and it does not detect recombination in data sets where recombination most likely has occurred. However, it detects recombination in data sets where recombination seems unlikely. Interestingly, the Runs Test showed the worst performance of all methods in previous computer simulations (Posada and Crandall 2001 ).
The bootscanning implementation, Simplot, seems very conservative when a 95% threshold is used. When the bootscanning threshold was dropped from 95% to 70%, it detected recombination in the HIV data sets (data not shown). However, in simulations it was observed that even a bootscanning threshold of 90% resulted in high false positive rates. In addition, it infers recombination in the VertebCOI data set, which may be a false positive.
The Sneath Test seems to work well, except for the likely wrong detection of recombination in Perom12S and InsectaCOII. Because the Sneath Test works in a pairwise manner, this inference could reflect some specific pattern not recognized in the data as a whole.
Finally, Triple does infer what seem to be false positives (e.g., Perom12S), while failing to reject what seem to be clear recombinant data sets (e.g., HGVgenome).
Discussion
Recombination Detection Patterns
The different recombination methods explored in this study showed different behavior. Conclusions here depend on a good knowledge of which data sets are recombinant and which ones are not. Obviously, we do not know the true recombination status of these data sets, but it seems reasonable to assume that in some of the data sets recombination has occurred, whereas in others it is absent.
To summarize inferences obtained by the different methods (see fig. 1 ), data sets were ordered by the strength of the recombination inference, from least to most, using an arbitrary recombination score (R-score) that ranges from 0 to 1, defined as R-score = ([number of methods inferring recombination with a P-value lesser than 0.01 × 6 + number of methods inferring recombination with a P-value greater than 0.01 but lesser than 0.05 × 3 + number of methods inferring recombination with a P-value greater than 0.05 but lesser than 0.10]/[24 × 6]). For each data set, a 50% consensus decision (yes or no) about the presence of recombination was obtained by considering whether the R-score was greater than 0.5. Because recombination was inferred in Fusarium3 by most methods, the 50% consensus for FusariumTriq101, which is a subset of the Fusarium3 data set, including the putative breakpoint, was also scored as yes. In addition, to assess the accuracy and false rate for each recombination method, a consensus power was calculated as the percentage of time a method inferred recombination for a data set with an R-score > 0.5 (i.e., the inference consensus for the data set was “yes” and the method inferred “yes”). A consensus false rate was also calculated as the percentage of time a method inferred recombination for a data set with an R-score < 0.5 (i.e., the inference consensus for the data set was “no” and the method inferred “yes”).
Given the statistics described previously, Geneconv, MaxChi2, Reticulate, and Sneath Test, were the most powerful methods, followed by Chimaera, Pist, Rdp, and Triple. Among the methods with least power were Homoplasy Test, RecPars, and Simplot. Regarding false positives, Chimaera, MaxChi2, PhyPro, Rdp, RecPars, and Simplot showed the lowest values (<0.10), whereas Pist, Runs Test, and Triple showed the highest false positive rates (>0.4). These results suggest that substitution and compatibility methods seem to be more powerful than the phylogenetic or the distance methods. Among all methods, MaxChi2 performed the best.
Recombination seems to be easier to detect, in general, with increasing levels of divergence. However, recombination was inferred in Candida-mtDNA by most recombination methods, even though there is not much variation in this data set (<1%). No relationship was found between the number of sequences, sites, segregating sites or informative sites, and power to detect recombination, but this is most likely a product of the low number of data sets evaluated. Nevertheless, there is a clear indication that the number of sites influences the power to detect recombination. A recombination event was suggested to have occurred in FusariumTri101 data set (O'Donnell et al. 2000 ), but all methods, except one (Geneconv) did not detect recombination in this data set. However, when two other genes (in which there is no evidence of recombination) situated on both sides of the Tri101 gene are added to build the Fusarium3 data set, most methods detect recombination. In fact, some methods identify the breakpoint in the Tri101 gene only in the Fusarium3 data set.
The detection patterns observed here are in good agreement with those observed in the computer simulations. The best performing methods here (considering power and false positive error rates) were also the best in previous computer simulations (Posada and Crandall 2001 ). Also, the worst performing method here (Runs Test) was the worst performing method in the computer simulations. Likewise, recombination methods seem to be more powerful with increased levels of divergence, both with simulated and real data sets. In addition, methods confounded by the presence of high rate variation among sites in simulations here inferred recombination from putative nonrecombinant data sets that showed high levels of rate among sites (e.g., Homoplasy Test with WolfCR).
Reevaluation of Previous Recombination Inferences
The results of the recombination analyses carried out in this study do not completely agree with previous inferences. In some data sets where recombination has been claimed (PetuniaS-RNase) or assumed to be absent (Candidula16S), inferences were very ambiguous, and the disagreement with previous studies cannot be easily elucidated. For other data sets, however, inferences here are congruent enough to suggest that previous beliefs should be reconsidered.
Although it has been claimed that recombination has occurred in MammPGK and MammPDH (Fitzgerald et al. 1996 ) on the basis of phylogenetic incongruence, the evidence for recombination in those data sets is very weak here, and it is only strongly suggested by a few methods (Pist, Plato, RecPars, Triple). Interestingly, all these methods are phylogenetic methods. For divergent data sets with some rate variation among sites (as in MammPGK and MammPDH), these phylogenetic methods showed some power without inferring false positives in the computer simulations (Posada and Crandall 2001 ). It is possible that the recombination events in these data sets are very old, and that the substitution pattern has been obscured since by repeated mutation; therefore, they are no longer identifiable by the substitution methods. However, the inference in the original paper has some potential problems. In particular, the authors measured phylogenetic incongruence by considering bootstrap support for different clades in different regions of the alignment. Bootstrap values are not designed to compare phylogenetic hypotheses (trees); for this purpose, adequate methods exist (Shimodaira and Hasegawa 1999 ). Using such methods in a maximum likelihood framework, the PDH trees regarded as different by Fitzgerald et al. (1996) do not seem to be significantly different. There is thus reasonable doubt about the presence of recombination in these data sets.
Recombination has also been claimed to be present in the Armillaria-mtDNA data set (Saville, Yoell, and Anderson 1996 ; Saville, Kohli, and Anderson 1998 ), whereas here only the Homoplasy Test, Plato, and the Sneath Test infer its presence. Armillaria-mtDNA is a low-divergence data set (<1%) with extreme rate variation among sites. It is precisely in this situation where the Homoplasy Test shows 90% false positives (Posada and Crandall 2001 ). However, Plato and the Sneath Test do not show an excess of false positives in computer simulations under these conditions, but they do infer recombination in some mitochondrial data sets where recombination seems very unlikely (e.g., Perom12S, InsectaCOII, VertebCOI). Of course, there may be some aspects of real data sets that may induce false positives that were not explored in the computer simulations, like nonindependence among sites. The presence of recombination in this data does not seem conclusive but cannot be discarded.
The analysis here suggests that recombination has occurred in the mitochondria of Boletales (BoletalesATP6). This finding is even more interesting when we consider that this data set is composed of divergent species. Recombination in the mitochondria of fungi also seems to be very evident in the Candida-mtDNA data set.
Although the HIV(B)EnvNR (only subtype B sequences) and HIVEnvNR (several subtypes represented) data sets do not include known circulating recombinant forms (http://hiv-web.lanl.gov/CRFs/CRFs.html), this analysis indicates that recombination is prevalent in those sequences. This finding suggests that the frequency of recombination in HIV-1 is currently underestimated, especially within subtypes. Indeed, this result has very serious implications for vaccine development and HIV-1 dynamics, and it implies that coinfection might be more common than currently thought. In addition, many past inferences based on HIV-1 evolutionary history assumed no recombination, especially when dealing with within-subtype sequences. A thorough analysis of the known HIV-1 sequences is imperative to evaluate the impact of recombination on our current understanding of this virus.
Evaluating Methods for Detecting Recombination
The performance of methods for detecting recombination could be evaluated using several criteria. Here the only criterion employed—and the most basic—was the ability to detect, qualitatively, the presence of recombination. Other possible performance criteria relate to the characterization of the recombination events, like the identification of the parental and recombinant sequences involved or the estimation of the recombinational breakpoints. However, these criteria are less adequate for the purpose of comparing these 14 recombination methods. First, there are not many well-characterized recombination events (but see Drouin et al. 1999 ). Second, several of the data sets studied here presumably have been subject to numerous, and overlapping, recombination events, making the characterization of each recombination event cumbersome. Third, not all methods evaluated here are designed to characterize the recombination events (see table 1 ).
Recombination methods detect recombination either directly (Yes-No answer; e.g., Bootscanning) or calculate a P-value for the null hypothesis of no recombination (P-value obtained from permutations of the data, from Monte Carlo simulation, or from a statistical distribution). To evaluate these methods I have used the criteria offered by each program, trying to reproduce the behavior of a researcher using these same programs or methods. In the case of RecPars, an arbitrary decision was made. The output of RecPars consists of one tree or more for different regions of the alignment. Here, recombination was inferred when more than one tree was estimated for a given alignment. This kind of statistic (0/1) is not very powerful, and it could explain why RecPars, as implemented here, did not detect recombination in the Candida-mtDNA or Fusarium3 data sets. It is possible that permutations of the data would be a reasonable approach to build a proper null distribution of this test statistic. However, the program RecPars does not currently provide this possibility. A potential problem with methods that simulate the Monte Carlo P-value (Plato and Pist) is that in the generated data sets the amount of diversity is not the same as in the original data set. This might help explain why these methods did not perform among the best.
Choosing a Method to Detect Recombination
A researcher planning to scan his or her data for recombination might select the most appropriate method, according to different criteria. One of the first things to consider is what the purpose of the study is. If the researcher just wants to detect the presence of recombination, any of the methods evaluated here could potentially be used. However, if the characterization of the recombination events (identification of parental and recombinant sequences or recombination breakpoints [or both]) is of interest, only a few methods are appropriate (see table 1 ). If the interest is to measure the amount of recombination, among those evaluated here only methods like the Homoplasy Test and Pist might be used. Second, the methods contemplated here are able to deal with alignments of homologous sequences. Therefore, whether the sequences analyzed are orthologs from the same locus or members of a multigene family seems irrelevant for the selection of a recombination method. Third, in some cases, a researcher could be interested only in analyzing two sequences where there are some reasons to believe that recombination or gene conversion has occurred. The minimum number of sequences that a method requires is indicated in table 1 . Fourth, in order to maximize performance, that is, increase power and decrease false positives, data sets with very low divergence (1%) could be analyzed with the Homoplasy Test (as long as a high level of rate variation among sites does not exist; this conclusion is also based on the computer simulations), MaxChi2, or Rdp. For higher levels of divergence (>1%), the Homoplasy Test is not adequate, and methods like MaxChi2 (the best performing method), Geneconv, Reticulate, or Rdp could be used (see also Posada and Crandall 2001 ). However, it does not seem that one should rely too much on a single method.
Brandon Gaut, Reviewing Editor
Keywords: detection of recombination mtDNA recombination HIV-1 recombination
Address for correspondence and reprints: David Posada, Variagenics, 60 Hampshire Street, Cambridge, Massachusetts 01239. dposada@variagenics.com .
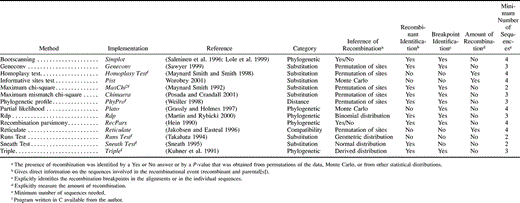
Table 2 Empirical Data Sets Evaluated for the Presence of Recombination (Available from the Author)
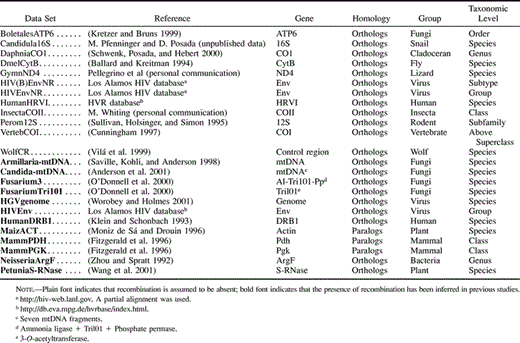
Table 2 Empirical Data Sets Evaluated for the Presence of Recombination (Available from the Author)
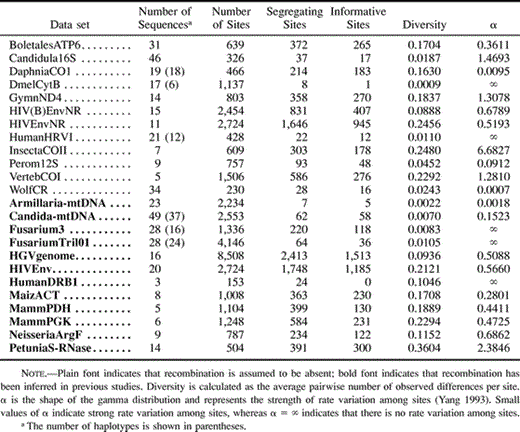
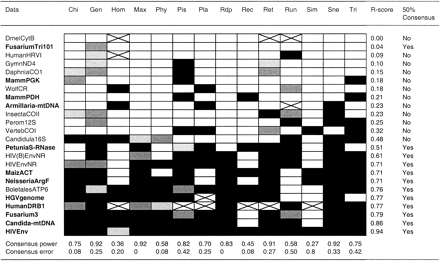
Fig. 1.—Recombination inference. Data sets in plain font indicate that recombination was assumed to be absent. Data sets in bold font indicate that recombination has been suggested. Cells in black indicate recombination was detected. Dark gray indicates that recombination was detected but with a P-value marginally significant (0.05 > P-value > 0.01). Light gray indicates recombination was not detected but the P-value was marginally nonsignificant (0.10 > P-value > 0.05). Cells in white indicate that recombination was not detected. Crossed cells indicate that inference was not possible. Data sets were ordered by the strength of the recombination inference, from least to most, using an arbitrarily defined recombination score (R-score). The R-score, the 50% consensus, and the consensus power and false positive error rates are described in the text. Methods abbreviations are—Chi: Chimaera; Gen: Geneconv; Hom: Homoplasy Test; Max: MaxChi2; Phy: PhyPro; Pis: Pist; Pla: Plato; Rdp: Rdp; Rec: RecPars; Ret: Reticulate; Run: Runs Test; Sim: Simplot; Sne: Sneath Test; Tri: Triple
Thanks to Keith A. Crandall for discussion. Brandon Gaut and two anonymous reviewers provided very helpful comments. Special thanks to Darren Martin and Stuart Ray for modifying their programs. Thanks to Andrew Rambaut and Mike Worobey for giving access to Pist before it was published and to Mary Kuhner for providing the Triple fortran code. Mike Whiting, Katia Pellegrino, Markus Pfenninger, Mike Worobey, Jim Anderson, and Kerry O'Donnell generously sent their data sets. This work was supported by a Brigham Young University Graduate Studies Award and by a NSF Doctoral Dissertation Improvement Grant (NSF DEB 0073154).
References
Anderson J. B., C. Wickens, M. Khan, L. E. Cowen, N. Federspiel, T. Jones, L. M. Kohn,
Ballard J. W. O., M. Kreitman,
Brown C. J., E. C. Garner, K. A. Dunker, P. Joyce,
Cunningham C. W.,
Drouin G., F. Prat, M. Ell, G. D. Paul Clark,
DuBose R. F., D. E. Dykhuizen, D. L. Hartl,
Fitzgerald J., H.-H. M. Dahl, I. B. Jakobsen, S. Easteal,
Grassly N. C., E. C. Holmes,
Hein J.,
———.
Hillis D. M., J. P. Huelsenbeck,
Hillis D. M., B. K. Mable, C. Moritz,
Holmes E. C., M. Worobey, A. Rambaut,
Jakobsen I. B., S. Easteal,
Jakobsen I. B., S. E. Wilson, S. Easteal,
Klein J., C. Schonbach,
Kretzer A. M., T. D. Bruns,
Kuhner M. K., D. A. Lawlor, P. D. Ennis, P. Parham,
Lole K. S., R. C. Bollinger, R. S. Paranjape, D. Gadkari, S. S. Kulkarni, N. G. Novak, R. Ingersoll, H. W. Sheppard, S. C. Ray,
Maddison D. R., W. P. Maddison,
Martin D., E. Rybicki,
Maynard Smith J., N. H. Smith,
Moniz de Sá M., G. Drouin,
O'Donnell K., H. C. Kistler, B. K. Tacke, H. H. Casper,
Posada D., K. A. Crandall,
———.
Salminen M. O., J. K. Carr, D. S. Burke, F. E. McCutchan,
Saville B. J., Y. Kohli, J. B. Anderson,
Saville B. J., H. Yoell, J. B. Anderson,
Sawyer S. A.,
Schwenk K., D. Posada, P. D. N. Hebert,
Shimodaira H., M. Hasegawa,
Sneath P. H. A.,
———.
Sneath P. H. A., M. J. Sackin, R. P. Ambler,
Stephens J. C.,
Sullivan J., K. E. Holsinger, C. Simon,
Swofford D. L.,
Swofford D. L., G. J. Olsen, P. J. Waddell, D. M. Hillis,
Takahata N.,
Vilá C., I. R. Amorim, J. A. Leonard, D. Posada, J. Castroviejo, F. Petruccci-Fonseca, K. A. Crandall, H. Ellegren, R. K. Wayne,
Wang X., A. L. Hughes, T. Tsukamoto, T. Ando, T. Kao,
Weiller G. F.,
Wiuf C., T. Christensen, J. Hein,
Worobey M.,
Worobey M., E. C. Holmes,
Yang Z.,

{kind=link}